

Abstract
Transglutaminases (TGases) catalyze the cross-linking of peptides and proteins by the formation of γ-glutamyl-ɛ-lysyl bonds. Given the implication of tissue TGase in various physiological disorders, development of specific tissue TGase inhibitors is of current interest. To aid in the design of peptide-based inhibitors, a better understanding of the mode of binding of model peptide substrates to the enzyme is required. Using a combined kinetic/molecular modeling approach, we have generated a model for the binding of small acyl-donor peptide substrates to tissue TGase from red sea bream. Kinetic analysis of various N-terminally derivatized Gln-Xaa peptides has demonstrated that many CBz-Gln-Xaa peptides are typical in vitro substrates with KM values between 1.9 mM and 9.4 mM, whereas Boc-Gln-Gly is not a substrate, demonstrating the importance of the CBz group for recognition. Our binding model of CBz-Gln-Gly on tissue TGase has allowed us to propose the following steps in the acylation of tissue TGase. First, the active site is opened by displacement of conserved W329. Second, the substrate Gln side chain enters the active site and is stabilized by hydrophobic interaction with conserved residue W236. Third, a hydrogen bond network is formed between the substrate Gln side chain and conserved residues Y515 and the acid-base catalyst H332 that helps to orient and activate the γ-carboxamide group for nucleophilic attack by the catalytic sulphur atom. Finally, an H-bond with Y515 stabilizes the oxyanion formed during the reaction.
Keywords: tissue transglutaminase, molecular modeling, docking, enzyme kinetics, peptide substrate, autodock, TGase
Transglutaminases (TGases, EC 2.3.2.13) are enzymes that catalyze the cross-linking of peptides and proteins by the formation of isopeptide bonds between the γ-carboxamide group of a glutamine side chain and the ɛ-amino group of a lysine side chain. This reaction is known to occur via a modified ping-pong mechanism (Folk 1969) in which a glutamine-containing protein or peptide, the acyl-donor substrate, reacts with the enzyme’s catalytic cysteine residue to form a thioester bond which generates the covalent acyl-enzyme intermediate with concomitant release of ammonia (Scheme 1 ▶). This intermediate then reacts with a second substrate, the acyl-acceptor, which can be almost any primary amine (Aeschlimann and Paulsson 1994), to yield the amide product and free enzyme. The acyl-enzyme intermediate can also be hydrolyzed in the absence of primary amine but at a slower rate. A conserved Cys-His-Asp catalytic triad that is similar to that of cysteine proteases catalyzes these reactions. Although TGases exhibit high specificity towards the side chain of L-Gln as the acyl-donor substrate (Asn and D-Gln are not recognized), their specificity towards the acyl-acceptor is lower, and many primary amines can be recognized (Clarke et al. 1959; Folk 1983).
Scheme 1.

Catalytic mechanism of tissue TGase.
TGases are divided into nine classes (Chen and Mehta 1999), of which the most abundant class is the ubiquitous tissue transglutaminase (Fesus and Piacentini 2002) found in all vertebrates (Wada et al. 2002). Tissue TGases are intracellular, monomeric enzymes that exhibit a calcium-dependent transglutaminase activity that is inhibited by GDP/GTP binding. Their structure comprises four domains (Fig. 1 ▶): the N-terminal β-sandwich domain, the catalytic core, the barrel 1 domain, and the C-terminal barrel 2 domain. Two crystal structures of tissue transglutaminase have been deposited in the Brookhaven Protein Data Bank. These are the structures of red sea bream tissue TGase (1G0D; Noguchi et al. 2001) and of human tissue transglutaminase with GDP bound at its allosteric binding site (1KV3; Liu et al. 2002). However, no TGase has yet been cocrystallized with ligand bound at the active site, limiting our understanding of the mode of substrate binding, although kinetic experiments have been performed that give clues as to the structural requirements for acyl-donor and acyl-acceptor substrates of tissue TGase (Clarke et al. 1959; Folk and Chung 1973). Folk and Cole (1965) made numerous observations with respect to the structural requirements for small peptide acyl-donor substrates of guinea pig liver TGase. They reported that Gln alone does not act as a substrate, nor do the peptides Gln-Gly and Gly-Gln-Gly. However, CBz-Gln-Gly, CBz-Gln-Gly ethyl ester, and benzoyl-Gly-Gln-Gly act as substrates. CBz-Gln shows poor but detectable activity. Folk and Cole concluded that for a glutamine on a small peptide to be recognized by tissue TGase it must be at least the next-to-last residue and the peptide should bear an N-terminal CBz group. For this reason, the commercially available CBz-Gln-Gly peptide serves as one of the most common nonproteic acyl-donor substrates for TGases in the literature.
Figure 1.

Crystal structure of red sea bream TGase (Noguchi et al. 2001). (A) Red sea bream TGase with active site residues circled. Front view. The P356–G369 loop is represented in ribbon diagram. (B) Top view.
Although there is no consensus as to their physiological role, tissue TGases have been reported to act in endocytosis (Abe et al. 2000), apoptosis (Huo et al. 2003), extracellular matrix modification (Priglinger et al. 2003), and cell signaling (Singh et al. 2003). Tissue TGases have also been implicated in various physiological disorders such as Alzheimer’s disease (Kim et al. 1999), cataract formation (Shridas et al. 2001), and more recently, in Celiac sprue (Shan et al. 2002). As a result, development of inhibitors specific to tissue TGases (as opposed to type 1 or 3 TGases; Chen and Mehta 1999) is of current interest. However, to aid in inhibitor design, we need to gain a better understanding of the mode of binding of model peptide substrates to the enzyme. To this end, we performed a combined experimental/molecular modeling strategy consisting of (1) the synthesis of a series of CBz- or Boc-derivatized peptides and the measurement of their KM values, and (2) molecular modeling of their binding at the active site. Correlating the kinetic results with molecular modeling of the ligands bound at the active site cavity has allowed us to gain a better understanding of the requirements for productive binding of small peptide acyl-donor substrates.
Results
Kinetic characterization of the N-terminally derivatized peptides
The synthesis of the derivatized peptide substrates has been described (Gagnon et al. 2002). The kinetic parameters KM and kcat of guinea pig liver TGase were determined for a series of CBz-derivatized Gln-Xaa peptides as well as for the Boc-derivatized Gln-Gly dipeptide ( Table 1). The seven CBz-peptides assayed gave similar KM and kcat values, whereas the Boc-dipeptide did not appear to act as a donor substrate, showing no reactivity at concentrations up to 40 mM, the limit of its solubility. The similar KM and kcat values obtained for all the CBz-derivatized Gln-Xaa peptides indicate that the identity of the C-terminal amino acid does not significantly affect productive binding or turnover. Thus, these substrates result in a similar catalytic efficiency. Because amino acids having side chains with widely differing chemical and structural attributes were assayed and because we observed that the nature of these side chains does not affect substrate recognition or efficiency, we can conclude that substrate binding does not depend on the nature of the C-terminal amino acid. However, the nature of the N-terminal functional group, Boc or CBz, has an important effect on substrate recognition by the enzyme. Namely, Boc-Gln-Gly does not serve as a donor substrate, whereas Folk and Cole (1966) observed that CBz-Gln does act as a substrate of tissue type TGase, albeit a poor one, although Gln-Gly does not. Our kinetic data confirm that the presence of the CBz group contributes importantly to recognition of dipeptide substrates by tissue TGase, presumably by allowing better binding to the enzyme.
Table 1.
Kinetic constants for guinea pig liver TGase with various N-terminally derivatized peptides
| Substrate | kcat (min−1)a | KM (mM) | kcat/KM (min−1 mM−1)a |
| CBz-Gln-Gly | 96 | 3.2 ± 0.5 | 30 |
| CBz-Gln-Ala | 51 | 1.9 ± 0.6 | 26 |
| CBz-Gln-Val | 71 | 4.4 ± 0.4 | 16 |
| CBz-Gln-Leu | 127 | 7 ± 1 | 18 |
| CBz-Gln-Phe | 54 | 2.7 ± 0.8 | 20 |
| CBz-Gln-Ser | 66 | 2.9 ± 0.6 | 23 |
| CBz-Gln-Gly-Gly | 102 | 9.4 ± 0.5 | 11 |
| Boc-Gln-Gly | — | >40b | — |
a Values determined using the extinction coefficient measured for the CBz-Gln-Gly anilide. The standard deviation on these values is ±20%.
b No reaction was observed for concentrations up to 40 mM, the limit of solubility for this dipeptide.
Modeling of substrate binding
To obtain more information regarding the mode of binding of acyl-donor substrates on tissue TGase, we attempted to model substrate binding using the coordinates of the crystal structure of tissue TGase from red sea bream that were obtained at 2.5 Å resolution. This structure, although lacking the activating calcium ion, was chosen over the crystal structure of human tissue TGase because the latter was cocrystallized with GDP and is therefore in the inactive state (Liu et al. 2002). The percentage of identity of the catalytic domains of red sea bream TGase with guinea pig liver TGase is >55%, and the percentage of homology is ~70%. Furthermore, all of the active site residues (W236, C272, H300, W329, H332, and Y515 of red sea bream tissue TGase; Fig. 1 ▶) are conserved in all sequenced tissue TGases, justifying the use of the coordinates of fish transglutaminase for our modeling in the absence of coordinates for the guinea pig liver TGase. As a first step toward modeling the acyl-donor substrate binding on tissue TGase, we created all of the substrates given in Table 1 in silico with the BIOPOLYMER module of InsightII, and their structures were energy-minimized. All adopted a “Y”-shaped extended conformation, with the vertical line representing the Gln side chain and the two arms pointing upwards being the peptide backbone, with the CBz or Boc group on one side and on the other, the C-terminal amino acid. These conformations were used as starting structures for automated and manual docking at the active site of tissue TGase.
The crystal structure of red sea bream TGase presents a shallow, narrow cleft running diagonally on its surface, passing over the active site (Fig. 2 ▶). We identified this cleft as the possible binding site of the small peptide substrate’s backbone and N-terminal functional group, as it permitted maximal Gln side chain insertion into the active site, bringing the γ-carboxamide group into close proximity with the catalytic triad. Visual inspection suggests that it would be difficult for it to be located in any significantly different manner because of steric clashes.
Figure 2.

Proposed binding cleft for the peptide substrate N-terminal functional group at the surface of red sea bream TGase. The diagonal cleft running over the active site cavity is in yellow; the Gln side chain of the CBz-Gln-Gly substrate that enters the active site cavity away from the viewer is represented by ×. Negative charges are in red; positive are in blue. The four positions tested for manual docking of the N-terminal CBz group are numbered. The CBz group is drawn to scale.
To test this hypothetical binding mode, we performed automated docking experiments using AutoDock 3.0 software (Morris et al. 1998). Preliminary docking experiments were performed using the PDB file 1G0D. We observed that the CBz-Gln-Gly and Boc-Gln-Gly substrates never entered the active site during the 50 runs performed. Indeed, the active site of the enzyme is visibly blocked by a Trp residue, W329 (Fig. 1 ▶) which is conserved in all tissue TGases as well as in Factor XIII (Yee et al. 1994), epidermal (Ahvazi et al. 2002), and keratinocyte TGases. This apparently results from the structure having been resolved in the absence of substrate. Because automated docking is performed without movement of the protein atoms, it is unlikely to succeed in identifying a binding mode for the substrate Gln side chain inside the active site. Thus, we manually increased the accessibility of the active site of the structure of red sea bream TGase by introducing torsions in the Cα–Cβ (−83.13°) and Cβ–Cγ (+48.52°) bonds of residue W329. These torsions opened the active site cavity while minimizing steric clashes between W329 and the rest of the protein. With this starting structure, the AutoDock procedure resulted in several structures with the substrate Gln entering the active site (Table 2). This indicates that W329 may prevent entry to the active site, acting as a ‘gate’ that could regulate TGase activity. Of the 50 runs performed with CBz-Gln-Gly, only four generated a structure where the substrate’s Gln side chain had entered the active site (Table 2). This low number of hits is expected because the substrate was allowed a high degree of conformational flexibility. Three of the four bound structures positioned the CBz group at position 0; the last one positioned the CBz at position 1 (Fig. 2 ▶). With the Boc-Gln-Gly substrate, only six of the 50 runs generated a structure where the Gln side chain had entered the active site. Of these six structures, two had the Boc at position 2, three at position 0, and one at position 1. It should be noted that these positions are not precisely defined but represent restricted zones within which positioning was observed. These AutoDock results allowed us to establish three potential binding sites at the surface of the enzyme for the N-terminal functional group. Position 0, which is in the diagonal cleft, appears to be favored for binding. This suggests that positioning of the N-terminal functional group in the cleft is favorable to productive binding of the Gln side chain inside the active site cavity. The low number of runs that resulted in a structure where Gln was bound in the active site reflects the generally weak affinity of tissue TGase for these dipeptide substrates. This in turn is manifested in their millimolar KM values. We also observed that after minimization of these 10 structures using InsightII, 75% (3/4) of the CBz-Gln-Gly structures formed an H-bond with conserved residue Y515 of the active site, whereas only 16% (1/6) of the Boc-Gln-Gly structures had formed this H-bond ( Table 2).
Table 2.
Automated docking results for the docking of CBz-Gln-Gly and Boc-Gln-Gly on red sea bream TGase
| Ligand | Na | Interaction energyb (kcal/mole) | FG positionc | H-bond with Y515 |
| CBz-Gln-Gly | 4 | −56.6 | 1 | √ |
| −21.9 | 0 | × | ||
| +23.3 | 0 | √ | ||
| −8.4 | 0 | √ | ||
| Boc-Gln-Gly | 6 | −19.9 | 2 | × |
| +10.8 | 0 | × | ||
| +3.1 | 0 | × | ||
| −13.9 | 0 | √ | ||
| −6.3 | 2 | × | ||
| −58.1 | 1 | × |
a Number of runs out of 50 that generated docking of the substrate with the Gln side chain entering the active site.
b Total interaction energy between the enzyme and the ligand after minimization (see Materials and Methods).
c N-terminal functional group position according to Fig. 2 ▶.
To further test the hypothesis that the binding site of the N-terminal functional group is in the cleft defined by positions 0 and 2 and to identify the preferred position for the N-terminal functional group in the cleft, we manually docked the acyl-donor substrate at four different positions on the enzyme. First, the reacting Gln side chain was positioned inside the active site respecting the conditions described in the Materials and Methods section, while the N-terminal functional group was positioned in one of the four different positions on the surface of the enzyme (Fig. 2 ▶). These positions place the N-terminal functional group alternatively in the cleft (positions 0 and 2), in a small cavity perpendicular to the cleft (position 1), or directly on top of residue W236 (position 3). Steric clashes were more important in position 3, which was chosen as a negative control for the molecular modeling procedure. Then, steric clashes between the enzyme and the substrate were energy-minimized while the enzyme/substrate distance was constrained, as described in Materials and Methods. This caused the active site to ‘open up’ as residue W329 was displaced, indicating that this residue is very mobile, because it readily moves away from the active site after merely a minimization.
A 10-picosecond molecular dynamics simulation was then performed on the generated enzyme-substrate complexes to explore the immediate conformational space. One hundred conformers were thus generated. The conformer with the lowest energy was further energy-minimized, and the resulting structure was analyzed. This resulting structure with each substrate was always of lower energy than the structure generated before the molecular dynamics simulation. The backbone of the enzyme was maintained fixed during Trial 1 of manual docking (Table 3) to determine the most plausible binding site for the N-terminal functional group in order for the small CBz-Gln-Gly substrate to adopt a conformation that best fit the enzyme’s structure and not the opposite. This experiment was also performed two more times with varying conditions, including independent manual substrate positioning, a 25 Å or 40 Å layer of water covering the active site, and with no constraint on the enzyme backbone, which generated similar results (Table 3). In all three independent trials with CBz-Gln-Gly, the enzyme-substrate interaction energy was the lowest and was consistently negative when the CBz was positioned in the upper left part of the cleft (position 0; Fig. 2 ▶), indicating that it is the only one of the four positions tested that is energetically favorable and reproducible for binding the CBz group for a small peptide substrate. Thus, it appears that the preferred position for docking the CBz group on the surface of the enzyme is located in the upper left corner of the diagonal cleft (position 0). This position also allows better insertion of the reacting Gln side chain into the active site based on the anti-conformation of the Gln side chain in the active site compared to the other three positions (data not shown). Manual docking experiments therefore gave us similar results to those obtained with automated docking, while providing additional insights into the preferred position of the N-terminal functional group. The remainder of the docking experiments were performed with manual docking of the substrates on tissue TGase.
Table 3.
Manual docking results for CBz-Gln-Gly on red sea bream TGase
| Trial | FG positiona | Interaction energyb (kcal/mole) |
| 1c | 0 | −21.1 |
| 1 | +36.5 | |
| 2 | +48.0 | |
| 3 | +94.4 | |
| 2d | 0 | −32.9 |
| 1 | +38.2 | |
| 2 | −26.6 | |
| 3 | −30.0 | |
| 3e | 0 | −22.8 |
| 1 | +28.8 | |
| 2 | +15.1 | |
| 3 | +74.8 |
a N-terminal functional group position as defined in Fig. 2 ▶.
b Total interaction energy between the enzyme and the ligand after minimization (see Materials and Methods).
c 25 Å layer of water covering the active site; protein backbone was fixed.
d 25 Å layer of water covering the active site; no fix on the protein backbone.
e 40 Å layer of water covering the active site; no fix on the protein backbone.
Following the docking studies on the putative positioning of the CBz group using CBz-Gln-Gly, binding of the CBz-Gln-Xaa substrates was modeled with the N-terminal functional group in position 0. The Boc-Gln-Gly substrate was modeled as a control to ensure that the modeling results are consistent with its lack of reactivity as a substrate (Table 1). For each test, the substrate was manually positioned in an independent manner. During the calculations, only the residues including atoms within 20 Å of the catalytic Sγ atom of C272, the water molecules, and the substrate were mobile. Constraints were applied as described in Materials and Methods to retain the substrate Gln proximal to the catalytic residues, thus increasing the likelihood of modeling productive enzyme/substrate complexes. After the minimization/molecular dynamics methodology was performed, the interaction energy between the enzyme and each substrate was analyzed. For the CBz-Gln-Gly substrate, analyzed in 13 tests, we found that the interaction energy (ΔG) between ligand and protein was negative, thus favorable, in all but one case ( Table 4). In the single case where the interaction energy was positive, the electrostatic component of the interaction energy between enzyme and substrate was very high (+158.9 kcal/mole). In this case, the carboxylate of CBz-Gln-Gly was found to be in closer proximity than usual to a conserved, negatively charged residue of red sea bream TGase, Glu360, which could result in such an unfavorable electrostatic energy. Nonetheless, the average of the 13 data sets suggests that there is a favorable interaction between the enzyme and the CBz-Gln-Gly substrate. The average van der Waals energy between the enzyme and the CBz-Gln-Gly ligand was −53 ± 5 kcal/mole. While examining the interactions between the enzyme and CBz-Gln-Gly, we found that each of the 13 structures generated showed an H-bond between the Oη proton of Y515 and the Oδ of Gln from CBz-Gln-Gly. Also, 77% of the structures (10/13) displayed an H-bond between Nɛ of H332 and Hɛ of Gln side chain. The proximity and orientation displayed in this H-bond pairing resembles the interaction that takes place during catalysis when the acid/base catalyst, H332, protonates the ammonia leaving group. The high prevalence of these H-bonds suggests that they are important in binding the glutaminyl residue at the active site.
Table 4.
Manual docking results for CBz-Gln-Gly, CBz-Gln-Xaa, and Boc-Gln-Gly
| Ligands | H-bond with Y515 (%) | H-bond with H332 (%) | van der Waals E (kcal/mole) | E ≥ 0 kcal/molea (%) |
| CBz-Gln-Gly | 100 (13/13) | 77 (10/13) | −53 ± 5 | 8 (1/13) |
| CBz-Gln-Xaab | 100 (7/7) | 86 (6/7) | −60 ± 5 | 0 (0/7) |
| Boc-Gln-Gly | 69 (9/13) | 62 (8/13) | −44 ± 4 | 38 (5/13) |
Ligands were positioned with the N-terminal functional group at position 0, as defined in the text.
a Total interaction energy between the enzyme and the ligand that is positive, thus unfavorable.
b Xaa = Ala, Val, Leu, Phe, Ser, or Gly-Gly. Ala was modeled twice.
For the CBz-Gln-Xaa substrates (Xaa = Ala, Val, Leu, Phe, Ser, or Gly-Gly), we obtained similar results. Interaction energies between the enzyme and the substrates were negative and thus favorable for all substrates, independently of the side chain of the C-terminal amino acid. Also, all of these molecules formed the same H-bond between the proton of Oη of Y515 and the Oδ of Gln, as observed for CBz-Gln-Gly, whereas 86% (6/7) of them formed an H-bond between Nɛ of H332 and Hɛ of the Gln side chain. These results indicate that the nature of the second amino acid does not significantly affect efficiency of binding to tissue TGase, which correlates well with the kinetic data from Table 1.
To evaluate the contribution of the N-terminal functional group to substrate binding, we compared the binding of Boc-Gln-Gly and CBz-Gln-Gly to tissue TGase. The Boc-Gln-Gly peptide was manually docked similarly to the CBz-Gln-Gly peptide with the N-terminal functional group at position 0 or position 2. The results at position 2 were similar to those for CBz-Gln-Gly at position 2; that is, they were of considerably higher energy and were not pursued (data not shown). The binding of Boc-Gln-Gly at position 0 did not generate reproducible results as CBz-Gln-Xaa did. Indeed, some results generated poor interaction energy between enzyme and substrate, whereas others gave as good an interaction as the CBz peptides. Of the 13 trials, 38% (5/13) generated a positive interaction energy, indicating unfavorable binding. Furthermore, the H-bond between the Boc-Gln-Gly substrate and Y515 was found in only 69% (9/13) of the trials, whereas the H-bond with H332 was found in 62% of the trials (8/13), which is significantly less than for the CBz-derivatized peptides. In addition, the average van der Waals energy is −44 ± 4 kcal/mole, which is more than 10 kcal/mole higher than for the average of all CBz-derivatized peptides (CBz-Gln-Gly and CBz-Gln-Xaa). Taken together, these results suggest that the presence of the Boc group does not allow for binding that is as constant and reproducible as for the CBz-Gln-Xaa substrates. It thus appears that the Boc group is deleterious to proper binding, because it does not allow a constant productive insertion of Gln side chain into the active site.
Modeling of the tetrahedral and acyl-enzyme intermediate
To minimize any potential bias arising from the manual docking undertaken during the modeling study and to investigate more thoroughly the importance of the H-bond network with Y515 and H332 identified for the binding of the CBz-peptides, we created in silico the tetrahedral and acyl-enzyme intermediates for the reaction of tissue TGase with CBz-Gln-Gly. These intermediates have a covalent bond between the Cδ atom of Gln from the donor substrate and Sγ of the catalytic Cys residue, thus eliminating the requirement for manual docking of the substrate. The acyl-enzyme and tetrahedral intermediates were constructed respecting the χ1–χ2 and χ2–χ3 plots dihedral angles (Janin et al. 1978) for the substrate Gln and C272. Two starting structures for the acyl-enzyme intermediate were constructed, one with the CBz group at position 0 and another at position 2. These dihedral angles still respected the χ1–χ2 and χ2–χ3 plots at the end of the simulation, indicating that the generated structure was coherent with regards to the conformation of the two residues involved in the covalent bond between enzyme and substrate (data not shown). After the simulation, a lower energy for the modeled system was obtained for the acyl-enzyme intermediate with the CBz group at position 0 compared to position 2 (−4513 kcal/mole versus −4461 kcal/mole). This is consistent with the hypothesis that position 0 represents the actual CBz binding site because it is energetically more favorable.
While analyzing the covalent structures generated, we observed that the acyl portion of the molecule adopted a conformation very similar to the model of TGase with the noncovalently docked substrate. Indeed, the RMSD of the Gln side chains of CBz-Gln-Gly in the Michaelis complex and the acyl portion of the acyl-enzyme and tetrahedral intermediates are very low (Table 5), indicating near-identical positioning of the Gln side chain inside the active site for these three modeled structures (Fig. 3A ▶–C). Also, the dihedral angles χ1 and χ2 are nearly identical among the three structures, which demonstrates that the conformation of the Gln side chain in the Michaelis complex is the same as in the tetrahedral and acyl-enzyme intermediates. This suggests that the insertion of the Gln side chain of the docked substrate is coherent and thus there was no bias in positioning during manual docking. The H-bond between the reactive Gln γ-carboxamide group and the enzyme Y515 residue was present in the acyl-enzyme intermediate. The H-bond between Nɛ of H332 and Hɛ of Gln side chain was not present because the Gln side chain in the acylenzyme intermediate no longer carries the NH2 group.
Table 5.
Comparison of the conformation of the Gln side chain of the acyl-donor substrate CBz-Gln-Gly in the modeled Michaelis complex, acyl-enzyme, and tetrahedral intermediates
| RMSD of Gln side chain (Å) | Dihedral angles of Gln | ||||
| Structure | Acyl-enzyme intermediate | Tetradedral intermediate | Michaelis complex | χ1 (°) | χ2 (°) |
| Acyl-enzyme intermed. | — | 0.12 | 0.10 | −175 | +172 |
| Tetrahedral intermed. | 0.12 | — | 0.02 | −178 | −169 |
| Michaelis complex | 0.10 | 0.02 | — | −176 | −171 |
Figure 3.
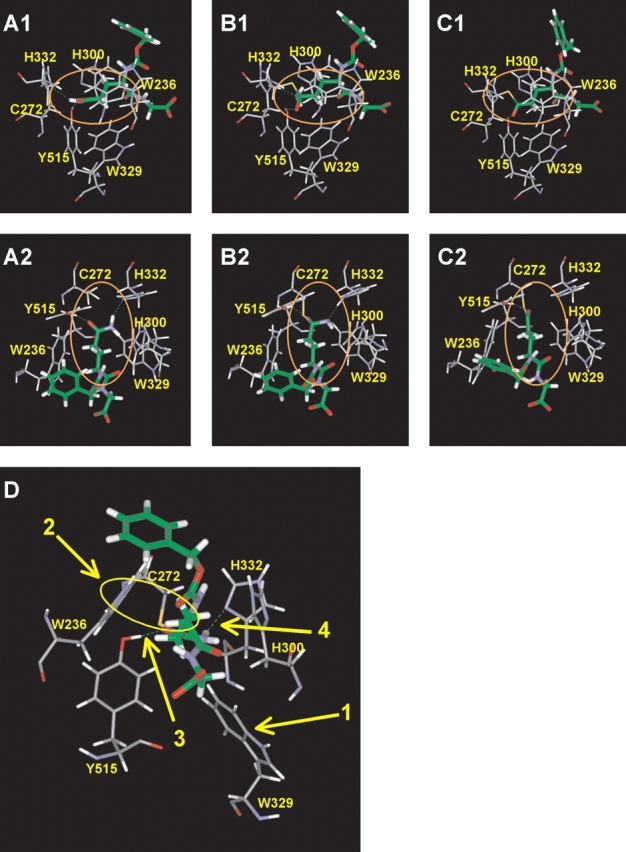
Model of TGase with the acyl-donor CBz-Gln-Gly. (A) Michaelis complex, (B) tetrahedral intermediate, and (C) acyl-enzyme intermediate. A1–C1, side view. A2–C2, top view. In C2, the NH3 molecule has been released. (D) Interactions proposed to be important in the modeled Michaelis complex of TGase with CBz-Gln-Gly: (1) Displacement of W329 toward the bottom to allow entrance of substrate Gln side chain into the active site; (2) Hydrophobic interaction of the indole group of W236 with the methylene groups of the Gln side chain; (3) H-bond of the Gln γ-carboxamide with Y515; (4) H-bond of the Gln γ-carboxamide with H332.
There are two H-bonds in the tetrahedral intermediate structure (Fig. 3B ▶) that are similar to the H-bonds between CBz-Gln-Gly and the enzyme in the Michaelis complex. The first H-bond occurs between the oxyanion generated by nucleophilic attack and the hydroxyl function of Y515. The second occurs between Nɛ of Gln and the acidic proton of protonated H332. This is consistent with the known mechanism of the enzyme, where the leaving group NH2 is protonated by the imidazolium group of H332. The H-bond formed between the oxyanion and Y515 could significantly stabilize the tetrahedral intermediate. These results, along with the docking study of CBz- or Boc-derivatized peptides, support the importance of these H-bonds.
Structural analysis of CBz-peptide binding
The 13 structures generated by molecular dynamics following manual docking of CBz-Gln-Gly peptides and the seven structures generated with the CBz-Gln-Xaa substrates were analyzed as a group. The Gln side chain enters the active site almost perpendicularly with respect to the center of mass of the enzyme. Figure 3D ▶ illustrates one of the trial structures, chosen because it was typical and representative. The conformation of the substrate Gln side chain is the low-energy anti-conformation. The χ1 and χ2 dihedral angles of the substrate Gln are near 180°, which gives a trans-conformation to the side chain that is common for amino acids. Three aromatic residues, W236, W329, and H300 that appear to form a tunnel leading to the catalytic residues, surround the side chain. As previously discussed, residue W329 is displaced by the insertion of the substrate Gln side chain into the active site so as to open the active site that it blocks in the unbound state. This displacement places the W329 side chain in a position very similar to that achieved manually as part of the automated docking experiments (Cα − Cβ = −95.53° and Cβ − Cγ = +82.4°). The roof of the tunnel is formed by a loop of residues P356–G369 (Fig. 1 ▶). The two methylene groups of the Gln side chain of the substrate are in contact with the indole group of residue W236. This maximizes the hydrophobic interaction between the indole group of W236 and the methylene groups of the substrate Gln side chain during the course of the simulation—as evidenced by the concerted movement of these two groups during the dynamics simulation. The substrate γ-carboxamide, as mentioned earlier, adopts a conformation allowing it to form H-bonds with residues Y515 and H332. In this conformation, the nucleophilic attack by the thiolate can only be achieved from the top or the bottom of the substrate γ-carboxamide, in keeping with an addition-displacement mechanism. If the attack occurs from the top, the oxyanion generated could form a stabilizing H-bond with the hydroxyl group of Y515. Such a bond is observed in the model of the tetrahedral intermediate.
Discussion
No existing crystal structures of tissue TGase (Noguchi et al. 2001; Liu et al. 2002) have been resolved with ligand bound at the active site, limiting our understanding of the binding of acyl-donor substrates. The only structural elements that have been shown to be important for productive binding of small peptide acyl-donor substrates are (1) the length of the side chain (Asn versus Gln), (2) the stereochemistry of the Gln side chain (D-Gln versus L-Gln), (3) the length of the peptide substrate (Clarke et al. 1959; Folk 1983), and (4) to a lesser extent, the position of the reactive Gln in the peptide/protein sequence (Ohtsuka et al. 2000). Thus, it is known that for small acyl-donor peptide substrates, the reactive Gln residue must be at least the second residue from the C terminus and the third from the N terminus (Folk and Cole 1965), where CBz can replace the first two amino acids. Molecules such as Gln and Gln-Gly do not act as substrates, whereas CBz-Gln-Gly is the most widely used substrate.
To gain insight into the binding of small peptide acyl-donor substrates on tissue TGase, we used a combined kinetic/molecular modeling approach. Kinetic tests were performed on guinea pig liver transglutaminase, because it is the most extensively characterized tissue TGase. Molecular modeling was performed on the crystal structure of red sea bream tissue TGase. The catalytic core domain of this enzyme has a sequence homology of ~70% to that of guinea pig liver, and all but four residues composing the cleft (Fig. 2 ▶) are conserved between the two enzymes, and thus the two structures can be considered very similar. All of the active site residues (W236, C272, H300, W329, H332, and Y515 of red sea bream tissue TGase) are conserved in all tissue TGases, including the human enzyme, of which the catalytic domain has 58% identity and 70% homology with the red sea bream enzyme. This justifies the use of the coordinates of red sea bream transglutaminase for our modeling in the absence of coordinates for the guinea pig liver TGase, as well as the pertinence of these studies to future drug design. Furthermore, CBz-Gln-Gly is an efficient acyl-donor sub-strate of both red sea bream TGase and guinea pig liver TGase (Ohtsuka et al. 2000) and is therefore appropriate for docking studies on this structure.
Molecular modeling suggests that there is a hydrophobic interaction between the indole group of the side chain of W236 and the methylene groups of the Gln side chain of the substrate. Molecular dynamics simulations also confirm the concerted movement of these two groups. Residue W236 (W241 in guinea pig liver TGase and human tissue TGase) is conserved in all eukaryotic TGases that have been sequenced. In site-directed mutagenesis studies of human tissue TGase, replacement of W241 with Ala, Gln, His, Phe, or Tyr resulted in a substantial loss of activity, demonstrating the importance of this Trp to catalysis (Murthy et al. 2002). Significantly, the mutants W241H, W241F, and W241Y, possessing aromatic side chains, showed low but readily measurable activities of approximately one-tenth that of wild type and around 20-fold higher than that of the non-aromatic mutants W241A and W241Q (Murthy et al. 2002). In consideration of these results and the contacts revealed by our modeling studies of red sea bream TGase, we propose that the acyl-donor substrate Gln residue is stabilized in the active site through hydrophobic interactions of the methylene groups of the substrate side chain with the aromatic indole group of residue W236. This could partly account for the fact that peptide- or protein-bound Asn is not a substrate of TGase because its shorter and less hydrophobic side chain, lacking one methylene unit compared to Gln, would not allow these stabilizing interactions.
Molecular modeling suggests that residue Y515 is crucial in acyl-donor substrate binding and reactivity. The presence of a ubiquitous H-bond between the hydroxyl group of Y515 and the carbonyl oxygen from Gln side chain for all of the efficient substrates (CBz-peptides) indicates that Y515 participates in stabilizing the substrate in the active site. This tyrosine residue is strictly conserved in all tissue TGases. It belongs to the barrel 1 domain but in the folded structure is located within the active site and in close proximity to the catalytic C272 in the core domain. Taken together, these data suggest that Y515 plays an important role in this enzyme. It has been proposed to form a regulatory H-bond with the catalytic sulphur atom, preventing nucleophilic attack by the sulphur atom on the substrate carboxamide group (Yee et al. 1994; Noguchi et al. 2001). However, our findings suggest that it could orient the substrate γ-carboxamide for nucleophilic attack by the thiolate and that the H-bond formation could activate the carbonyl towards this nucleophilic attack by rendering it more electrophilic. Our studies also reveal the presence of this H-bond in the structures of the acyl-enzyme and tetrahedral intermediates, further suggesting its involvement in stabilizing the oxyanion generated during catalysis. We are currently undertaking mutagenesis of guinea pig liver TGase at this position to verify our hypothesis.
The H-bond network identified in our modeling study, involving residues Y515 and H332 of red sea bream tissue TGase and the γ-carboxamide group of the donor substrate, could be critical for transglutaminase activity. This network is highly prevalent in the structures generated by modeling with efficient substrates (CBz-peptides) and is present at a significantly lower percentage in the structures generated with the unreactive dipeptide Boc-Gln-Gly. This H-bond network could catalyze acylation by orienting the γ-carboxamide group relative to the two catalytic residues C272 and H332, activating the γ-carbonyl group to nucleophilic attack by C272 by rendering it more electrophilic and stabilizing the oxyanionic transition state. Molecular modeling showed that the H-bond between the Oη proton of Y515 and the Oδ of Gln from CBz-Gln-Gly was present 100% of the time with all efficient substrates as well as in the acyl-enzyme and tetrahedral intermediates. However, this H-bond was not always present when a poor substrate, Boc-Gln-Gly, was modeled.
Residue W329 blocks the active site in the crystal structure of red sea bream TGase and human TGase (W332 in human tissue TGase), preventing substrate entry. Indeed, automated docking experiments with AutoDock 3.0 using the crystal structure of the red sea bream enzyme whose active site is blocked by residue W329 did not generate structures with substrate in the active site. However, we have shown by molecular dynamics simulation over 10 psec that this residue readily moves to allow insertion of the side chain into the active site. This tryptophan residue is also strictly conserved in all of the tissue TGases and is located in a loop comprising residues S321 to W329. It could therefore act as a ‘gate’ that could close the active site and exclude water. W329 could then move to open the active site upon substrate binding, allowing substrates to enter. The opening of the active site by displacement of this residue may be mediated by allosteric calcium binding, which is essential for TGase activity. This hypothesis is indirectly supported by results from crystallographic studies of TGase 3, showing that calcium binding induces conformational changes that expose the corresponding W327 in a way that should increase its dynamic mobility and interactions with incoming substrates (Ahvazi et al. 2002).
The difference in recognition reflected by the varied KM values for Gln-Gly dipeptides having different N-terminal functional groups, Boc or CBz, could be attributed to various factors. First, the van der Waals energy of interaction between the enzyme and the N-terminally derivatized Gln-Gly dipeptide substrate varies by almost 10 kcal/mole, depending on the N-terminal functional group. This difference in interaction energy correlates well with the poorer binding of the Boc-derivatized dipeptides. Furthermore, modeling of the binding of Boc-derivatized dipeptides gave unfavorable, positive interaction energies far more frequently than CBz-derivatized dipeptides. Finally, the aforementioned H-bond network is frequently absent in the structures generated with the Boc-dipeptides. These differences in binding could be the result of differences in the shape and size between the CBz and Boc groups, because the CBz is planar whereas the Boc is globular. The Boc group is also shorter in that the bulky tert-butyl group is attached directly to the carbamate oxygen, whereas the bulky phenyl group of the CBz group is attached to an additional methylene unit, increasing its length and conformational flexibility. It is also possible that good recognition of CBz-Gln-Gly by tissue TGase is conferred by the aromaticity of the CBz group. We are currently undertaking kinetic studies of various substrates containing differing N-terminal functional groups to explore the importance of the nature of this group to binding affinity.
Summarized in Table 6 and Scheme 2 ▶ are the interactions that we propose for the acylation of tissue TGase by small peptide substrates. First, the active site is opened by displacement of residue W329. This displacement could occur after calcium binding and/or upon binding of acyl-donor substrate to the enzyme. The Gln side chain then enters the active site. Its insertion is favored and stabilized by hydrophobic interactions between the methylene groups of the Gln side chain and the indole group of residue W236. This residue could also be partly responsible for the specificity towards Gln rather than Asn. The γ-carboxamide group of the substrate Gln then forms the H-bond network with residues Y515 and H332. This network orients the γ-carboxamide group for nucleophilic attack by the catalytic thiol and increases electrophilicity of the γ-carboxamide, making it more reactive. After nucleophilic attack, the resulting oxyanion is stabilized by the H-bond formed with the hydroxyl group of Y515, while the NH2 group of the γ-carboxamide forms an H-bond with the acidic proton of the H332 imidazolium. Then, ammonia is released on formation of the acyl-enzyme intermediate.
Table 6.
Proposed interactions between acyl-donor substrates and tissue TGase
| Interaction | Possible role |
| Steric obstruction by W329 | • ‘Gate’ to the active site |
| Hydrophobic interaction with W236 | • Stabilization of Gln side chain insertion into active site |
| • Specificity for Gln side chain vs. Asn | |
| H-bond with H332 | • Orientation of substrate γ-CONH2 group for nucleophilic attack |
| H-bond with Y515 | • Orientation of substrate γ-CONH2 group for nucleophilic attack |
| • Activation of the substrate γ-carbonyl toward nucleophilic attack | |
| • Stabilization of the tetrahedral intermediate |
Scheme 2.

Acylation step of the TGase mechanism.
Using a combined kinetic/molecular modeling approach, we generated a model for the binding of small acyl-donor peptide substrates on tissue TGase, and identified possible structural elements important for productive binding. Our findings, while correlating well with the known mechanism and specificity of tissue TGases, increase our understanding of the binding of acyl-donor substrates on tissue TGase. While raising new hypotheses regarding the importance of the N-terminal functional group of small peptide substrates, our model should be helpful in the future development of inhibitors for this class of enzymes, implicated in various diseases.
Materials and methods
Synthesis of the N-terminally derivatized peptide substrates
Peptide substrates were synthesized according to a published procedure based on the activation of CBz- or Boc-protected amino acids as their corresponding p-nitrophenyl esters, followed by reaction with unprotected amino acids (Gagnon et al. 2002).
Kinetic measurements
Kinetic parameters were measured using a kinetic protocol previously developed in our laboratory (de Macédo et al. 2000). According to this method, N,N-dimethyl-1,4-phenylene diamine (DMPDA) is used as an acceptor substrate for the TGase-mediated transamidation from Gln-containing donor substrates. For CBz-Gln-Gly, the anilide resulting from the transamidation reaction with DMPDA has been shown to have an extinction coefficient of 8940 M−1cm−1 at 278 nm. Although the specific extinction coefficients of each of the corresponding anilides formed from the other substrates used in this study were not determined, they were assumed to not differ significantly. This was corroborated by the observation that similar concentrations of different anilide products gave similar absorbance values, and the kcat values shown in Table 1 were calculated based on this assumption. A KM value was measured for each substrate using this method at 37°C and pH 7.0,over substrate concentrations that ranged from 0.2- to fivefold KM. By way of comparison, a value of 3.2 mM was thus determined for the KM of CBz-Gln-Gly, in accordance with that determined in the literature by various methods ( Folk and Chung 1985; Day and Keillor 1999; de Macédo et al. 2000).
Computational methods
Computations were performed using the InsightII package (version 2000, Accelrys). The BIOPOLYMER module was used to build or modify molecular structures, and all energy minimizations and molecular dynamics calculations were performed with the DISCOVER module using the consistent valence force field (CVFF). Interaction energies between the enzyme and the ligands were calculated with the DOCKING module of InsightII. Automated docking was performed with AutoDock 3.0 software (Scripps).
Preparation of the protein structure
The starting coordinates were taken from the PDB file 1G0D of red sea bream tissue transglutaminase (Noguchi et al. 2001). The crystallographic water molecules as well as the sulfate ion were removed. Hydrogen atoms were added using the BIOPOLYMER module at the normal ionization state of the amino acids at pH 7.0. Two dihedral angle torsions involving hydrogen atoms were introduced in the protein structure in order to render it more consistent with the catalytic process. The first involves the hydrogen atom of the hydroxyl group of Y515, which was turned towards the sulphur atom of the catalytic C272. This torsion was introduced according to the suggested existence of a regulatory H-bond between the catalytic Cys and Y515 of red sea bream tissue TGase (Noguchi et al. 2001). The second involves the hydrogen atom of the thiol group of C272, which was turned towards Nδ of H332. This torsion was introduced according to the known catalytic mechanism of the enzyme in which the general acid-base catalyst, H332, deprotonates the catalytic Cys residue prior to or during catalysis (Case and Stein 2003). Energy minimization of the added hydrogen atoms was then performed to remove bad contacts in the protein structure. One thousand steps of steepest descents minimization were performed, followed by a conjugate gradients minimization until convergence of 0.01 kcal/mole/Å. Both minimizations were performed while keeping the heavy atoms of the protein fixed and using a dielectric constant of 1. This generated the structure g0d300.
Computational construction of substrate molecules
Substrates were built using fragments from the BIOPOLYMER module fragment library. The resulting structures were energy-minimized using 1000 steps of steepest descents minimization followed by a conjugate gradients minimization until convergence of 0.01 kcal/mole/Å with a distance-dependent dielectric constant of 80 to mimic implicit water. These resulting molecules served as starting structures for the docking experiments.
Automated docking of substrates on TGase
AutoGrid was used to generate geometry-centered grid maps with 0.375 Å spacing based on a region that contained all residues including atoms within a 15 Å radius centered about the catalytic sulphur atom in the structure g0d300. A distance-dependent dielectric constant of 4.0 was used to mimic the interior of the protein. The genetic algorithm implemented in AutoDock 3.0 (Morris et al. 1998) was applied with a starting population size of 50. Fifty runs were performed for each ligand (CBz-Gln-Gly and Boc-Gln-Gly) with a maximum number of energy evaluations of 250,000 and a maximum number of generations of 27,000. Auto-Tors was used to define the available torsions for the ligands. However, we restricted the torsions of the Gln side chain (Cα–Cβ, Cβ–Cγ and Cγ–Cδ) to define an extended conformation that maximizes its length. The torsions of the methyl groups of the Boc functional group were also deleted, as they do not affect the structure of the molecule. There remained seven possible torsions for CBz-Gln-Gly and six for Boc-Gln-Gly. After automated docking, the resulting enzyme/substrate structures were energy-minimized using the DISCOVER module of InsightII. A simulation zone containing all residues including atoms within 20 Å of the sulphur atom of the catalytic cysteine residue was used, with the remainder of the protein being fixed. An assembly was created between the enzyme and the substrate, and a layer of 25 Å of explicit water was added around the substrate. This layer of water completely covered the active site region. A nonbond cutoff of 11 Å was used to speed up calculations, and a distance restraint was applied to maintain the substrate’s reactive group (γ-carboxamide group of Gln) at a maximal distance of 3 Å from the catalytic Sγ of C272 and Nδ of H332. A dielectric constant of 1 was applied. A multistage minimization was performed on the enzyme/substrate complex to further refine the complex. One thousand iterations of steepest descents minimization was performed, followed by a conjugate gradients minimization until convergence of 0.001 kcal/mole/Å.
Manual docking of substrates on TGase
Manual docking of the minimized substrates on TGase was performed as follows. First, the reactive Gln side chain of the rigid substrates was inserted into the well-known active site cavity (Yee et al. 1994) perpendicularly to the center of mass of the protein so as to maximize the depth of insertion while maintaining all protein atoms fixed. The side chain was placed in a sterically unhindered position that is consistent with Gln side chain insertion into the active site cavity. Second, the distance between the reactive atoms of the enzyme and the substrates (Sγ of C272 with Cδ of the Gln side chain and Nɛ of Gln side chain with Nδ of H332) was kept at values lower than 3 Å. Third, the peptide backbone and the N-terminal functional group of the substrates were placed in a sterically unhindered position on the surface of the enzyme, with the functional group in one of the four tested positions (Fig. 2 ▶). Finally, an assembly was created between the enzyme and the substrate, and a layer of either 25 Å or 40 Å of explicit water was added around the substrate. This layer of water completely covered the active site region.
Calculations on the enzyme/substrate complex
For all reaction steps, a simulation zone containing all residues including atoms within 20 Å of the sulphur atom of the catalytic C272 residue was used, with the remainder of the protein being fixed. A nonbond cutoff of 11 Å was used to speed up calculations, and a distance restraint was applied to maintain the substrate’s reactive group (γ-carboxamide group of Gln) close to the catalytic residues at a maximal distance of 3 Å from Sγ of C272 and Nδ of H332. For the molecular dynamics experiments, an additional restraint of 100 kcal/mole on the oxygen atoms of the water molecules was added to prevent them from “boiling off.” A dielectric constant of 1 was applied.
A multistage minimization was performed on the enzyme/substrate complex to remove bad contacts between the substrate and the enzyme. First, 1000 iterations of steepest descents minimization was performed, followed by a conjugate gradients minimization until convergence of 0.001 kcal/mole/Å. This first minimization was followed by a molecular dynamics simulation in which the molecular system was allowed to equilibrate at 300 K for 1 psec, followed by the actual simulation to explore conformational space for 10 psec while maintaining the same temperature. Snapshots were taken every 0.1 psec, generating 100 different conformers. The conformer with the lowest energy was energy-minimized with 100 steps of steepest descents followed by a conjugate gradients minimization to convergence of 0.001 kcal/mole/Å. At this point, the structures were analyzed.
Building of the acyl-enzyme intermediate and the tetrahedral intermediate
As a starting point for the energy calculations, the acyl-enzyme and tetrahedral intermediates were built using the BIOPOLYMER module. While building these molecules, we introduced torsions at the χ1, χ2, and χ3 dihedral angles of the catalytic C272 and the reactive Gln of the substrate. These angles (χ1 = −30° and χ2 = −60° for Cys, χ1 = −160°, χ2 = 180°, and χ3 = 0° for Gln) were selected from published χ1–χ2 and χ2–χ3 plots for amino acid side chains (Janin et al. 1978). The angles allowed the positioning of the N-terminal functional group in position 0 as defined in Figure 2 ▶, whereas the angles χ1 = −45° and χ2 = −45° for Cys, χ1 = 180°, χ2 = 180°, and χ3 = 180° for Gln allowed the positioning of the N-terminal functional group in position 2. For the tetrahedral intermediate, the catalytic His residue was protonated, a formal charge of −1 was introduced at the oxyanion, and the partial charges were calculated using the CVFF force field. These molecules were then minimized and subjected to a molecular dynamics simulation in the same manner as the enzyme/substrate intermediates.
Acknowledgments
We thank Dr. Andreea Schmitzer for assistance in molecular modeling. R.A.C. and P.G. were recipients of Fonds de recherche sur la nature et las technologies (FQRNT) fellowships. This research was funded by FQRNT Grant 80357 (jointly held by J.N.P. and J.W.K.).
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Article published online ahead of print. Article and publication date are at http://www.proteinscience.org/cgi/doi/10.1110/ps.03433304.
References
- Abe, S., Yamashita, K., Kohno, H., and Ohkubo, Y. 2000. Involvement of transglutaminase in the receptor-mediated endocytosis of mouse peritoneal macrophages. Biol. Pharm. Bull. 23 1511–1513. [DOI] [PubMed] [Google Scholar]
- Aeschlimann, D. and Paulsson, M. 1994. Transglutaminases: Protein cross-linking enzymes in tissues and body fluids. Thromb. Haemost. 71 402–415. [PubMed] [Google Scholar]
- Ahvazi, B., Kim, H.C., Kee, S.H., Nemes, Z., and Steinert, P.M. 2002. Three-dimensional structure of the human transglutaminase 3 enzyme: Binding of calcium ions changes structure for activation. EMBO J. 21 2055–2067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Case, A. and Stein, R.L. 2003. Kinetic analysis of the action of tissue transglutaminase on peptide and protein substrates. Biochemistry 42 9466–9481. [DOI] [PubMed] [Google Scholar]
- Chen, J.S. and Mehta, K. 1999. Tissue transglutaminase: An enzyme with a split personality. Int. J. Biochem. Cell Biol. 31 817–836. [DOI] [PubMed] [Google Scholar]
- Clarke, D.D., Mycek, M.J., Neidle, A., and Waelsh, H. 1959. The incorporation of amines into proteins. Arch. Biochem. Biophys. 79 338–354. [DOI] [PubMed] [Google Scholar]
- Day, N. and Keillor, J.W. 1999. A continuous spectrophotometric linked enzyme assay for transglutaminase activity. Anal. Biochem. 274 141–144. [DOI] [PubMed] [Google Scholar]
- de Macédo, P., Marrano, C., and Keillor, J.W. 2000. A direct continuous spectrophotometric assay for transglutaminase activity. Anal. Biochem. 285 16–20. [DOI] [PubMed] [Google Scholar]
- Fesus, L. and Piacentini, M. 2002. Transglutaminase 2: An enigmatic enzyme with diverse functions. Trends Biochem. Sci. 27 534–539. [DOI] [PubMed] [Google Scholar]
- Folk, J.E. 1969. Mechanism of action of guinea pig liver transglutaminase. VI. Order of substrate addition. J. Biol. Chem. 244 3707–3713. [PubMed] [Google Scholar]
- ———. 1983. Mechanism and basis for specificity of transglutaminase-catalyzed epsilon-(γ-glutamyl) lysine bond formation. Adv. Enzymol. Relat. Areas Mol. Biol. 54 1–56. [DOI] [PubMed] [Google Scholar]
- Folk, J.E. and Chung, S.I. 1973. Molecular and catalytic properties of transglutaminases. Adv. Enzymol. Relat. Areas Mol. Biol. 38 109–191. [DOI] [PubMed] [Google Scholar]
- ———. 1985. Transglutaminases. Methods Enzymol. 113 358–375. [DOI] [PubMed] [Google Scholar]
- Folk, J.E. and Cole, P.W. 1965. Structural requirements of specific substrates for guinea pig liver transglutaminase. J. Biol. Chem. 240 2951–2960. [PubMed] [Google Scholar]
- ———. 1966. Transglutaminase: Mechanistic features of the active site as determined by kinetic and inhibitor studies. Biochim. Biophys. Acta 122 244–264. [DOI] [PubMed] [Google Scholar]
- Gagnon, P., Huang, X., Therrien, E., and Keillor, J.W. 2002. Peptide coupling of unprotected amino acids through in situ pnitrophenyl ester formation. Tetrahedron Lett. 43 7717–7719. [Google Scholar]
- Huo, J., Metz, S.A., and Li, G. 2003. Role of tissue transglutaminase in GTP depletion-induced apoptosis of insulin-secreting (HIT-T15) cells. Biochem. Pharmacol. 66 213–223. [DOI] [PubMed] [Google Scholar]
- Janin, J., Wodak, S., Levitt, M., and Maigret, B. 1978. Conformation of amino acid side-chains in proteins. J. Mol. Biol. 125 357–386. [DOI] [PubMed] [Google Scholar]
- Kim, S.Y., Grant, P., Lee, J.H., Pant, H.C., and Steinert, P.M. 1999. Differential expression of multiple transglutaminases in human brain. Increased expression and cross-linking by transglutaminases 1 and 2 in Alzheimer’s disease. J. Biol. Chem. 274 30715–30721. [DOI] [PubMed] [Google Scholar]
- Liu, S., Cerione, R.A., and Clardy, J. 2002. Structural basis for the guanine nucleotide-binding activity of tissue transglutaminase and its regulation of transamidation activity. Proc. Natl. Acad. Sci. 99 2743–2747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris, G.M., Goodsell, D.S., Halliday, R.S., Huey, R., Hart, W.E., Belew, R.K., and Olson, A.J. 1998. Automated docking using a lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 19 1639–1662. [Google Scholar]
- Murthy, S.N.P., Iismaa, S., Begg, G., Freymann, D.M., Graham, R.M., and Lorand, L. 2002. Conserved tryptophan in the core domain of transglutaminase is essential for catalytic activity. Proc. Natl. Acad. Sci. 99 2738–2742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noguchi, K., Ishikawa, K., Yokoyama, K., Ohtsuka, T., Nio, N., and Suzuki, E. 2001. Crystal structure of red sea bream transglutaminase. J. Biol. Chem. 276 12055–12059. [DOI] [PubMed] [Google Scholar]
- Ohtsuka, T., Ota, M., Nio, N., and Motoki, M. 2000. Comparison of substrate specificities of transglutaminases using synthetic peptides as acyl donors. Biosci. Biotechnol. Biochem. 64 2608–2613. [DOI] [PubMed] [Google Scholar]
- Priglinger, S.G., May, C.A., Neubauer, A.S., Alge, C.S., Schönfeld, C.L., Kampik, A., and Welge-Lussen, U. 2003. Tissue transglutaminase as a modifying enzyme of the extracellular matrix in PVR membranes. Invest. Ophthalmol. Vis. Sci. 44 355–364. [DOI] [PubMed] [Google Scholar]
- Shan, L., Molberg, Ø., Parrot, I., Hausch, F., Filiz, F., Gray, G.M., Sollid, L.M., and Khosla, C. 2002. Structural basis for gluten intolerance in celiac sprue. Science 297 2275–2279. [DOI] [PubMed] [Google Scholar]
- Shridas, P., Sharma, Y., and Balasubramanian, D. 2001. Transglutaminase-mediated cross-linking of α-crystallin: Structural and functional consequences. FEBS Lett. 499 245–250. [DOI] [PubMed] [Google Scholar]
- Singh, U.S., Pan, J., Kao, Y.L., Joshi, S., Young, K.L., and Baker, K.M. 2003. Tissue transglutaminase mediates activation of RhoA and MAP kinase pathways during retinoic acid-induced neuronal differentiation of SH-SY5Y cells. J. Biol. Chem. 278 391–399. [DOI] [PubMed] [Google Scholar]
- Wada, F., Nakamura, A., Masutani, T., Ikura, K., Maki, M., and Hitomi, K. 2002. Identification of mammalian-type transglutaminase in Physarum polycephalum. Evidence from the cDNA sequence and involvement of GTP in the regulation of transamidating activity. Eur. J. Biochem. 269 3451–3460. [DOI] [PubMed] [Google Scholar]
- Yee, V.C., Pedersen, L.C., Le Trong, I., Bishop, P.D., Stenkamp, R.E., and Teller, D.C. 1994. Three-dimensional structure of a transglutaminase: Human blood coagulation factor XIII. Proc. Natl. Acad. Sci. 91 7296–7300. [DOI] [PMC free article] [PubMed] [Google Scholar]