

Abstract
The transcriptional activity of an in vitro assembled human interferon-β gene enhanceosome is highly synergistic. This synergy requires five distinct transcriptional activator proteins (ATF2/c-JUN, interferon regulatory factor 1, and p50/p65 of NF-κB), the high mobility group protein HMG I(Y), and the correct alignment of protein-binding sites on the face of the DNA double helix. Here, we investigate the mechanisms of enhanceosome-dependent transcriptional synergy during preinitiation complex assembly in vitro. We show that the stereospecific assembly of the enhanceosome is critical for the efficient recruitment of TFIIB into a template-committed TFIID-TFIIA-USA (upstream stimulatory activity complex) and for the subsequent recruitment of the RNA polymerase II holoenzyme complex. In addition, we provide evidence that recruitment of the holoenzyme by the enhanceosome is due, at least in part, to interactions between the enhanceosome and the transcriptional coactivator CREB, cAMP responsive element binding protein (CBP). These studies reveal a unique role of enhanceosomes in the cooperative assembly of the transcription machinery on the human interferon-β promoter.
Complex patterns of gene expression are achieved with a relatively small number of transcription factors, due primarily to the combinatorial organization of transcriptional enhancers and promoters (1). Most natural enhancers consist of DNA-binding sites for multiple, distinct transcription factors. Thus, the number and arrangement of these sites, and the presence or absence of the corresponding transcription factors, determine the selective and synergistic activation of genes in response to extracellular signals or developmental queues. An additional level of complexity and specificity in enhancer function can be provided by the assembly of a three-dimensional nucleoprotein complex (enhanceosome) consisting of transcriptional activators and one or more architectural proteins (1, 2). Interactions between transcriptional activator proteins and the transcriptional machinery have been studied almost exclusively with simple synthetic promoters containing multiple copies of transcription activator-binding sites. By comparison, relatively little is known about the mechanisms by which complex multi-component transcriptional enhancers synergistically activate transcription.
An excellent model for studying complex enhancers is provided by the virus-inducible enhancer of the human interferon-β (IFN-β) gene (3). Recently, we assembled a functional IFN-β enhanceosome by using the purified recombinant transcriptional activator proteins ATF2/c-JUN, interferon regulatory factor 1 (IRF1), and p50/p65 of NF-κB (4). Under conditions in which the transcriptional activator proteins were limiting, the assembly of the enhanceosome on the wild-type (WT) IFN-β enhancer was highly cooperative, and this assembly required the presence of the high mobility group protein HMG I(Y), an architectural protein. Once assembled, the enhanceosome was extraordinarily stable. Cooperative assembly and enhanced stability of the enhanceosome were not observed when the enhancer contained insertions of a half-helical turn of DNA, which alters the relative positions of protein-binding sites on the face of the DNA double helix (4).
The assembly of the IFN-β enhanceosome with purified recombinant proteins provides a unique opportunity to investigate the detailed mechanisms of transcriptional synergy. In this paper, we use in vitro transcription and protein recruitment assays with immobilized DNA templates to analyze the mechanisms of enhanceosome-dependent transcriptional synergy. We find that the enhanceosome facilitates transcription factor (TF)IIB recruitment into a template-committed TFIID-TFIIA-USA (upstream stimulatory activity) complex and recruits purified RNA polymerase II holoenzyme complex to the promoter through contacts with CREB [cAMP responsive element binding protein] binding protein (CBP). Maximal levels of enhanceosome-dependent recruitment of RNA polymerase II initiation complexes require the correct alignment of enhancer-binding factors on the face of the DNA double helix.
MATERIALS AND METHODS
Purification of Basal and IFN-β Enhanceosome Factors.
Basal transcription factors TFIIA, TFIIE/F/H and USA were fractionated from a HeLa cell nuclear extract (NE) by phosphocellulose (P11) chromatography and purified as described (5). RNA polymerase II was purified from HeLa nuclear pellets as described (5, 6). Flag-tagged TFIID was purified by affinity chromatography from stably transfected HeLa cells (6). Recombinant TATA box-binding protein (TBP) and TFIIB were expressed as a hexahistidine-tagged protein in bacteria and were purified by nickel affinity chromatography (7, 8). To purify RNA polymerase II holoenzyme, a HeLa cell NE was fractionated by a P11 column and eluted with 300–500 mM KCl, followed by a DEAE-cellulose (DE52) column. Fractions were collected and analyzed for RNA polymerase II and CBP by immunoblotting assays. The fraction enriched with both proteins (eluted with ≈300 mM KCl from DE52) was used for affinity chromatography with an anti-RAP74 antibody and eluted with 800 mM KCl as described (9). CBP-RNA polymerase II complexes were further affinity purified by antibodies against CBP. A HeLa cell NE was depleted of TBP and TFIIB, respectively, with a mild heat treatment and with an anti-TFIIB antibody (10). IFN-β enhanceosome factors (ATF2, c-JUN, HMG I(Y), IRF1, p50, and p65) containing a hexahistidine were purified from bacterial cell lysates by nickel affinity chromatography as described (4).
In Vitro Transcription and Protein Recruitment Assays.
Immobilized DNA templates were prepared as described (10) with dyna-magnetic beads M-280 (11). Briefly, DNA fragments containing IFN-β enhancer plus TATA-promoter regions were isolated from −110 IFN-β-chloramphenicol acetyl transferase and attached to streptavidin-conjugated dynabeads via a biotin moiety. After incubation with the assembled enhanceosome followed by appropriate transcription components, the beads were pelleted and extensively washed with transcription buffer containing no nucleoside triphosphates (NTPs). For transcription assays, washed beads were incubated in transcription buffer with NTPs and indicated protein components, and then transcripts were analyzed by a primer extension assay (4). Recruitment of factors on purified promoter complexes was determined by immunoblotting assays with specific antibodies (10).
RESULTS
The IFN-β Enhanceosome Is Required for the Efficient Recruitment of TFIIB into the Preinitiation Complex.
To analyze the effect of the in vitro assembled IFN-β enhanceosome on preinitiation complex (PIC) assembly, we carried out in vitro transcription experiments with biotinylated DNA templates immobilized on streptavidin magnetic beads (10–12). Immobilized DNA containing the intact −110 IFN-β gene enhancer/promoter was incubated in a HeLa cell NE in the presence or absence of purified enhanceosome components (ATF2, c-JUN, IRF1, HMG I(Y), p50, and p65) (4). The assembled complexes then were extensively washed to remove unbound factors and tested for transcriptional activity by using a primer extension assay. As shown in Fig. 1A, when the immobilized DNA was preincubated in a HeLa cell NE in the absence of enhanceosome components, only a low level of basal transcription was observed (lanes 1 and 2). Addition of enhanceosome components after the promoter complexes were washed on beads did not rescue the transcriptional activity (lane 2). By contrast, when the enhanceosome components were present during the preincubation period, a high level of transcription was observed (lanes 3 and 4), but this transcription was not further increased by the subsequent addition of enhanceosome components after washing (lane 4). Thus, the IFN-β enhanceosome acts during PIC assembly.
Figure 1.

The IFN-β enhanceosome facilitates the recruitment of TFIIB during preinitiation complex assembly. (A and B) A HeLa NE was incubated with immobilized DNA templates in the absence or presence of the IFN-β enhanceosome, which was assembled by using purified recombinant proteins ATF2/c-JUN, IRF1, p50/p65 of NF-κB, and HMG I(Y). Promoter complexes were purified by washing and analyzed for transcription in the presence of NTPs and the enhanceosome components as indicated above each lane. (B) Factors added to the reaction after the washing step are indicated immediately above the autoradiogram. −TBE NE: An NE in which TBP was inactivated with a mild heat treatment. −TBP/IIB NE: An NE in which TBP was inactivated with a mild heat treatment, and TFIIB was depleted with an anti-TFIIB antibody. −TBP/−IIB NE + IIB: purified recombinant TFIIB (IIB) protein was added to an NE lacking TBP and TFIIB. The amounts of template-bound TBP and TFIIB were determined by immunoblotting assays after washing promoter complexes (bottom).
To identify the steps in PIC assembly that require the IFN-β enhanceosome, in vitro transcription reactions were carried out with an immobilized IFN-β DNA template in untreated NE or in NE depleted of TBP and/or TFIIB (Fig. 1B). TBP was inactivated by a mild heat treatment, whereas TFIIB was depleted by using an anti-TFIIB antibody (10). When the IFN-β promoter was preincubated in an untreated HeLa cell NE in the absence of the enhanceosome, washed, and then incubated with the enhanceosome in the presence of a TBP-depleted NE (−TBP NE), a high level of transcription was observed (lane 3). The level of activated transcription was indistinguishable from that observed when the promoter was preincubated in a HeLa cell NE in the presence of enhanceosome components (lane 3 vs. 2). This result suggests that TBP can bind to the promoter during the preincubation step, even in the absence of the IFN-β enhanceosome. Thus, at least in vitro, TBP recruitment is not limiting for enhanceosome-dependent transcriptional synergy.
When the in vitro transcription reaction was carried out with the IFN-β enhanceosome in the presence of extracts in which both TBP and TFIIB were depleted (−TBP/−IIB NE), activated transcription (lane 3) was reduced to the basal level (lane 4). However, high levels of transcription could be reconstituted by the addition of purified TFIIB recombinant protein (lane 5). Thus, TFIIB, but not TBP, recruitment can be facilitated by the IFN-β enhanceosome in vitro.
Further evidence for this conclusion was provided by determining the levels of TBP and TFIIB recruited to DNA by the IFN-β enhanceosome (Fig. 1B). The amounts of TBP or TFIIB bound to the promoter were directly measured by immunoblotting assays after washing the promoter complexes assembled in the absence or presence of the enhanceosome (lanes 1 and 2). When the −110 IFN-β promoter was incubated in an NE containing both TBP and TFIIB in the absence of the enhanceosome (lane 1), only a basal level of transcription was observed (Fig 1B, Top) and TBP, but not TFIIB, was detected in the promoter complexes in the immunoblots (Fig 1B, Bottom). By contrast, when the enhanceosome components were present during the preincubation in NE, high levels of both activated transcription and recruited TFIIB were observed (lane 2). However, the level of TBP bound to the promoter was the same regardless of the presence of the IFN-β enhanceosome (cf. lanes 1 and 2). Thus, consistent with experiments with TBP/TFIIB-depleted extracts, the IFN-β enhanceosome is required for the recruitment of TFIIB into the PIC.
A Helical Phasing Mutation in the IFN-β Enhancer Interferes with the Enhanceosome-Dependent Recruitment of TFIIB.
To determine whether the stereospecific assembly of the IFN-β enhanceosome is required for the assembly of TFIIB into the PIC, we examined the effect of a helical phasing mutation on TFIIB recruitment. This helical phasing mutant DNA (PRDI/II 6) has a half helical turn (6 bp) of DNA inserted between PRDI and PRDII in the IFN-β enhancer. This mutation was previously shown to significantly decrease transcriptional synergy of the IFN-β gene promoter both in vivo (13) and in vitro (ref. 4; also see Fig. 3). As shown in Fig. 2A, the amount of TBP bound to the promoter was unaffected by the presence or absence of the WT or mutant (PRDI/II 6) enhancer (lanes 1–8). In contrast, the amount of TFIIB recruited to the promoter with the WT IFN-β enhancer was significantly greater than that observed with the enhanceosome assembled on DNA containing the PRDI/II helical phasing mutation (cf. lanes 4 and 8). We note that this difference in the level of TFIIB recruitment was observed with NEs (lane 4 vs. 8) but not with purified TBP and TFIIB (TB) proteins (lane 2 vs. 6). Thus, the correct positioning of enhancer-binding factors on the face of the DNA helix is required for high levels of TFIIB recruitment, but additional factors present in the NE are necessary for efficient TFIIB recruitment.
Figure 3.
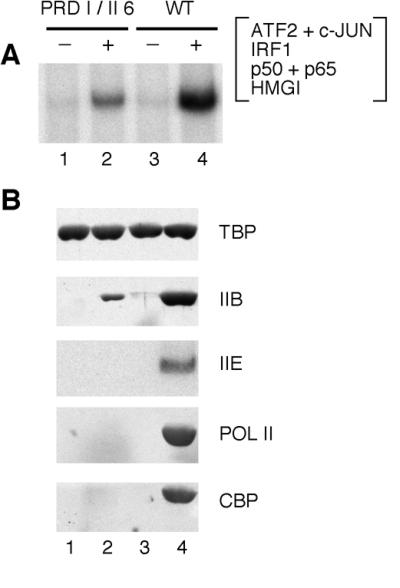
The IFN-β enhanceosome recruits high levels of TFIIE, RNA polymerase II, and CBP. (A and B) A HeLa NE was incubated with immobilized templates by using WT and helical phasing mutant (PRDI/II 6) IFN-β enhancer DNAs in the absence or presence of the enhanceosome. IFN-β enhanceosome was assembled by using purified recombinant proteins ATF2/c-JUN, IRF1, p50/p65 of NF-κB, and HMG I(Y). (A) Promoter complexes were purified by washing and analyzed for transcription in the presence of NTPs. (B) The amounts of template-bound factors TBP, TFIIB (IIB), TFIIEα (IIE), RNA polymerase II (POL II), and CBP were determined by immunoblotting assays after washing promoter complexes.
Figure 2.

High levels of TFIIB recruitment require both the stereospecific assembly of the IFN-β enhanceosome and the template-committed TFIID-TFIIA-USA complex. (A and B) The IFN-β enhanceosome was assembled in vitro with purified recombinant proteins ATF2/c-JUN, IRF1, p50/p65 of NF-κB, and HMG I(Y). The amounts of template-bound TBP and TFIIB (IIB) were determined by immunoblotting assays after isolating promoter complexes and washing. (A) HeLa NE and TBP/TFIIB proteins (TB) were incubated with immobilized templates by using WT and helical phasing mutant (PRDI/II 6) IFN-β enhancer DNAs in the absence or presence of the enhanceosome. (B) Specific sets of general transcription factors as indicated were incubated with immobilized templates by using WT and helical phasing mutant (PRDI/II 6) IFN-β enhancer DNAs in the absence or presence of the enhanceosome. T, TBP; B, TFIIB; A, TFIIA; USA, upstream stimulatory activity; D, TFIID; EFH, TFIIE, TFIIF and TFIIH; POLII, RNA polymerase II.
TFIID, TFIIA, and USA Are Required for Maximal Levels of TFIIB Recruitment.
To identify the factors required for high levels of TFIIB recruitment by the IFN-β enhanceosome, specific sets of purified basal transcription factors were incubated with the immobilized DNA templates in the absence or presence of the WT IFN-β enhanceosome (Fig. 2B). Incubation in the presence of TFIID, TFIIA, TFIIB, and USA resulted in high levels of TFIIB recruitment (lane 16). However, this high level of recruitment was not detected when any one of the components tested (TFIID, TFIIA, TFIIB, and USA) was left out of the reaction mixture (lanes 1–14). Thus, at least three separate basal (co)factors (TFIID, TFIIA, and USA) are required for high levels of TFIIB recruitment into the PIC by the IFN-β enhanceosome. The involvement of multiple factors suggests that cooperative assembly of the PIC is important for synergistic transcriptional activation by the enhanceosome (4).
The cooperative assembly of TFIIB into the TFIID-TFIIA-USA-promoter complex requires the WT enhanceosome because the level of TFIIB recruitment was significantly less when the enhanceosome was assembled on the IFN-β promoter containing the PRDI/II 6 helical phasing mutation (e.g., WT vs. PRDI/II 6 in lane 16). These results are consistent with previous observations showing that the WT, but not mutant IFN-β, enhanceosome promotes the highly cooperative assembly of a stable TFIID-TFIIA-TFIIB-USA complex, which is resistant to disruption by sarkosyl detergent (4).
The IFN-β Enhanceosome Efficiently Recruits CBP, RNA Polymerase II, and other PIC Components to the Promoter.
Next, we determined the effects of the enhanceosome on the assembly of PIC components subsequent to the binding of TFIIB. Immobilized templates were incubated with HeLa cell NEs in the absence or presence of the WT or mutant (PRDI/II 6) enhanceosomes (Fig. 3). As expected, assembly of a WT IFN-β enhanceosome resulted in high levels of in vitro transcription, compared with those observed with the enhanceosome assembled on the PRDI/II 6 DNA (Fig. 3A). In addition, TFIIB was more efficiently recruited to the IFN-β promoter by the WT enhanceosome compared with the PRDI/II 6 enhanceosome (Fig. 3B), consistent with results in Fig. 2. Under these conditions, the levels of TFIIE and RNA polymerase II recruited to the IFN-β promoter by the PRDI/II 6 enhanceosome were too low to be detected in our immunoblots (Fig. 3B; data not shown). By contrast, the presence of the WT enhanceosome dramatically increased the levels of both TFIIE and RNA polymerase II recruited to the IFN-β promoter (lane 4). Thus, as with TFIIB, the stereospecific assembly of the enhanceosome is required for the efficient recruitment of downstream PIC components such as TFIIE and RNA polymerase II.
The transcriptional coactivator CBP interacts with and promotes the activity of many transcription factors (14). In addition, CBP has been implicated in the synergistic activation of transcription by the IFN-β enhanceosome (15, 16). Thus, we examined the PIC complexes assembled in the presence of the WT enhanceosome for the presence of CBP (Fig. 3B). We found that, like TFIIE and RNA polymerase II, CBP was efficiently recruited to the IFN-β promoter. By contrast, CBP was not detected in complexes assembled with the mutant (PRDI/II 6) enhanceosome (cf. lanes 2 and 4). Thus, the presence of CBP is not sufficient for high levels of transcription because specific interactions between the enhanceosome and CBP are required for efficient CBP recruitment and transcriptional synergy.
Role of CBP-Enhanceosome Interactions in the Recruitment of RNA Polymerase II Holoenzyme.
Previous studies have shown that yeast and human RNA polymerase II can be isolated as a high molecular weight complex containing general transcription factors and other components of the PIC (17). This RNA polymerase II holoenzyme also contains CBP (18, 19). We therefore carried out experiments to test the possibility that interactions between the enhanceosome and CBP are required for the recruitment of the holoenzyme to the IFN-β promoter. To this end, we purified an RNA polymerase II holoenzyme containing CBP (9) and carried out experiments to determine whether an antibody against the N terminus of CBP can interfere with the enhanceosome-dependent recruitment of the holoenzyme (Fig. 4). Previous studies have shown that the N terminus of CBP can interact with IFN-β enhanceosome components including c-JUN, IRF1, and p65 (16, 20–22). Purified RNA polymerase II holoenzyme was incubated with TFIID, TFIIA, TFIIB, and USA in the absence or presence of the enhanceosome with increasing amounts of the anti-CBP antibody (Fig. 4). As shown in Fig. 4A, addition of the anti-CBP antibody inhibited enhanceosome-mediated activation of the IFN-β promoter but had no effect on the basal level of transcription (lanes 1–3 vs. 4–6). A concomitant decrease in the level of CBP recruitment was observed as the amount of anti-CBP antibody was increased (Fig. 4B, lanes 4–6). We also observed decreased levels of enhanceosome-dependent recruitment of other components of the RNA polymerase II holoenzyme, including TFIIE, RNA polymerase II, and SRB7 (23). In sharp contrast, the levels of TFIIB and TBP were not significantly affected by the addition of the anti-CBP antibody (Fig. 4B). These data indicate that specific interactions between CBP and the IFN-β enhanceosome are required for efficient recruitment of the RNA polymerase II holoenzyme by the enhanceosome.
Figure 4.
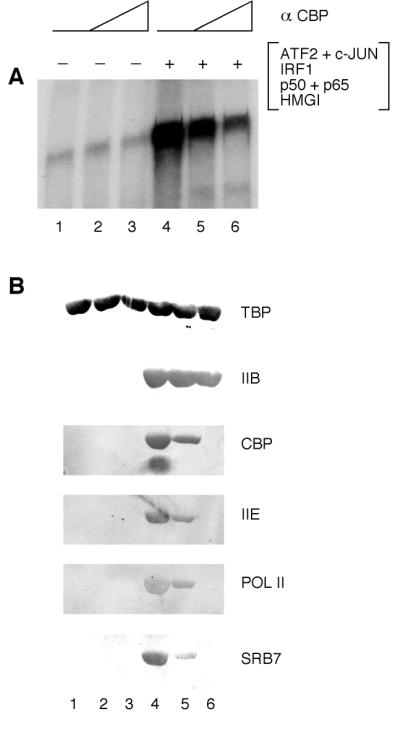
Enhanceosome-CBP interactions are required for the efficient recruitment of the RNA polymerase II holoenzyme to the preinitiation complex. (A and B) Immobilized IFN-β enhancer DNA template was incubated with TFIID, TFIIA, TFIIB, USA and RNA polymerase II holoenzyme in the absence or presence of an IFN-β enhanceosome assembled by using purified recombinant proteins ATF2/c-JUN, IRF1, p50/p65 of NF-κB and HMG I(Y). Increasing amounts of the anti-CBP antibody (α CBP) were preincubated with the RNA polymerase II holoenzyme before mixing with all the other components. (A) Promoter complexes were purified and analyzed for transcription in the presence of NTPs. (B) The amounts of template-bound factors TBP, TFIIB (IIB), CBP, TFIIEα (IIE), RNA polymerase II (POL II), and SRB7 were determined by immunoblotting assays after washing promoter complexes.
DISCUSSION
Recruitment of TFIIB into the TFIID-TFIIA-USA Complex by the Enhanceosome.
In a previous study, we showed that IFN-β enhanceosome-dependent transcriptional activity in vitro requires the basal transcription factors TFIID, TFIIA, and TFIIB and the cofactor USA (4). Analysis of enhanceosome stability in vitro by using sarkosyl inhibition or DNA competition experiments suggested that cooperative interactions between the IFN-β enhanceosome and the PIC led to the mutual stabilization of those two complexes (4).
Here, we use an immobilized DNA template assay to show that TBP binds to the IFN-β promoter in the absence or presence of the WT enhanceosome, whereas efficient recruitment of TFIIB requires the WT enhanceosome (Figs. 1 and 2). Efficient recruitment of TFIIB was not observed when the enhanceosome was assembled on an enhancer containing a helical phasing mutation (Fig. 2). Thus the presence of enhanceosome components is not sufficient for cooperative TFIIB recruitment; these components must be assembled into a stereospecific complex.
Results obtained with the natural IFN-β enhancer are strikingly different from those obtained with artificial promoters containing one or more GAL4-binding sites. In both cases, TBP stably associates with the promoter in the presence or absence of activators, but the recruitment of TFIIB requires the presence of activators (Figs. 1 and 2; also see refs. 10–12). However, a different result was obtained with the artificial promoters when activator cooperativity was analyzed. With the artificial promoter, an increase in the number of GAL4-binding sites resulted in a cooperative increase in TFIIE recruitment but very little increase in the recruitment of TFIIB (11). By contrast, with the IFN-β promoter, a direct correlation was observed between transcriptional synergy and the efficient recruitment of TFIIB, as well as TFIIE and other components of the RNA polymerase II holoenzyme (Figs. 2–4). Consistent with our earlier enhanceosome-dependent transcription studies (4), we find that the enhanceosome requires TFIID, TFIIA, and USA factors for high levels of TFIIB recruitment, whereas TBP itself is sufficient for TFIIB recruitment by GAL4 fusion protein activators (Fig. 2; data not shown; ref. 11). This difference may be a consequence of the highly cooperative interactions between the enhanceosome and the TFIID-TFIIA-TFIIB-USA complex in the intact IFN-β promoter (4). These cooperative interactions can be further facilitated by interactions with the RNA polymerase II holoenzyme (Figs. 3 and 4).
Role of CBP in Enhanceosome-Dependent Recruitment of the RNA Polymerase II Holoenzyme.
In this paper, we show that the IFN-β enhanceosome efficiently recruits TFIIE, RNA polymerase II, and CBP to the promoter and that the stereospecific alignment of transcription factor-binding sites is required for this cooperative recruitment (Fig. 3). The importance of the enhanceosome-CBP interactions was evidenced by the observation that an anti-CBP antibody inhibited the synergistic transcriptional activation by the IFN-β enhanceosome but had no effect on the basal level of transcription (Fig. 4). Moreover, the presence of this antibody had no effect on the amounts of TBP and TFIIB bound to the promoter but dramatically inhibited the recruitment of holoenzyme components such as TFIIE, RNA polymerase II, SRB7, and CBP. Thus, two apparently separable steps in the PIC assembly process are critical for transcriptional synergy of the IFN-β enhanceosome in vitro. One is the assembly of the TFIID-TFIIA-TFIIB-USA complex, and the other is the recruitment of the RNA polymerase II holoenzyme complex. The latter step involves, at least in part, direct interactions between CBP and the enhanceosome. Of course, it is likely that the IFN-β enhanceosome also interacts with other components of the RNA polymerase II holoenzyme. The fact that we were unable to completely inhibit enhanceosome-dependent transcription is consistent with this possibility (Fig. 4; data not shown).
Our studies reveal a critical and unique role of the enhanceosome for the cooperative recruitment of RNA polymerase II complexes. This stereospecific requirement of enhanceosome-dependent transcriptional synergy is consistent with the idea that the WT enhanceosome presents a specific three-dimensional activation surface that optimally interacts with the components of PIC including TFIIB and CBP (Figs. 2–4; also see refs. 4, 15). Cotransfection studies show that CBP can promote enhanceosome-dependent transcriptional synergy and that this synergy requires the correct positioning of transcription factors on enhancer DNA (15). Alterations in the order of the activation domains within the enhancer complex abrogated CBP-dependent transcriptional synergy. Taken altogether, the previous in vivo experiments and the in vitro studies presented here are consistent with the view that the precise alignment of contacts within the enhanceosome in three-dimensional space is required for high levels of transcriptional synergy.
Additional Effects of Virus-Inducible Phosphorylation of Enhanceosome Components.
The results presented here clearly demonstrate the importance of enhanceosome structure in transcriptional synergy, and they reveal new insights into the detailed mechanisms involved in this process with complex enhancers. Future studies are required to determine the role of virus-inducible protein modifications on enhanceosome function. For example, it is known that phosphorylation of the p65 subunit of NF-κB (24), ATF2 (25), and c-JUN (26) is required for efficient binding of these proteins to CBP. In addition, although IRF1 can synergize with other components of the IFN-β enhanceosome both in vivo and in vitro (4, 13, 15), recent studies have implicated other members of the IRF family in the virus induction of the IFN-β gene in vivo (16, 27–30). Specifically, virus infection results in the phosphorylation of IRF3 and IRF7 and their association with CBP/p300.
In vitro chromatin precipitation experiments revealed that IRF3 and IRF7, but not IRF1, associate with the endogenous IFN-β promoter in vivo upon virus infection (16). Thus, although IRF1 appears to be functionally interchangeable with IRF3 and IRF7 in transfection (13, 15) and in vitro transcription (4) assays, IRF3 and IRF7 proteins associate with the endogenous IFN-β promoter in vivo. However, the IRF family proteins are highly conserved, IRF1 binds specifically to the IFN-β promoter, and IRF1 synergizes with other IFN-β enhanceosome components. Thus, it seems likely that the basic mechanisms of transcriptional synergy revealed by studies of the IRF1 enhanceosome accurately reflect general mechanisms involved in enhanceosome-dependent synergy. We note, however, that the stronger interactions between enhanceosome components and CBP, mediated by the phosphorylation of transcriptional activator proteins upon virus infection, are likely to substantially increase the level of transcriptional synergy. Thus, the level of synergy observed with the IRF1 enhanceosome is likely to be a minimum estimate of the synergy achieved with IRF3 and IRF7.
Acknowledgments
We thank members of the Maniatis laboratory for helpful discussions and Danny Reinberg and Richard Young for antibodies. This work was supported by National Institutes of Health Grant AI20642 (to T.M.) and by the Creative Research Initiatives of the Korean Ministry of Science and Technology (to T.K.K.). T.K.K. also was supported by the Cancer Research Fund of the Damon Runyon-Walter Winchell Foundation (DRG-1329).
ABBREVIATIONS
- IFN
interferon
- USA
upstream stimulatory activity
- PIC
preinitiation complex
- NE
nuclear extract(s)
- WT
wild-type
- TBP
TATA box-binding protein
- NTP
nucleoside triphosphate
- HMG I(Y)
high mobility group protein
- TF
transcription factor
- CBP
CREB (cAMP responsive element binding protein) binding protein
- PRD
positive regulatory domain
- IRF
interferon regulatory factory
References
- 1. Tjian R, Maniatis T. Cell. 1994;77:5–8. doi: 10.1016/0092-8674(94)90227-5. [DOI] [PubMed] [Google Scholar]
- 2.Carey M. Cell. 1998;92:5–8. doi: 10.1016/s0092-8674(00)80893-4. [DOI] [PubMed] [Google Scholar]
- 3.Maniatis, T., Falvo, J. V., Kim, T. H., Kim, T. K., Lin, C. H., Parekh, B. S. & Wathelet, M. G. (1998) Cold Spring Harbor Symp. Quant. Biol. 63, in press. [DOI] [PubMed]
- 4.Kim T K, Maniatis T. Mol Cell. 1997;1:119–129. doi: 10.1016/s1097-2765(00)80013-1. [DOI] [PubMed] [Google Scholar]
- 5.Meisterernst M, Roy A L, Lieu H M, Roeder R G. Cell. 1991;66:981–993. doi: 10.1016/0092-8674(91)90443-3. [DOI] [PubMed] [Google Scholar]
- 6.Chiang C M, Ge H, Wang Z, Hoffmann A, Roeder R G. EMBO J. 1993;12:2749–2762. doi: 10.1002/j.1460-2075.1993.tb05936.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kim T K, Roeder R G. J Biol Chem. 1997;272:7540–7545. doi: 10.1074/jbc.272.11.7540. [DOI] [PubMed] [Google Scholar]
- 8.Kim T K, Zhao Y, Hui G, Roeder R G. J Biol Chem. 1995;270:10976–10981. doi: 10.1074/jbc.270.18.10976. [DOI] [PubMed] [Google Scholar]
- 9.Maldonado E, Shiekhattar R, Sheldon M, Cho H, Drapkin R, Rickert P, Lees E, Anderson C W, Linn S, Reinberg D. Nature (London) 1996;381:86–89. doi: 10.1038/381086a0. [DOI] [PubMed] [Google Scholar]
- 10.Kim T K, Roeder R G. Proc Natl Acad Sci USA. 1994;91:4170–4174. doi: 10.1073/pnas.91.10.4170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Choy B, Green M. Nature (London) 1993;366:531–536. doi: 10.1038/366531a0. [DOI] [PubMed] [Google Scholar]
- 12.Kim T K, Hashimoto S, Kelleher R J, Flanagan P M, Kornberg R D, Horikoshi M, Roeder R G. Nature (London) 1994;369:252–255. doi: 10.1038/369252a0. [DOI] [PubMed] [Google Scholar]
- 13.Thanos D, Maniatis T. Cell. 1995;83:1091–1100. doi: 10.1016/0092-8674(95)90136-1. [DOI] [PubMed] [Google Scholar]
- 14.Shikama N, Lyon J, Thangue N B. Trends Cell Biol. 1997;7:230–236. [Google Scholar]
- 15.Merika M, Williams A J, Chen G, Collins T, Thanos D. Mol Cell. 1998;1:277–287. doi: 10.1016/s1097-2765(00)80028-3. [DOI] [PubMed] [Google Scholar]
- 16.Wathelet M G, Lin C H, Parekh B S, Ronco L V, Howley P M, Maniatis T. Mol Cell. 1998;1:507–518. doi: 10.1016/s1097-2765(00)80051-9. [DOI] [PubMed] [Google Scholar]
- 17.Koleske A J, Young R A. Trends Biochem Sci. 1995;20:113–116. doi: 10.1016/s0968-0004(00)88977-x. [DOI] [PubMed] [Google Scholar]
- 18.Nakajima T, Uchida C, Anderson S F, Parvin J D, Montminy M. Genes Dev. 1997;11:738–747. doi: 10.1101/gad.11.6.738. [DOI] [PubMed] [Google Scholar]
- 19.Neish A S, Anderson S F, Schlegel B P, Wei W, Parvin J D. Nucleic Acids Res. 1998;26:847–853. doi: 10.1093/nar/26.3.847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Arias J, Alberts A S, Brindle P, Claret F X, Smeal T, Karin M, Feramisco J, Montminy M. Nature (London) 1994;370:226–229. doi: 10.1038/370226a0. [DOI] [PubMed] [Google Scholar]
- 21.Gerritsen M E, Williams A J, Neish A S, Moore S, Shi Y, Collins T. Proc Natl Acad Sci USA. 1997;94:2927–2932. doi: 10.1073/pnas.94.7.2927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Perkins N D, Felzien L K, Betts J C, Leung K, Beach D, Nabel G. Science. 1997;275:523–527. doi: 10.1126/science.275.5299.523. [DOI] [PubMed] [Google Scholar]
- 23.Chao D M, Gadbois E L, Murray P J, Anderson S F, Sonu M S, Parvin J D, Young R A. Nature (London) 1996;380:82–85. doi: 10.1038/380082a0. [DOI] [PubMed] [Google Scholar]
- 24.Zhong H, Voll R E, Ghosh S. Mol Cell. 1998;1:661–671. doi: 10.1016/s1097-2765(00)80066-0. [DOI] [PubMed] [Google Scholar]
- 25.Kawasaki H, Song J, Eckner R, Ugai H, Chiu R, Taira K, Shi Y, Jones N, Yokoyama K K. Genes Dev. 1998;12:233–245. doi: 10.1101/gad.12.2.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bannister A J, Oehler T, Wilhelm D, Angel P, Kouzarides T. Oncogene. 1995;11:2509–2514. [PubMed] [Google Scholar]
- 27.Lin R, Heylbroeck C, Pitha P M, Hiscott J. Mol Cell Biol. 1998;18:2986–2996. doi: 10.1128/mcb.18.5.2986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sato M, Tanaka N, Hata N, Oda E, Taniguchi T. FEBS Lett. 1998;425:112–116. doi: 10.1016/s0014-5793(98)00210-5. [DOI] [PubMed] [Google Scholar]
- 29.Schafer S L, Lin R, Moore P A, Hiscott J, Pitha P M. J Biol Chem. 1998;273:2714–2720. doi: 10.1074/jbc.273.5.2714. [DOI] [PubMed] [Google Scholar]
- 30.Yoneyama M, Suhara W, Fukuhara Y, Fukuda M, Nishida E, Fujita T. EMBO J. 1998;17:1087–1095. doi: 10.1093/emboj/17.4.1087. [DOI] [PMC free article] [PubMed] [Google Scholar]