

Abstract
Leukocytes roll along the endothelium of postcapillary venules in response to inflammatory signals. Rolling under the hydrodynamic drag forces of blood flow is mediated by the interaction between selectins and their ligands across the leukocyte and endothelial cell surfaces. Here we present force-spectroscopy experiments on single complexes of P-selectin and P-selectin glycoprotein ligand-1 by atomic force microscopy to determine the intrinsic molecular properties of this dynamic adhesion process. By modeling intermolecular and intramolecular forces as well as the adhesion probability in atomic force microscopy experiments we gain information on rupture forces, elasticity, and kinetics of the P-selectin/P-selectin glycoprotein ligand-1 interaction. The complexes are able to withstand forces up to 165 pN and show a chain-like elasticity with a molecular spring constant of 5.3 pN nm−1 and a persistence length of 0.35 nm. The dissociation constant (off-rate) varies over three orders of magnitude from 0.02 s−1 under zero force up to 15 s−1 under external applied forces. Rupture force and lifetime of the complexes are not constant, but directly depend on the applied force per unit time, which is a product of the intrinsic molecular elasticity and the external pulling velocity. The high strength of binding combined with force-dependent rate constants and high molecular elasticity are tailored to support physiological leukocyte rolling.
Adhesive interactions between cells play a central role in the function of the immune system. Unique among the cell–cell interactions of the immune system is the rolling of leukocytes on activated endothelium. This rolling under hydrodynamic shear forces is a first step in directing leukocytes out of the blood stream into sites of inflammation and is mediated by the selectins, a family of extended, modular, and calcium-dependent lectin receptors (1, 2). To fulfill their physiological function, the selectins and their ligands exhibit a unique combination of mechanical and biochemical properties. They associate very fast and with high affinity and can tether cells over distances of up to 100 nm by their long, chainlike structure. The selectin-ligand complexes withstand high tensile forces and terminate their binding in a controlled way to maintain rolling without themselves being pulled out of the cell membrane.
P-selectin, located on the endothelial cell wall, supports leukocyte rolling under hydrodynamic flow via interactions with its glycoprotein counterligand P-selectin glycoprotein ligand-1 (PSGL-1) expressed on microvilli of the leukocyte surface (2). P-selectin/PSGL-1 binding involves interacting sites that are located close to the amino-terminal portion of each molecule and furthest away from the cellular membrane (2). Both molecules are well characterized (2–6) and provide a prototypic system to study the kinetics and mechanics of adhesion processes at the molecular level. Here we have used recombinant proteins consisting of the extracellular portions of P-selectin and PSGL-1 expressed as chimeras with the human IgG1 constant region. To bind P-selectin, PSGL-1 requires several posttranslational modifications (3, 5), including appropriate terminal glycosylation containing the fucosylated and sialylated tetrasaccharide sialyl Lewisx (sLex) (7, 8). We show here that PSGL-1 expressed in 293 cells transfected with fucosyl transferase III (FTIII) (9) is fully functional and binds P-selectin with an affinity similar to the natural ligand.
We investigated the interaction of P-selectin with PSGL-1 under defined external forces by using atomic force microscopy (AFM) and directly measured the mechanical properties of single complexes. In AFM force-spectroscopy experiments, ligand molecules are immobilized on the tip of an AFM cantilever and their corresponding receptors on a counter surface. The AFM sensor is approached toward the surface where the molecules can bind and subsequently is retracted at a constant pulling velocity. By monitoring the cantilever deflection during such an approach-retraction cycle the binding, stretching, and rupture of receptor/ligand complexes can be investigated in terms of forces (10–16). As illustrated in Fig. 1, AFM simulates the binding, stretching, and unbinding of receptor/ligand complexes under rolling conditions. Recent theoretical considerations show that rupture forces in such experiments should depend on the pulling velocity of the force sensor (17). Here we present experimental data and some simple models on the velocity dependence of the AFM adhesion probability and rupture forces of a receptor/ligand system and we discuss their relation to kinetic and thermodynamic data obtained from standard biochemical assays.
Figure 1.

Schematic representation of selectin-ligand interaction during leukocyte rolling on venular endothelium and in AFM experiments. The approach and retraction of the AFM sensor simulates the physiological rolling process. After binding, the force on the complex increases continuously with increasing sensor-surface distance. After rupture, the chain-like molecules shrink to their equilibrium length, and their binding sites, both located close to the amino-terminal ends of the molecules, are separated.
MATERIALS AND METHODS
Production of Recombinant P-Selectin/IgG.
The cDNA for human P-selectin (18) up to and including the sixth complement repeat domain was isolated by PCR from a human lung lambda cDNA library (CLONTECH) by using primers Psel1 (GGT AAA TGC GGC CGC ACC ATG GCC AAC TGC CAA ATA GCC ATC) and Psel5 (GGC CTC TCA CCA ACT TTC TTG TCC ACT TCA CAC ATT GGT GGG GAG TCT GTC). Both primers have 5′ extensions containing either the restriction endonuclease recognition site for NotI (Psel1) or a sequence complementary to the end of the CH1 domain and part of the following intron of the human IgG1 heavy chain (Psel5). A DNA fragment encoding for genomic human IgG1 (19), encompassing the end of the CH1 domain up to and including the complete CH3 domain was PCR-amplified from plasmid 94/4.7HHB by using primers HG1CH1 (GTG GAC AAG AAA GTT GGT GAG AGG CC) and HG1CH3.NOT (GCT TGG CGG CCG CCG CAC TCA TTT ACC CGG AGA CAG). Both fragments were gel-purified and subsequently assembled by gene splicing by overlap extension PCR (20) using primers Psel1 and HG1CH3.NOT. The assembled P-selectin/IgG fragment was restricted with NotI and subcloned into the mammalian expression vector pcDNAI/neo (Invitrogen) to produce vector P6CRhg1.neo. The sequence of the P-selectin portion up to and including the sixth CR domain was verified by DNA sequencing.
CHO-K1 cells, maintained in MEMα (GIBCO) with 5% fetal bovine serum, were seeded overnight in 6-well plates and transfected with 4 μg of P6CRhg1.neo by using Lipofectamine (GIBCO) according to the manufacturer’s instructions. Cells were trypsinized and plated in three 96-well plates. After 24 hr, 0.5 mg/ml of G418 (GIBCO) was added for selection, and media were changed every few days. After 3–4 weeks the plates were checked for P-selectin/IgG production by using a human IgG ELISA (21). Cells from high-producing wells were further cloned by limiting dilution. In this manner, several P-selectin/IgG producing clones were obtained. For production, P-selectin/IgG producing clones were grown in T175 flasks including 0.5 mg/ml of G418 in the media. Media (30 ml/flask) were changed every 24 hr, pooled, and kept at −20°C.
P-selectin/IgG pooled cell supernatants were filtered and concentrated by using a YM100 membrane (Amicon). The concentrated supernatant was applied on a protein A Sepharose column (Pharmacia) and eluted by using the MAPS II buffer system (Bio-Rad). The pH of the eluate was quickly adjusted to pH 8 by the addition of 1 M Tris⋅HCl, pH 8.7. The eluate from the protein A column was concentrated on a YM100 membrane and dialyzed overnight against 20 mM Tris⋅HCl, pH 7.4. The solution then was loaded on a Mono Q HR10/10 column (Pharmacia), equilibrated with the same buffer, and eluted with a linear gradient to 1 M NaCl. Mono Q column fractions were analyzed by SDS/PAGE, pooled, concentrated, dialyzed against PBS, and stored at −20°C.
For labeling with biotin, P-selectin/IgG was diluted to 0.1 mg/ml in 100 mM borate buffer, pH 9.0, 100 mM NaCl. For every 1 mg of P-selectin/IgG, 20 μl of a 10 mg/ml dimethyl sulfoxide solution of N-(+)-biotinyl-6-aminocaproic acid N-succinimidyl ester (Fluka) was added to the solution. After a 4-hr incubation at room temperature on a rotating shaker, the unreacted biotin activated ester was quenched by the addition of 0.5 ml of 1 M Tris⋅HCl, pH 8.7. After concentration on a YM100 membrane, the solution was dialyzed against PBS, aliquoted, and stored at −20°C.
Construction of PSGL-1/IgG Chimera.
The complete cDNA for PSGL-1 was amplified by PCR from a HL-60 cDNA library. The PSGL-1 cDNA in pBluescript KS+ (Stratgene) was sequenced in its entirety. A single point mutation (A for G) at position 77 of the coding region of PSGL-1 was detected. Correction of the point mutation was performed by using the Chameleon Double-Stranded, Site-Directed Mutagenesis Kit (Stratagene) according to instructions. Sequencing of the resulting vector PSGL-1cor.ksp showed that the insert was identical to the published PSGL-1 sequence (3). For the construction of a carboxy-terminal IgG fusion of PSGL-1 we used an in-frame HincII recognition sequence located at position 942 of the published PSGL-1 cDNA sequence. An additional HincII site located in the multiple cloning site of PSGL-1cor.ksp was removed by restriction with XhoI and SalI and subsequent religation with T4 DNA ligase.
To allow insertion of desired fusion partners, we created the expression vector hum-cG1.neoI, which contains a cDNA copy encoding for a modified human IgG1 heavy chain, where valine at the carboxyl-terminal end of the CH1 region is replaced with isoleucine by the addition of an in-frame EcoRV recognition sequence. Vector hum-cG1.neoI was constructed by PCR amplification of the modified human IgG1 cDNA sequence with primers GGA TCC GAT ATC GAC AAG AAA GTT GAG CCC AAA TC and GGT GCT TGG CGG CCG CCG CAC TCA TTT ACC CGG AGA CAG by using first-strand cDNA obtained from mRNA of E-selectin/IgG producing cells (21) and subcloning the EcoRV- and NotI-restricted 0.72-bp DNA fragment into expression vector pcDNAI/neo (Invitrogen) cut with the same enzymes.
For the construction of a PSGL-1/IgG chimera expression vector, a 0.93-kb HindIII and HincII restricted DNA fragment was excised from modified PSGL1cor.ksp and subcloned into HindIII and EcoRV restricted vector hum-cG1.neoI. This vector, designated PSGL1hG1.neoI, contains an insert encoding for a fusion protein of the extracellular portion of human PSGL-1 up to and including valine-295 and a modified human IgG1 heavy chain.
Expression vector PSGL1hg1.hyg, allowing hygromycin selection of transfectants, was constructed by transfer of a 1.66-kb HindIII/XhoI-restricted DNA fragment containing the entire PSGL-1/IgG coding region from PSGL1hG1.neoI into HindIII/XhoI-restricted pcDNA(+).hyg. Expression vector pcDNA(+).hyg was constructed by inserting into the single NheI restriction endonuclease site present in pcDNAI/Amp (Invitrogen) a XbaI-restricted 1.68-kb DNA fragment encompassing the entire expression unit for hygromycin-B resistance. The fragment was obtained from vector pREP7 (Invitrogen) by PCR amplification by using primers Hyg1.Xba (GCT CTA GAG CGT TTG CTG GCG GTG TCC) and Hyg2.Xba (GCT CTA GAC CAT GGG TCT GTC TGC TCA GTC CA). In pcDNA(+).hyg both the hygromycin-B resistance gene and inserted genes/cDNAs are transcribed in the same direction.
Expression and Purification of PSGL-1 Chimera.
The complete cDNA of FTIII in vector pcDM7 (9), from J. Lowe (University of Michigan, Ann Arbor), was excised by using XhoI and subcloned into pcDNAI/neo (Invitrogen) to create the expression vector FT3.neo. Wild-type 293 cells seeded overnight in a 6-well plate were transfected with 4 μg of FT3.neo by using Lipofectamine. After 24 hr, cells were trypsinized into a T175 flask, and 0.5 mg/ml of G418 was added in the media. After selection for 4 weeks, cells were sorted by using the antibody CSLEX (Becton Dickinson). The high sLex-expressing cells were cultured for an additional 4 weeks, resorted, and cloned by using limiting dilution. Individual clones were analyzed by Fluorescence Assisted Cell Sorting by using CSLEX, and the highest sLex-expressing clone (293/FTIII) was used for production of PSGL-1/IgG.
For the construction of cell lines permanently producing PSGL-1/IgG, wild-type 293 or 293/FTIII cells were seeded overnight in 6-well plates and transfected with 4 μg of PSGL1hg1.hyg by using Lipofectamine. Cells were trypsinized into three 96-well plates, and 0.2 mg/ml of hygromycin was added in the media after 24 hr. After 4–5 weeks, wells were checked for the production of human IgG by using a human IgG ELISA (21). Clones were obtained by limiting dilution from wells with high levels of human IgG. Cells subsequently were maintained in selection antibiotic containing media.
Clones producing PSGL-1 (293/PSGL-1 and 293/FTIII/PSGL-1) were seeded in a hollow fiber cartridge (BR110, UniSyn, Tustin, CA) and maintained in a Cell-Pharm 1000 (UniSyn Technologies). MEMα with selection antibiotics was used for the circulation media, and the media inside the cartridge contained 10% ultra-low IgG fetal bovine serum. Media was harvested from the cartridge every 48 hr. After the first week of culture, selection antibiotics were withdrawn from the circulating media. PSGL-1/IgG was purified from pooled cell supernatants as indicated above for P-selectin/IgG. Purified PSGL-1/IgG was biotinylated as described above for P-selectin/IgG.
ELISA Assays.
Maxisorb assay plates (Nunc) were coated with 0.1 ml of P-selectin/IgG (4 μg/ml in PBS, overnight at 4°C) and then blocked with 0.25 ml of 2% BSA in PBS for 2 hr at room temperature. Wells then were incubated with various amounts of PSGL-1/IgG-biotin in 0.1 ml of ELISA buffer (Tris⋅HCl, pH 7.4 containing 100 mM NaCl, 2 mM CaCl2 including 0.05% Tween 20 and 0.5% BSA) for 45 min at room temperature. After washing three times with wash buffer (Tris⋅HCl, pH 7.4 containing 100 mM NaCl, 2 mM CaCl2 including 0.05% Tween 20), the plates were incubated with 0.1 ml of 1/5,000 dilution avidin/horseradish peroxidase (Jackson ImmunoResearch) in ELISA buffer for an additional 45 min. Plates then were washed six times with wash buffer, developed with 0.1 ml of 3,3′,5,5′-tetramethylbenzidine solution (Sigma) for 10 min and stopped with 0.05 ml of 1 M H2SO4.
Plasmon Resonance.
Experiments were performed on a BIAcore 2000 (Pharmacia Biosensor) at 20°C and a constant flow rate of 5 μl/min. The buffer used was Tris⋅HCl, pH 7.4 containing 100 mM NaCl, 2 mM CaCl2 (assay buffer) including 0.05% Tween 20. Biotinylated PSGL-1/IgG was captured on a sensor chip SA5 containing immobilized streptavidin on its surface. For each experimental cycle, a defined concentration of P-selectin/IgG was injected for 10 min, followed by a 10-min dissociation phase, where only buffer was passed over the surface. Regeneration of the PSGL-1/IgG surface was done with 10 mM glycycrhinic acid in assay buffer, a known inhibitor of the P-selectin/PSGL-1 interaction (22). Data were analyzed by using the BIAevaluation program (V2.1; Pharmacia Biosensor).
AFM.
Standard glass coverslips and Si3N4 AFM sensors were silanized with mercaptomethyl-dimethyl-ethoxysilane (ABCR, Karlsruhe, Germany) in gas phase at 50°C and subsequently incubated with avidin for 2 hr (2 mg/ml in PBS, pH 7, Sigma). The sensor was incubated with an excess of biotinylated PSGL-1/IgG and the coverslip with an excess of biotinylated P-selectin/IgG for 2 hr. The remaining free binding sites of the unreacted avidin were blocked by subsequently incubating both surfaces with biotin for 1 hr (1 mg/ml in PBS, pH 7, Sigma). A Nanoscope III Multimode AFM (Digital Instruments, Santa Barbara, CA) fitted with a liquid cell containing assay buffer was used for measurements. For negative controls, the assay buffer was supplemented with 5 mM EDTA instead of 2 mM Ca2+.
The spring constants of the Si3N4 AFM sensors are specified to about flever = 0.06 N m−1 (Digital Instruments). The presented data were obtained by two different sensors with spring constants of 0.064 N m−1 and 0.058 N m−1 with an accuracy of 15%. These values have been determined by measuring the individual sensor geometry by optical microscopy (length and width) and white light interferometry (thickness) (23) and verified by a careful analysis of the power spectrum density of the thermal fluctuations of the cantilevers (24). The minimum force Fdet, which can be detected by such a sensor is about 15 pN (Fdet= (kBT⋅flever)1/2). During contact between AFM tip and surface of up to 40 ms, the maximum compressive force was limited to 500 pN. Approach and pulling velocities were identical in all experiments and varied from 200 nm s−1 to 4,000 nm s−1.
RESULTS AND DISCUSSION
PSGL-1 produced in FTIII-transfected 293 cells contains high amounts of sLex (data not shown) and binds to P-selectin with high affinity, whereas PSGL-1 produced in wild-type 293 cells contains no detectable sLex and does not bind to P-selectin (Fig. 2a). As measured by ELISA, recombinant PSGL-1, under equilibrium (nonflow) conditions, exhibits a binding constant of 60 nM for P-selectin (Fig. 2b), coinciding with the 100 nM reported for the natural P-selectin/PSGL-1 interaction (25). The kinetic rates of the P-selectin/PSGL-1 interaction were determined by plasmon resonance with a kon = (4 ± 2) × 105 M−1 s−1 and an apparent koffapp = (3 ± 1) × 10−4 s−1. In these experiments no external forces are present, and molecular mobility is only diffusion limited. Therefore, the off-rate is dominated by avidity effects (26) and may be considered only as a lower limit of an intrinsic molecular off-rate.
Figure 2.

Binding of recombinant PSGL-1 to P-selectin. (a) ELISA format assay of PSGL-1/IgG-biotin binding to P-selectin/IgG-coated plates, showing the dependence of binding on fucosylation with the tetrasaccheride sLex. The production of fucosylated and nonfucosylated PSGL-1/IgG is described in Materials and Methods. (b) Inhibition curve by using a constant amount of PSGL-1/IgG-biotin and variable amounts of PSGL-1/IgG as inhibitors in the same ELISA assay to determine the binding constant. (c) An overlay of sensorgrams from plasmon resonance experiments used for determination of kinetic rate constants. The indicated amount of P-selectin/IgG was injected over the sensor surface with captured biotinylated PSGL-1/IgG. During the following dissociation phase of 10 min only buffer was injected over the sensor surface. As a control, the highest P-selectin/IgG concentration was passed over the sensor surface with captured nonfucosylated PSGL-1/IgG.
For AFM measurements, P-selectin and PSGL-1 were immobilized on glass coverslips and Si3N4 AFM tips, respectively, and probed in Ca2+ containing assay buffer. The dependence of the observed adhesion between AFM tip and surface on the formation of P-selectin/PSGL-1 complexes was verified by two controls, both of which are standard controls for the physiological selectin interaction. Binding could always be abolished by removing Ca2+ and adding EDTA to the assay buffer. The same tip/surface combination showed again normal binding events when the assay solution was replaced again with Ca2+ containing assay buffer. Additionally, in three independent experiments, tips functionalized with inactive PSGL-1/IgG produced in wild-type 293 cells, and therefore lacking sLex, showed no adhesion events to a P-selectin functionalized surface. This finding is in agreement to the ELISA results (see Fig. 2). Rupture forces at specific pulling velocities always displayed a single peak distribution (see Fig. 3), implying the rupture of a single P-selectin/PSGL-1 complex and excluding a statistically distributed simultaneous rupture of several complexes. Measurements were reproducible over hours and typically for more than 1,000 approach-retract cycles, indicating a proper fixation of the molecules to their surface via multiple biotin-avidin bonds in parallel. This indication is supported by the fact that IgG antibodies reacted with excess biotin under conditions similar to those used here are on the average modified with 10 biotin moieties per antibody (27).
Figure 3.

(a) Histogram of 584 rupture force values of P-selectin/PSGL-1 complexes obtained from AFM experiments at a pulling velocity of 2,800 nm s−1. It shows a single peak distribution with a mean rupture force of 159 pN and a SD of 30 pN. The width of the distribution reflects the stochastic nature of the rupture process. (b) Histogram of 600 rupture forces from a Monte-Carlo simulation of the experiment in a showing a mean rupture force of (168 ± 23) pN (for details see text).
Molecular Elasticity.
Typical AFM force-distance curves are shown in Fig. 4a. Upon stretching a molecular complex by increasing the distance between the AFM sensor and surface (see Fig. 1), the intramolecular forces were found to increase nonlinearly as it is typical for a chain-like system (16). The elasticity of such a system can be described by different models of polymer elasticity (28). A modified freely jointed chain (FJC) model (29) describes well the force–extension relation of the complex in the high and low force regime. It accounts not only for the entropic elasticity of a molecular chain but also for the intrinsic enthalpic elasticity of the complex, which could be stretched over its equilibrium contour length, e.g., Fig. 4a. The modified FJC model can be described by:
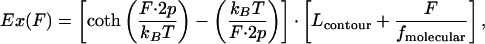 |
where kB, T, p, fmolecular, and Lcontour, respectively denote Boltzmann constant, temperature, persistence length, molecular spring constant, and molecular contour length.
Figure 4.

(a) Force-distance curves between an AFM sensor carrying PSGL-1 and a P-selectin functionalized surface. Curves at slow and fast pulling velocity, with multiple adhesion events (seen in less than 15% of curves) and with no adhesion (from top to bottom). Force values below the baseline correspond to tensile forces in the complex by retracting the AFM sensor (from left to right) until the binding ruptures and the sensor snaps back to its equilibrium position (see Fig. 1). (b) Fit of the force-extension relation of a P-selectin/PSGL-1 complex according to a model of molecular elasticity (modified FJC model, see text).
Fig. 4b shows a fit of a typical force curve to the FJC model with Lcontour = (100 ± 5) nm, fmolecular = (5.3 ± 1.5) pN nm−1 and p = (0.35 ± 0.05) nm. Here, the equilibrium contour length of the recombinant molecules was calculated with P-selectin = 27 nm (25), PSGL-1 = 50 nm (30), IgG fragment = 5 nm, and biotin/avidin complex = 5 nm. This calculation leads to a contour length of the whole complex of about 97 nm. The molecular spring constant corresponds to the slope of the force extension curve at large extensions just before rupture. A direct correlation between the obtained persistence length and the molecular structure is not straightforward because of the heterogeneous protein/carbohydrate composition of the complex. But in comparison to other molecular systems, the P-selectin/PSGL-1 complex exhibits a persistence length similar to that of cell adhesion proteoglycans (16), dextran (14), or a titin Ig domain (15) and a molecular spring constant comparable with that of a single-stranded DNA of 100 nm length (30).
Rupture Forces.
As can be deduced from Fig. 4a, the rupture forces have no constant value but continuously increase with increasing pulling velocity (see Fig. 5) up to values of 165 pN. This force value is below the estimated maximum force that can be sustained by P-selectin/PSGL-1 complexes when tethering neutrophils under flow conditions (31–33). But, compared with other AFM experiments, 165 pN is significantly higher than typical antigen/antibody binding forces of about 60 pN (12). The variation of rupture forces with pulling velocity is consistent with recent theoretical considerations (17). Upon modeling the rupture process of an individual complex as a random event, the dependence of rupture forces on pulling velocity can be described by a Monte-Carlo simulation (15). For every successive short time interval (Δt = 0.1 ms) of the stretching process, the actual force F(Ex(t)) was calculated via the FJC model by using the time-dependent extension Ex(t) = vpull⋅t, where vpull denotes the pulling velocity. By calculating the off-rate according to koff(F) = koff0 exp(−F(t)⋅spot/kBT) (15, 17), the probability of a rupture of the binding at a specific force was determined by Prupture = koff(F)⋅Δt. The status of the binding for every time interval was determined by comparing the rupture probability with the probability obtained from a random number generator (Ppoll). In case of Ppoll < Prupture, the actual force is defined as the rupture force of the complex at given pulling velocity. These calculated rupture forces at different pulling velocities were fitted to the experimentally determined rupture forces (see Fig. 5).
Figure 5.

Dependence of P-selectin/PSGL-1 rupture forces on AFM pulling velocity. Each data point represents a Gaussian fit to about 50 rupture force values. The error bars correspond to the SD of the rupture force distribution in Fig. 3. (Inset) The experimental rupture forces (data points) show a logarithmic dependence on pulling velocity and can be modeled by a Monte-Carlo simulation of the unbinding process (straight line, for details see text).
From such a fit an off-rate koff0 = 0.022 s−1 at zero external force and a mean width of the P-selectin/PSGL-1 binding potential spot = 0.25 nm was determined. By using these parameters and a pulling velocity of 2,800 nm s−1 for a Monte Carlo simulation, the distribution of rupture forces is in good agreement with the experimental distribution (Fig. 3). It is worth noting that rolling experiments with beads or cells can be described only in terms of a constant force over time and cannot account for the force dynamics of stretching processes. These experiments probably result in underestimation of spot and overestimation of koff0 (32). In contrast, the results from AFM experiments presented here take the detailed force per unit time relation and the stochastic nature of single molecule interaction events into account.
Adhesion Probability.
Another characteristic of the P-selectin/PSGL-1 binding, relevant to its biological function, was found by investigating the dependence of the adhesion probability between AFM tip and surface on the pulling velocity. The adhesion probability Padhesion is defined as the ratio between the number of force curves showing an adhesion event to the total number of curves. Interestingly, the adhesion probability in the P-selectin/PSGL-1 system increases with faster pulling velocities (Fig. 6, Inset). This phenomenon may be a general feature of selectin/ligand interactions and seems to be the underlying molecular property of the increase in leukocyte tethering probability with increased shear flow observed in L-selectin rolling experiments (34). In contrast, AFM experiments with biotin-avidin, antigen-antibody, or cell adhesion proteoglycans have shown a constant adhesion probability that is independent of pulling velocity (10–13).
Figure 6.

The time-dependent adhesion probability between a P-selectin functionalized surface and a PSGL-1-activated AFM tip reflects the fast forced off-rate of the P-selectin/PSGL-1 binding. The interaction time was varied upon changing the AFM pulling velocity (Inset). Each data point represents 30 approach-retraction cycles. The decrease in adhesion probability after long interaction times corresponds to a lifetime of the complex in the AFM experiment of about 70 ms.
In general, the adhesive interaction of an AFM tip to a surface depending on a specific biomolecular reaction can be described by the differential equation dPadhesion(t)/dt = −krupture Padhesion(t) + kbind [1 − Padhesion(t)], where Padhesion(t) denotes the probability of an adhesion event between AFM tip and surface during retraction. krupture and kbind are the molecular rupture and binding rate of the interaction under AFM conditions. krupture directly corresponds to a forced off-rate koff, and kbind can be converted into an on-rate via an effective concentration according to kbind = kon⋅ceff(d). The effective concentration ceff describes the number of binding partners within the intersection volume of the accessible space for molecules on the AFM tip and on the surface and depends on the distance (d) between both. Because our system has a very fast on-rate and shows adhesion probabilities up to nearly 100% at fast pulling velocities, the binding is considered to be instantaneously initiated by contact between tip and surface. A rebinding after rupture of the stretched complex is inhibited because the molecules shrink to their equilibrium length and their binding sites are separated (see Fig. 1). Therefore, immediately after rupture, ceff equals zero. The above-mentioned differential equation can be simplified for the P-selectin/PSGL-1 interaction in AFM experiments according to dPadhesion(t)/dt = −koff Padhesion(t). By using the boundary condition Padhesion (t = 0) = 1 the instantaneous binding at contact between tip and surface is taken into account and the missing kbind term accounts for the inhibited rebinding.
To describe its kinetics, a time scale of the AFM experiment has to be estimated. As can be calculated by the FJC model, forces in the complex during the first 60 nm of its extension are below the AFM detection limit of about 15 pN. Therefore, a rupture event can be detected only after a certain interaction time tint = sdet/vpull, defined by the distance traveled by the tip (sdet) after contact to this 15 pN detection limit divided by the pulling velocity (vpull). Consequently, the slower the pulling velocity the more time a complex has to unbind without being stretched significantly, leading to a decrease of observable adhesion events with longer interaction times (see Fig. 6). Using this timescale, the velocity dependence of the adhesion probability of a P-selectin/PSGL-1 complex can be converted into a time-dependent adhesion probability. It can be described as a first-order exponential decay according to the simplified differential equation (see above) with an off-rate of koff = (15 ± 2) s−1. This fast off-rate accounts for a “forced” unbinding process under the externally applied forces of the AFM experiment.
Energetics.
The kinetic and mechanical data can be directly compared by estimating the energies involved. The unforced binding constant KD = koff0/kon = 55 nM, obtained from the Monte Carlo simulation (koff0) and plasmon resonance (kon), is consistent with the equilibrium binding constant of 60 nM from ELISA and corresponds to a binding energy of 41 kJ mol−1. The energy required to change the off-rate koff = koff0 exp(ΔG/kBT) from the unforced to the forced value is 16 kJ mol−1. As can be calculated by integrating the force-extension function this energy is considerably smaller than the mechanical energy of 200 kJ mol−1 required to stretch a complex during the interaction time. This finding indicates that most of the mechanical energy is absorbed by working against the thermal motion of the chain-like P-selectin/PSGL-1 complex and only a fraction of the mechanical energy influences the binding kinetics. This fraction, however, is sufficient to account for the three orders of magnitude change from unforced to forced off-rates.
CONCLUSIONS
We have shown that the rupture of an adhesive interaction between single molecules can be well described by a stochastic, nondeterministic process. Description of a receptor/ligand binding strength either in terms of kinetics or from a purely mechanical point of view is incomplete, because both are coupled via the time dependence of the involved forces. Therefore, the rupture of a receptor/ligand complex can occur at variable forces and lifetimes, either by resting a long time at small forces or within a short time under high forces. Our Monte-Carlo simulation shows that rupture forces are mainly influenced by the off-rate of the receptor ligand interaction. As a consequence longer lifetimes of a complex should cause higher rupture forces. The on-rate seems to have no influence on rupture forces but can influence the adhesion probability. Therefore, rupture processes and forces are not adequately described by a thermodynamic equilibrium constant but by a kinetic rate constant. In general, changes in adhesion probabilities and forces in AFM experiments with pulling velocity are expected for interactions where the kinetic rate constants are on a time scale comparable to that of AFM experiments of about milliseconds to minutes.
Previous studies on selectin systems did not allow the direct determination of force, distance, and time parameters of single binding events. Although in the AFM experiment the adhesion molecules are immobilized on a hard surface instead of anchored in a cell membrane, this setup allows determination of their intrinsic molecular properties that are the basis for the physiological rolling process. Compared with other ligand/receptor systems studied under external forces the P-selectin/PSGL-1 system exhibits a fast forced off-rate and a high chain-like elasticity. Even at small extensions entropic forces of thermal motion accelerate the unbinding of the complex. After rupture the molecules immediately shrink to their equilibrium length and their binding sites separate. The physical separation prevents a rebinding of the same complex and can explain why selectins are not able to mediate firm adhesion of leukocytes to endothelium (31, 33). Furthermore, the weak logarithmic dependence of the molecular binding forces on pulling velocity (see Fig. 5) can explain the weak dependence of leukocyte rolling velocities on flow rates (35). The long contour length of the complex together with its high molecular elasticity reduces the mechanical loading on the receptor/ligand binding and allow leukocyte rolling even at high shear rates.
Leukocyte rolling is a good example of how the mechanical properties of single molecules determine the characteristics of cell-cell adhesion. Here we show that such processes do not depend only on solution affinities, but are also strongly influenced by mechanical stability, applied forces, and topology at the molecular level. Molecular probing techniques, such as AFM, provide the opportunity to manipulate and investigate single molecules or cells and obtain crucial information about adhesion processes not obtainable by solution methods.
Acknowledgments
We thank J. Lowe for providing the FTIII gene, H. Towbin for the use of the BIAcore 2000, and L. Scandella and E. Schürmann for technical help. Additional support from H.-J. Güntherodt and a critical reading of the manuscript by J. Engel is gratefully acknowledged.
ABBREVIATIONS
- AFM
atomic force microscopy
- FJC
freely jointed chain
- FTIII
fucosyl transferase III
- PSGL-1
P-selectin glycoprotein ligand-1
- sLex
sialyl Lewis x
References
- 1. Kansas G S. Blood. 1996;88:3259–3287. [PubMed] [Google Scholar]
- 2.McEver R P, Moore K L, Cummings R D. J Biol Chem. 1995;270:11025–11028. doi: 10.1074/jbc.270.19.11025. [DOI] [PubMed] [Google Scholar]
- 3.Sako D, Chang X-J, Barone K M, Vachino G, White H M, Shaw G, Veldman G M, Bean K M, Ahern T J, Furie B, et al. Cell. 1993;75:1179–1186. doi: 10.1016/0092-8674(93)90327-m. [DOI] [PubMed] [Google Scholar]
- 4.Moore K L, Patel K D, Bruehl R E, Fugang L, Johnson D A, Lichenstein H S, Cummings R D, Bainton D F, McEver R P. J Cell Biol. 1995;128:661–671. doi: 10.1083/jcb.128.4.661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sako D, Comess K M, Barone K M, Camphausen R T, Cumming D A, Shaw G D. Cell. 1995;83:323–331. doi: 10.1016/0092-8674(95)90173-6. [DOI] [PubMed] [Google Scholar]
- 6.Pouyani T, Seed B. Cell. 1995;83:333–343. doi: 10.1016/0092-8674(95)90174-4. [DOI] [PubMed] [Google Scholar]
- 7.Moore K L, Eaton S F, Lyons D E, Lichenstein H S, Cummings R D, McEver R P. J Biol Chem. 1994;269:23318–23327. [PubMed] [Google Scholar]
- 8.Li F, Erickson H P, James J A, Moore K L, Cummings R D, McEver R P. J Biol Chem. 1996;271:3255–3264. [PubMed] [Google Scholar]
- 9.Kukowska-Latallo J F, Larsen R D, Nair R P, Lowe J B. Genes Dev. 1990;4:1288–1303. doi: 10.1101/gad.4.8.1288. [DOI] [PubMed] [Google Scholar]
- 10.Moy V T, Florin E-L, Gaub H E. Science. 1994;266:257–259. doi: 10.1126/science.7939660. [DOI] [PubMed] [Google Scholar]
- 11.Dammer U, Popescu O, Wagner P, Anselmetti D, Güntherodt H-J, Misevic G N. Science. 1995;267:1173–1175. doi: 10.1126/science.7855599. [DOI] [PubMed] [Google Scholar]
- 12.Dammer U, Hegner M, Anselmetti D, Wagner P, Dreier M, Huber W, Güntherodt H-J. Biophys J. 1996;70:2437–2441. doi: 10.1016/S0006-3495(96)79814-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hinterdorfer P, Baumgartner W, Gruber H J, Schilcher K, Schindler H. Proc Natl Acad Sci USA. 1996;93:3477–3481. doi: 10.1073/pnas.93.8.3477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rief M, Oesterhelt F, Heymann B, Gaub H E. Science. 1997;275:1295–1297. doi: 10.1126/science.275.5304.1295. [DOI] [PubMed] [Google Scholar]
- 15.Rief M, Gautel M, Oesterhelt F, Fernandez J M, Gaub H E. Science. 1997;276:1109–1111. doi: 10.1126/science.276.5315.1109. [DOI] [PubMed] [Google Scholar]
- 16.Fritz J, Anselmetti D, Jarchow J, Fernandez-Busquets X. J Struct Biol. 1997;119:165–171. doi: 10.1006/jsbi.1997.3887. [DOI] [PubMed] [Google Scholar]
- 17.Evans E, Ritchie K. Biophys J. 1997;72:1541–1555. doi: 10.1016/S0006-3495(97)78802-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Johnston G I, Cook R G, McEver R P. Cell. 1989;56:1033–1044. doi: 10.1016/0092-8674(89)90636-3. [DOI] [PubMed] [Google Scholar]
- 19.Ellison J W, Berson B J, Hood L E. Nucleic Acids Res. 1982;10:4071–4079. doi: 10.1093/nar/10.13.4071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Horton R M, Hunt H D, Ho S N, Pullen J K, Pease L R. Gene. 1989;77:61–68. doi: 10.1016/0378-1119(89)90359-4. [DOI] [PubMed] [Google Scholar]
- 21.Kolbinger F, Patton J T, Geisenhoff G, Aenis A, Li X, Katopodis A G. Biochemistry. 1996;35:6385–6392. doi: 10.1021/bi9524528. [DOI] [PubMed] [Google Scholar]
- 22.Rao B N N, Anderson M B, Musser J H, Gilbert J H, Schaefer M E, Foxall C, Brandley B K. J Biol Chem. 1994;269:19663–19666. [PubMed] [Google Scholar]
- 23.Neumeister J M, Ducker W A. Rev Sci Instr. 1994;65:2527–2531. [Google Scholar]
- 24.Hutter J L, Bechhoefer J. Rev Sci Instr. 1993;64:1868–1873. [Google Scholar]
- 25.Ushiyama S, Laue T M, Moore K L, Erickson H P, McEver R P. J Biol Chem. 1993;268:15229–15237. [PubMed] [Google Scholar]
- 26.Nieba L, Krebber A, Plueckthun A. Anal Biochem. 1996;243:155–165. doi: 10.1006/abio.1996.0067. [DOI] [PubMed] [Google Scholar]
- 27.Bayer E A, Wilchek M. Methods Enzymol. 1990;184:146–147. doi: 10.1016/0076-6879(90)84257-h. [DOI] [PubMed] [Google Scholar]
- 28.Wang M D, Yin H, Landick R, Gelles J, Block S. Biophys J. 1997;72:1335–1346. doi: 10.1016/S0006-3495(97)78780-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Smith S B, Cui Y, Bustamante C. Science. 1996;271:795–799. doi: 10.1126/science.271.5250.795. [DOI] [PubMed] [Google Scholar]
- 30.Li F, Erickson J A, James J A, Moore K L, Cummings R D, McEver R P. J Biol Chem. 1996;271:6342–6348. doi: 10.1074/jbc.271.11.6342. [DOI] [PubMed] [Google Scholar]
- 31.Lawrence M B, Springer T A. Cell. 1991;65:859–873. doi: 10.1016/0092-8674(91)90393-d. [DOI] [PubMed] [Google Scholar]
- 32.Alon R, Hammer D A, Springer T A. Nature (London) 1995;374:539–542. doi: 10.1038/374539a0. [DOI] [PubMed] [Google Scholar]
- 33.Chen S, Alon R, Fuhlbrigge R C, Springer T A. Proc Natl Acad Sci USA. 1997;94:3172–3277. doi: 10.1073/pnas.94.7.3172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Alon R, Chen S, Puri K D, Finger E B, Springer T A. J Cell Biol. 1997;138:1169–1180. doi: 10.1083/jcb.138.5.1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Atherton A, Born G V R. J Physiol. 1973;233:157–165. doi: 10.1113/jphysiol.1973.sp010303. [DOI] [PMC free article] [PubMed] [Google Scholar]