

Abstract
Most genetic regulatory mechanisms involve protein–DNA interactions. In these processes, the classical Watson–Crick DNA structure sometimes is distorted severely, which in turn enables the precise recognition of the specific sites by the protein. Despite its key importance, very little is known about such deformation processes. To address this general question, we have studied a model system, namely, RecA binding to double-stranded DNA. Results from micromanipulation experiments indicate that RecA binds strongly to stretched DNA; based on this observation, we propose that spontaneous thermal stretching fluctuations may play a role in the binding of RecA to DNA. This has fundamental implications for the protein–DNA binding mechanism, which must therefore rely in part on a combination of flexibility and thermal fluctuations of the DNA structure. We also show that this mechanism is sequence sensitive. Theoretical simulations support this interpretation of our experimental results, and it is argued that this is of broad relevance to DNA–protein interactions.
In many protein–DNA complexes, the double helix is deformed severely from the classical Watson–Crick B-form by being bent, stretched, and untwisted (1–6). Although it is often presumed that protein binding can induce such deformations, the mechanisms by which this occurs are understood poorly (7, 8). To address this question, we have performed micromanipulation studies of the binding kinetics of RecA, a bacterial protein involved in homologous recombination (9, 10), to a single double-stranded DNA (dsDNA). This particular protein was chosen because of its nonspecific binding to dsDNA, which lengthens the dsDNA by a factor of 1.5. It is possible with this model system to monitor the binding kinetics by measuring this lengthening in a micromanipulation experiment and to use force to probe the mechanism by which dsDNA is deformed on protein binding.
The double helix under external tension exhibits a transition to an overstretched state (S-DNA) 1.7 times longer than its B-DNA length (11, 12). It is also known that, in specific conditions (involving the presence of a nonhydrolysable analog of ATP:ATP-γS), RecA polymerizes along and lengthens dsDNA by a factor of 1.5 (13). Thus, RecA-dsDNA complexes present a situation in which DNA conformational changes caused by protein binding can be coupled to an external force. Using this idea, we studied the rate of RecA polymerization onto a single dsDNA under fixed tension, grafted between an optical fiber, acting as a force transducer, and a latex bead manipulated with a micropipette mounted on piezoelectric transducers.
MATERIALS AND METHODS
Experimental Setup for Force Measurements.
Force–extension and kinetics measurements used a microscope built on carbon-epoxy composite structure board (Newport, Fountain Valley, CA) and Invar mounts to reduce thermal expansion. To measure forces in the piconewton range, the diameter of the optical fiber had to be reduced down to ≈5 to 15 μm. This was achieved through chemical etching in 40% hydrofluoric acid at 60°C for 15 to 20 min. Experiments were performed in plastic cuvettes into which the optical fiber was introduced horizontally; a vertical micropipette mounted on a PZT stage (Physik Instrument, Waldbronn, Germany) was used for bead manipulation. A laser diode (λ = 670 nm) was coupled through an optical isolator (made with a polarizer and a quarter-wave plate) into the optical fiber, the tip of which was imaged through a 40× objective (Zeiss) onto a two-quadrant photodiode (Hamamatsu, Middlesex, NJ) used as a position detector. The currents delivered by the two quadrants were converted into voltages; these voltages were amplified differentially with a low noise voltage amplifier (SR560, Stanford Research, Sunnyvale, CA), which fed a digital data acquisition board (AT-MIO16X, National Instruments, Austin, TX) plugged into a personal computer. Fiber deflection was calibrated by positioning the micropipette on the fiber and recording a voltage–position curve; experimental data were corrected for nonlinearities of the position detector. The fiber stiffness was determined by using the equipartition theorem applied to measurements of thermal displacements of its end. The experiment was computer-controlled by using labview software (National Instruments). Force–extension data acquisition was done at 300 Hz for, typically, 60 sec. For kinetic measurements, force was fixed by using a feedback loop programmed under labview. Drifts (≈1 μm/h from distortion of fiber position with respect to detector) were eliminated by relaxing the DNA molecule back to a random coil conformation every ≈200 sec and taking the measured voltage in this position as a new zero for force measurements. By doing this, force was maintained constant to within 5 pN during the 2–3 h of a typical kinetic run.
Preparation of DNA Molecules.
λ-EMBL3 phage DNA was purified by using standard techniques (Qiagen Lambda Kit). An adenonine- and thymidine-rich (AT-rich) dsDNA (156Gmac) was obtained from a pUXI3 plasmid (pUXI3 was derived from pUC19 by the insertion of a macronuclear DNA sequence, 156Gmac, from Paramecium primaurelia at the BamHI site of the polylinker) and was purified (Qiagen Plasmid Kit). Our labeling procedure was based on the ligation of two short dsDNA fragment (732 bp) containing labeled nucleotides at multiple positions to the ends of the DNA molecule. These fragments were obtained in the following way. PCR synthesis of a biotin or digoxygenin labeled fragment (732 bp in length) was performed in the presence of biotin or digoxygenin-dUTP in the reaction mixture. PCR was performed by using pUXI3 plasmid as a template. One of the primers was chosen so as to contain an EcoRI restriction site, and the second one was chosen so that this EcoRI site was unique in the amplified sequence that was part of 156Gmac. This particular sequence (156Gmac), had been chosen because of its high AT content (70%), which allowed maximization of the number of incorporated labels. The molar ratio of biotin (or digoxygenin)-dUTP to dTTP is 1/9. For a 700-bp DNA segment, the expected number of labels per molecule was 50 on the average. After PCR synthesis, the fragment was purified from the primers and nucleotides by using QIAquick PCR purification kit (Qiagen) and then was concentrated by ethanol precipitation. The PCR fragment then was cut with the restriction enzyme EcoRI, was purified by using a QIAquick PCR purification kit, and was concentrated by ethanol precipitation. The label (biotin, but more frequently dig) may have caused the inhibition of some enzymes, especially if the labeled nucleotide dUTP was present in the recognition site. This effect was diminished strongly because the EcoRI site was chosen in the primer. Indeed, in this case, only one strand of the recognition site was labeled because the other was part of the primer itself (and therefore unlabeled).
In the case of λ-EMBL3, an adapter consisting of two oligonucleotides (Genset) that hybridize over part of their length was ligated by the EcoRI end to one of the two PCR labeled fragments (dig- or biotin-labeled). The adapter-ligated PCR fragment was purified from the adapter (QIAquick PCR purification kit), was concentrated by ethanol precipitation, and was ligated to the left end of λ-DNA. The final product was purified from a low melting point agarose gel by using standard agarase treatment. An analogous procedure was used to multilabel the right end with the other labeled fragment.
In the case of 156Gmac, the procedure was somewhat different. First, 156Gmac was excised from the pUXI3 plasmid by BamHI restriction, followed by a low melting point agarose gel purification. A labeled EcoRI-digested PCR fragment similar to those described above then was ligated to the two BamHI extremities of the 156Gmac with the use of linker oligonucleotides and then was purified from a low melting point agarose gel. One of the two labeled fragments was removed by a PfIM1 restriction; this enzyme had a unique restriction site in the 156Gmac close to the BamHI site. Again, this was followed by low melting point agarose gel purification. The second labeled PCR fragment then was ligated to this free end of the 156Gmac with the use of linker oligonucleotides and finally was purified from a low melting point agarose gel.
Grafting of DNA to Bead and Optical Fiber.
Amino-coated 3-μm-diameter latex beads (Polysciences) were coated with streptavidin (Boehringer Mannheim) by using covalent coupling by glutaraldehyde (Electron Microscopy Grade, Sigma). The fiber was coated with aminosilane (Sigma) in 90% ethanol and finally was coated with antidigoxygenin (Boehringer Mannheim) by the same coupling method used for the beads.
Streptavidin-coated beads and dsDNA with digoxygenin- and biotin-labeled ends were incubated for 1 h to allow attachment of some dsDNA to the beads; after this incubation, the stoichiometry was approximately three dsDNA per bead as determined by agarose gel. The DNA-covered beads were injected into the measurement cell, where some of them became tethered to the antidigoxygenin-coated optical fiber via a single dsDNA. All experiments were performed at 26 (±1)°C. After 1 h of incubation, the cell was washed with RecA polymerization buffer (50 mM Tris⋅HCl/10 mM MgCl2/20 mM NaCl/100 μM EDTA, pH 7.5) and a force–extension curve was obtained to ensure that a single DNA molecule was anchored between the fiber and the bead (Fig. 1A, solid line).
Figure 1.

(A) Force vs. extension for λ-EMBL3, both before (solid line) and after (dashed line) complete RecA polymerization, showing elongation of the dsDNA. The difference in low-force behaviors (<10 pN) of the two curves shows that the nucleofilament has a larger persistence length than the initial dsDNA molecule. In the case of the nucleofilament, such force–extension curves are independent of the force applied during RecA polymerization. (B) Time dependence of elongation of a single λ-EMBL3 during RecAbinding for various fixed forces in presence of 1 mM ATP-γS. The initial time dependence of the elongation is highly reproducible, but the late time behavior (i.e., beyond 80% of the full elongation caused by RecA binding) is much more variable from one experiment to another. This might be because of nicks that, as RecA binds, become covered, making progressively more of the dsDNA twist-blocked and thereby limiting the rate of the final stages of polymerization.
RecA (New England Biolabs) and ATP-γS (final concentrations, 40 ng/μl RecA and 1 mM ATP-γS) then were injected into the cell while the DNA molecule was relaxed. Immediately after RecA injection, the dsDNA was subjected to a fixed force through a feedback loop (acting on the micropipette displacement), which kept the fiber deflection constant. RecA polymerization was monitored through measurement of the displacement of the micropipette necessary to maintain the force constant. Once polymerization was complete, another force–extension curve was obtained that characterized the elastic behavior of the complete RecA-dsDNA complex (Fig. 1A, dashed line).
RESULTS AND DISCUSSION
RecA Binding to λ-EMBL3 DNA.
Experiments performed on λ-EMBL3 (48,502 bp, 16.5-μm B-form contour length) showed RecA binding kinetics (followed by measuring DNA extension) to be affected strongly by the force applied (Fig. 1B): At 75 pN, the reaction was 20× faster than at 15 pN. Below 55 pN, RecA polymerization elongated B-DNA whereas above 55 pN, RecA polymerization shortened the initial S-DNA. In this latter case RecA does work against the external force. For each force, 10 different molecules were used to study RecA binding kinetics, and for extension <80% of that of the final RecA-dsDNA product, the kinetics were reproducible (see legend of Fig. 1B).
These results deserve several comments. First of all, one should note that RecA polymerization is affected by the supercoiling state of dsDNA and unnicked circular dsDNA plasmids are not fully covered by RecA (14). In our experiments, care was taken to work only with nicked linear dsDNA. Unnicked molecules with multiple grafting sites on both strands at each end do not exhibit a sharp overstretching transition near 50–60 pN (15). Experiments described here were performed only on molecules exhibiting the sharp overstretching transition (Fig. 1A), which is a signature of the presence of nicks along the molecule. Though our force–extension data prove the existence of at least one nick along the molecule used for kinetic studies, the possibility that the initiation of RecA polymerization occurs predominantly at nicks can be ruled out by two arguments. First, RecA is known to polymerize along unnicked, closed circular plasmids (14); therefore, nicks are not needed for nucleation of RecA polymerization along dsDNA. Second, the reproducibility of our experiments is not compatible with such a hypothesis: In the case of predominant nucleation at nicks, because there is no reason for each investigated molecule to have the same number of nicks, the kinetics would be strongly nonreproducible, at least in their initial behaviors.
Finally, it is interesting to note that force–extension curves (Fig. 1A) show that the dsDNA-RecA filament has a much larger persistence length (LP) than the bare dsDNA. It was very difficult with our setup (force resolution of ≈1 pN) to give a precise estimate of this persistence length in the case of the RecA-dsDNA complex. Attempts to fit our force–extension data to a continuum elasticity model (17), taking into account the Young modulus of the molecule, gave LP = 75 nm for λ-EMBL3 and a crude estimate of 800 nm for the dsDNA-RecA complex; this latter value is consistent with previous measurement by electron microscopy (LP = 600 nm) (16).
It is difficult to compare the kinetics under controlled extension to the kinetics in the absence of force. The kinetics at zero force could be monitored by measuring, at various time intervals, the force–extension curves of the complex, but, as shown here, as soon as we stretch the dsDNA, we speed up the binding kinetics, and this is not a true zero-force kinetic measurement. We should mention that, in the course of our kinetic experiments, we found tethered DNA molecules with no RecA bound even after 12 h of incubation in the RecA polymerization buffer at 26°C. It is then possible to infer that, in our conditions, the zero-force polymerization rate is much slower than the one determined under tension.
Thermal Conformation Fluctuations of dsDNA Play a Role in RecA Binding.
Understanding the phenomena described above starts with the recognition that dsDNA is a flexible molecule. Force–extension data (Fig. 1A) show the free-energy difference between B and S-DNA to be only 3kBT per bp. One may therefore expect dsDNA to exhibit thermal fluctuations of its structure. These fluctuations are characterized by a probability distribution P(u) for local stretching, u, of DNA under tension [Fig. 2; obtained from fitting a continuum elasticity model (17) to the force–extension curve of bare DNA given in Fig. 1A]. For B-DNA, the mean local stretching is 〈u〉 = 0, and, for S-DNA, 〈u〉 = 0.7. The probability of the state with stretching comparable to that in the RecA-dsDNA complexes, P(u = 0.5), increases with increasing force and reaches its maximum for forces above the overstretching transition (17). We propose that RecA binds to thermally stretched dsDNA; therefore, its binding and polymerization rely on coupling of DNA conformational fluctuations to protein–DNA and protein–protein interactions. This explains how external force accelerates RecA binding and polymerization, as well as the lengthening of B-DNA and the shortening of S-DNA on RecA binding.
Figure 2.

Probability distribution P(u) for local stretching, u, of a dsDNA molecule for various applied forces. The bimodal behavior observed for 55 pN is caused by the coexistence of B-DNA and S-DNA on the transition plateau (see force–extension data in Fig. 1A). The asymmetry in P(u) for 75 pN arises because of the steric limit, u ≈ 0.9, where the dsDNA phosphodiester backbones are stretched fully. Inset shows P(u) close to u = 0.5.
Monte Carlo Simulations of RecA Polymerization Controlled by Conformation Fluctuations of dsDNA.
This mechanism can be illustrated by using a simple kinetic model in which DNA stretching is described by using a one-dimensional Ising model with interactions chosen to reproduce the overstretching plateau. A string of integers, di, represents the DNA state (di = 0 for B-DNA; di = 1 for S-DNA). Each integer “site” represents three bp of DNA and therefore one RecA monomer-binding site. Force is coupled to DNA length, to which each site contributes 1.0 or 1.7 nm for di = 0 or di = 1 for bare DNA. RecA binding is described by additional integers ni (ni = 0 for bare DNA; ni = 1 for RecA bound); a DNA site with bound RecA is taken to be 1.5 nm in length. The energy of the DNA-RecA degrees of freedom is:
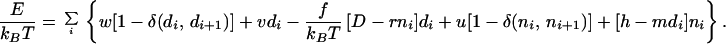 |
The first three terms describe DNA overstretching; the parameters for cooperativity of the B-DNA to S-DNA transition (w = 2.0), free energy difference (in units of kBT) between B and S-DNA (ν = 11.5), length increase per site during overstretching (D = 0.7 nm), and reduction of this length increase by RecA binding (r = 0.2 nm) are determined by bare dsDNA force extension curves (Fig. 1A). The final three terms describe RecA binding cooperativity (13) (u = 3.0), RecA binding free energy (including translational entropy loss) in the absence of DNA stretching (h = 3.0), and a strong shift in binding energy when DNA is stretched (m = 20.0). Precise estimates of the net binding free energy of RecA-dsDNA binding are not available, but our interactions are compatible with available data for enthalpy of RecA-ssDNA binding (18, 19). At zero force, a two-site (6 bp), RecA-stretched DNA region has a binding energy of -kBT and is therefore stable whereas an isolated RecA on stretched DNA has positive energy and is not thermodynamically stable.
Metropolis Monte Carlo dynamics with random choices for site updates were used (20). DNA structural fluctuations occurred on a timescale (nanoseconds) short compared with the time between successive DNA-RecA monomer encounters (microseconds to milliseconds); thus the dis were updated 104 times more often than the nis. Apart from this separation of timescales, details of the dynamics are unimportant as long as they are local and satisfy detailed balance.
Fig. 3 shows simulation results for RecA binding to DNA for this model, which includes only thermal fluctuations and attractive protein-DNA and protein-protein interactions. According to these simulations, polymerization begins at nucleation islands, which form when thermally stretched regions of DNA bind RecA and which then grow as additional RecAs bind to thermally stretched DNA at the island ends as observed in previous electron microscopy experiments (13). Above 40 pN, the increase in probability for local DNA stretching enhances both the nucleation and growth processes. This enhancement with increasing force continues up to 100 pN. Above 70 pN, there is no thermal barrier for DNA stretching in the model (the DNA is already stretched by the applied force), and polymerization occurs at a rate limited only by access of RecA to DNA (in the high force regime, other processes, such as DNA unwinding, not included in the present model are likely to become rate-limiting). By only taking into account variations of thermally activated DNA structural fluctuation with the applied force, this simple model qualitatively reproduces experimental results both below and above the transition to S-DNA.
Figure 3.

Time dependence of elongation of a dsDNA molecule obtained from an Ising-type model for various applied forces.
Enhancement of the Kinetics of RecA Binding by Sequence Dependence of dsDNA Elasticity.
To further test this picture of protein–dsDNA interactions mediated via DNA conformational fluctuations, we performed the same experiments on a different DNA molecule (156Gmac, 15,742-bp dsDNA, 5.35-μm contour length in B-form). This dsDNA is 70% AT (λ-EMBL3 is ≈50% AT). Bare DNA force–extension curves (Fig. 4A) show that 156Gmac is “softer” than λ-EMBL3 (see legend of Fig. 4A for an explanation of this “softness”). This softness of 156Gmac means that this molecule is easier to deform than λ-EMBL3; therefore, there will be larger thermally activated deviations from B-form and consequently accelerated RecA binding, as evidenced by comparison of the RecA binding kinetics for 156Gmac to those for single and dimer λ-EMBL3 (Fig. 4B). This sequence-dependence is in agreement with our theoretical picture; also, the fact that single and dimer λ-EMBL3 undergo the same reaction time course agrees with what is to be expected for homogeneous nucleation of RecA-dsDNA, that is, existence of a characteristic distance between nuclei and therefore a characteristic reaction time independent of DNA length. Such a sequence-dependent elasticity can be included in our model, and we anticipate that this will lead to nucleation of dsDNA-RecA complexes at soft AT-rich regions of the molecule.
Figure 4.

(A) Force vs. extension data for λ-EMBL3 (solid line) and 156Gmac (dotted line). These two curves were obtained from a single preparation by using a single fiber, thus avoiding any complication of comparison caused by slight changes in calibration of fiber displacement and/or stiffness. Two sets of beads (each one carrying either λ-EMBL3 or 156Gmac) were injected in the cuvette. Thus, two different types of beads became grafted to the fiber via either λ-EMBL3 or 156Gmac. Force–extension diagrams shown here result from successive experiments performed on the two different sets of beads. Extensions are given as fractions of B-form fully extended length to allow comparison of the two different molecules. A fit to the extensible worm-like-chain model (21) gives the persistence length A and the stretching modulus γ for the two molecules. For 156Gmac, A = 55 nm and γ = 270 nm−1; for λ-EMBL3, A = 75 nm and γ = 450 nm−1. The transition to S-DNA starts at 50 pN for 156Gmac instead of 55 pN for λ-EMBL3, and the transition width (in terms of force) is larger for 156Gmac than for λ-EMBL3. Finally, the overstretching of 156Gmac (u = 0.75) is larger than the overstretching of λ-EMBL3 (u = 0.65). According to these data, 156Gmac should exhibit stronger local structural thermal fluctuations than λ- EMBL3. (B) Comparison of RecA binding kinetics to λ-EMBL3, λ-EMBL3 dimer, and 156Gmac for a constant externally applied force of 45 pN. Extension is plotted as a fraction of B-form length to allow comparison of the three different molecules. Comparison of λ-EMBL3 and λ-EMBL3 dimer shows that the faster RecA binding in the case of 156Gmac is caused by the differing base content and is not an artifact of differing molecule lengths.
CONCLUSION
The mechanism proposed here for RecA polymerization is extremely simple, yet it captures all of the essential features of experimental kinetics measurements. In particular, it explains very simply how RecA can bind and polymerize along S-DNA as well (and even faster) as along B-DNA. Other mechanisms can be proposed to describe our observations: RecA first could bind to dsDNA and then stretch it, or RecA could bind dsDNA via induced-fit interactions. Although such mechanisms may seem plausible a priori, they are not fully satisfying for various reasons. In the first alternative mechanism, one has to assume that RecA binds at least as well to B-DNA as to S-DNA, though their conformation is likely very different (21). For both alternative mechanisms, it is difficult to explain with simple energetic considerations how RecA binds S-DNA faster than B-DNA because we have no precise knowledge of the energy landscape of the various dsDNA conformations involved in this problem; indeed, the final structure of dsDNA in the dsDNA-RecA complex is neither B- nor S-DNA, and studies of bp orientations even suggest that it could be closer to B form than to S form (7, 21). On the contrary, in our proposed mechanism, we only assume that dsDNA conformation suitable for RecA binding arises naturally because of thermal fluctuations, and we immediately infer from this the main trends of our kinetic experiments. Needless to say, we do not pretend that our description is complete; indeed, one may imagine that, in the final stages of RecA-dsDNA binding, further fine tuning (different from the ones arising from spontaneous thermal fluctuations) of the structures of both RecA and dsDNA may occur.
The nonspecificity of RecA binding to DNA, resulting in macroscopic DNA stretching, makes it an excellent candidate for single-molecule study of the fluctuation–selection binding mechanism. However, our results suggest what might be an important intrinsic property of dsDNA for its interaction with proteins: its flexibility and structural fluctuations. In the formation of other protein–DNA complexes, DNA fluctuations might play a key role, and those fluctuations might help to understand how deformed DNA structures arise in protein–DNA complexes (1–6).
Acknowledgments
This work is dedicated to the memory of the late François Caron. We acknowledge fruitful discussions with K. Adzuma, R. Lavery, T. Duke, G. V. Shivashankar, T. Ebbesen, and all of the members of the “Groupe Paramécie” at Laboratoire de Génétique Moléculaire de l’ENS. Acknowledgement is made to the Fondation pour la Recherche Médicale, to Direction des Recherches Etudes et Techniques–Ministère des Armées, to Centre National de la Recherche Scientifique Mission pour la Physique et de Chimie du Vivant, to North Atlantic Treaty Organization, to the Donors of The Petroleum Research Fund administered by the American Chemical Society for partial support of this research, to The Whitaker Foundation for partial support through a Biomedical Engineering Research Grant, and to the Research Corporation for partial support through a Research Innovation Award.
ABBREVIATION
- dsDNA
double-stranded DNA
- AT-rich
adenonine- and thymidine-rich
References
- 1. Kim Y, Geiger J H, Hahn S, Sigler P B. Nature (London) 1993;365:512–520. doi: 10.1038/365512a0. [DOI] [PubMed] [Google Scholar]
- 2.Juo Z S, Chiu T K, Leiberman P M, Baikalov I, Berk A J, Dickerson R E. J Mol Biol. 1996;261:239–254. doi: 10.1006/jmbi.1996.0456. [DOI] [PubMed] [Google Scholar]
- 3.Love J L, Li X, Case D A, Giese K, Grosschedl R, Wright P E. Nature (London) 1995;376:791–795. doi: 10.1038/376791a0. [DOI] [PubMed] [Google Scholar]
- 4.Schumacher M A, Choi K Y, Zalkin H, Brennan R G. Science. 1994;266:763–770. doi: 10.1126/science.7973627. [DOI] [PubMed] [Google Scholar]
- 5.Werner M H, Gronenborg A M, Clore G M. Science. 1996;271:778–784. doi: 10.1126/science.271.5250.778. [DOI] [PubMed] [Google Scholar]
- 6.Werner M H, Huth J R, Gronenborg A M, Clore G M. Cell. 1995;81(5):705–714. doi: 10.1016/0092-8674(95)90532-4. [DOI] [PubMed] [Google Scholar]
- 7.Takahashi M, Norden B. Adv Biophys. 1994;30:1–35. doi: 10.1016/0065-227x(94)90009-4. [DOI] [PubMed] [Google Scholar]
- 8.Lebrun A, Shakked Z, Lavery R. Proc Natl Acad Sci USA. 1997;94:2993–2998. doi: 10.1073/pnas.94.7.2993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Clark A J, Margulies A D. Proc Natl Acad Sci USA. 1965;53:451–459. doi: 10.1073/pnas.53.2.451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Travers A A. Curr Opin Struct Biol. 1992;2:71–77. [Google Scholar]
- 11.Cluzel P, Lebrun A, Heller C, Lavery R, Viovy J-L, Chatenay D, Caron F. Science. 1996;271:792–794. doi: 10.1126/science.271.5250.792. [DOI] [PubMed] [Google Scholar]
- 12.Smith S B, Cui Y, Bustamante C. Science. 1996;271:795–799. doi: 10.1126/science.271.5250.795. [DOI] [PubMed] [Google Scholar]
- 13.Stasiak A, Di Capua E, Koller T. J Mol Biol. 1981;151:557–564. doi: 10.1016/0022-2836(81)90010-3. [DOI] [PubMed] [Google Scholar]
- 14.Stasiak A, Di Capua E. Nature (London) 1982;299:185–186. doi: 10.1038/299185a0. [DOI] [PubMed] [Google Scholar]
- 15.Cluzel P. Ph.D. thesis. Université Paris 6; 1996. [Google Scholar]
- 16.Egelman E H, Stasiak A. J Mol Biol. 1986;191:677–697. doi: 10.1016/0022-2836(86)90453-5. [DOI] [PubMed] [Google Scholar]
- 17.Marko J F. Phys Rev E Stat Phys Plasmas Fluids Relat Interdiscip Top. 1998;57:2134–2149. [Google Scholar]
- 18.Takahashi M. J Biol Chem. 1989;264:288–295. [PubMed] [Google Scholar]
- 19.Wittung P, Ellouze C, Maraboeuf F, Takahashi M, Norden B. Eur J Biochem. 1997;245:715–719. doi: 10.1111/j.1432-1033.1997.00715.x. [DOI] [PubMed] [Google Scholar]
- 20.Binder K. Application of the Monte-Carlo Method in Statistical Physics. Berlin: Springer; 1987. pp. 1–36. [Google Scholar]
- 21.Lebrun A. Ph.D. thesis. Université Paris 6; 1997. [Google Scholar]