

Abstract
As a step toward understanding their functional role, the low frequency vibrational motions (<300 cm−1) that are coupled to optical excitation of the primary donor bacteriochlorophyll cofactors in the reaction center from Rhodobacter sphaeroides were investigated. The pattern of hydrogen-bonding interaction between these bacteriochlorophylls and the surrounding protein was altered in several ways by mutation of single amino acids. The spectrum of low frequency vibrational modes identified by femtosecond coherence spectroscopy varied strongly between the different reaction center complexes, including between different mutants where the pattern of hydrogen bonds was the same. It is argued that these variations are primarily due to changes in the nature of the individual modes, rather than to changes in the charge distribution in the electronic states involved in the optical excitation. Pronounced effects of point mutations on the low frequency vibrational modes active in a protein-cofactor system have not been reported previously. The changes in frequency observed indicate a strong involvement of the protein in these nuclear motions and demonstrate that the protein matrix can increase or decrease the fluctuations of the cofactor along specific directions.
Any fundamental description of a biological process must ultimately encompass an account of small and possibly large scale changes in the nuclear geometry of the participating molecules. In particular, a central element in the determination of reaction efficiencies is the structure and accessibility of the transition state, which is the highest point on the free energy barrier that opposes changes in nuclear geometry as the system moves from reactant to product state. For proteins, atoms are most easily displaced along the “soft” directions—the delocalized, low frequency modes—and the study of such motion during a reaction is therefore of particular interest.
In recent years, a new aspect of low frequency motion has been revealed by the observation of coherent nuclear motion, persisting on the picosecond (ps) timescale following impulsive optical excitation, and manifested as oscillations in the transient optical properties of the protein cofactors. The discovery of this phenomenon in a variety of protein-cofactor systems, including the bacterial reaction center (RC) (1, 2), bacteriorhodopsin (3), and heme proteins (4), implies that ultrafast biological processes can occur on a timescale that is faster than vibrational relaxation (5). Therefore the possibility of coherent (in addition to thermal) motion along the reaction coordinate must be included in any complete description of a reaction. In addition, the measurement of oscillations by using femtosecond (fs) transient spectroscopy provides a new and convenient method for probing the spectrum of low frequency, nuclear vibrations that are set in motion by a physiological trigger. This study is aimed at assessing the factors that influence the character of such nuclear motions, by exploiting the possibility of changing the local structure of the protein through site-directed mutagenesis. We show that the mutation of a single amino acid residue can bring about a large change in the frequencies of the vibrational modes that are set in motion by impulsive, optical excitation, indicating that the nature of the low frequency motions can be unexpectedly sensitive to the specific local geometry.
The model system used is the RC (6) from the purple bacterium Rhodobacter sphaeroides. This protein uses light to drive the transfer of an electron across the dielectric of the bacterial inner membrane, establishing a transmembrane potential difference that is ultimately used to drive energy-requiring reactions such as the synthesis of ATP. The reaction is initiated by excitation of the primary donor of electrons (P), a dimer of bacteriochlorophyll (Bchl) located close to the periplasmic face of the RC. Following creation of the first singlet excited state of P (P*), an electron is transferred to a molecule of bacteriopheophytin (HL) located ≈17 Å from P and half way across the membrane. This reaction is extremely rapid, taking place in ≈3 ps at room temperature and ≈1 ps at 10 K (7). In the present study, our primary interest is in the vibrational modes that are coupled to the P→P* transition because they give rise to coherent, directed motions that persist on the timescale of electron transfer (8). These vibrations are most easily observed as oscillations in the stimulated emission from the P* state, with a phase change of π for wavelengths on opposite sides of the emission maximum of P* (2). Vibrations in the ground-state of P also may be excited, through ground-state depletion or by impulsive stimulated Raman scattering, but can be distinguished from those in P* because the oscillations corresponding to ground-state vibrations change phase at the absorption maximum of P (9).
The molecular “identity” of the nuclear motions associated with formation of the P* state is essentially unknown. Their low frequency (mostly <250 cm−1) suggests that they are delocalized over the protein environment to a certain extent. However, Raman-active low frequency modes have been interpreted as being essentially localized in the Bchl macrocycle (10, 11) and the coordinating histidine axial ligand (12). We previously have studied coherent nuclear motion in RCs with mutations in the residue Tyr-M210 (13), which is located within van der Waals distance of P, HL, and the intervening monomeric Bchl, BL. This residue is not directly bonded to any of these cofactors but is thought to influence their electronic interaction. Mutation to His, Leu, or Trp did not affect the frequencies of the nuclear modes. In the present work, we have examined the effects of modification of the bonding between cofactor and protein by studying mutants designed to add or remove hydrogen bonds between the protein and the Bchls of P.
The Bchls of the P dimer (denoted PL and PM) both possess one acetyl carbonyl group and one keto carbonyl group that can in principle accept a hydrogen bond from the surrounding protein. The structure of the wild-type (wt) RC is such that only one of the four possible hydrogen bonds is formed, between His-L168 and the 2-acetyl carbonyl group of PL (14–16). Fig. 1 shows the PL and PM Bchls, together with the residues that have been mutated in this work. Extensive studies carried out by Allen and coworkers (17) have established that the hydrogen bond between His-L168 and the 2-acetyl carbonyl group of PL can be broken by mutation of this residue to Phe (mutation denoted HL168F). In the reverse manner, a hydrogen bond can be formed to the 2-acetyl carbonyl of PM by mutation of the symmetry-related residue Phe-M197 to His (FM197H) (18), and to the 9-keto carbonyl groups of PL and PM by mutation of residues Leu-L131 and Leu-M160 respectively to His (LL131H and LM160H) (19, 20). In all four of these mutants, high frequency Fourier transform (FT)-Raman spectroscopy shows the expected changes in spectrum that indicate the formation or breakage of the particular hydrogen bond (21).
Figure 1.

Stereoview of the two Bchl molecules, PL and PM, that form the primary donor of electrons in the bacterial RC, together with the residues that have been altered. The dotted line represents the hydrogen bond between HL168 and PL present in wt RCs.
A number of studies have shown that the electronic states of P are affected strongly by the pattern of hydrogen bonding to the surrounding protein, and the consequences for the electrochemical properties of the P dimer and the rate of electron transfer have been examined (18, 22–28). Here we have investigated how the nuclear dynamics of the P dimer are affected by modifications in the manner in which the dimer is attached to the protein medium. Three of the mutations specified above have been constructed, namely HL168F, FM197H, and LL131H, together with additional mutations to Leu and Asp at the L168 position (denoted HL168L and HL168D). All of these mutations bring about changes in the frequency of the observed vibrational modes.
METHODS
Preparation of Mutant RCs.
Single site mutations were introduced into the RC pufL and pufM genes by using mismatch oligonucleotides. For mutations LL131H, HL168F, HL168L, and HL168D, the template for mutagenesis was plasmid pALTXS-1, which consisted of a 849-bp XbaI–SalI restriction fragment encompassing the first 261 codons of the pufL gene, cloned into the plasmid pALTER-1 (Promega). For mutation FM197H, the template was plasmid pALTSB-1, which consisted of a 998-bp SalI–BamHI restriction fragment encompassing the last 22 codons of the pufL gene plus the whole of the pufM gene, also cloned into the plasmid pALTER-1. Base changes were confined to the target codon and were confirmed by DNA sequencing. Mutant pufL and pufM genes were transferred into the plasmid pRKEH10D for expression in the double deletion/insertion mutant strain DD13 as described in detail elsewhere (29–31). The resulting strains, named according to the mutation carried (FM197H and so on), were devoid of antenna complex and so contained the mutant RC as the sole pigment protein complex. The control strain was RCO2 (32), which lacks the antenna complexes but contains wt RCs. Experimental material consisted of intracytoplasmic membranes prepared from cells of each strain grown under semiaerobic conditions in the dark, as described (31).
Femtosecond Absorption Spectroscopy.
Intracytoplasmic membranes were suspended in 20 mM Tris buffer (pH 8.0) and were mixed with an equal volume of glycerol. The concentration of the sample was adjusted to an optical density of <0.4 at 880 nm (optical path length 1 mm). To prereduce the quinone electron acceptor QA, 50 mM DTT was added. The arrangement of the femtosecond spectrometer was as described (13). In brief, the sample was excited with ≈30 fs pulses centered at 880 nm, and the transient absorption was probed between 800 and 1000 nm by a continuum pulse, with parallel polarization of the two pulses and a variable time delay between excitation and measuring pulse. Absorption difference spectra ΔA(λ,t) were acquired with time steps of 33 fs on a 4-ps time scale. All experiments were performed at 18 K.
Noise Reduction by Singular Value Decomposition.
In general, the kinetic response of the sample at different wavelengths is correlated strongly, whereas some of the measurement noise is uncorrelated. This fact was used to reduce the noise level by performing singular value decomposition of the data matrix (33):
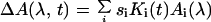 |
1 |
In this expression, Ki(t) and Ai(λ) are unit vectors representing kinetic traces and associated spectra, and si are monotonically decreasing singular values. Correlated signals can be described by a few terms with large singular values, whereas a large number of components, each with a small si, are required to account for the uncorrelated noise. Following singular value decomposition (33), the data matrix ΔA was reconstructed, retaining only the highest singular values. Typically, approximately eight components contained discernible signal (34).
FT of the Oscillatory Signal.
To separate out the oscillatory parts of the signal, the data at each wavelength were fitted to a single or double exponential decay and the oscillations obtained as the residual difference between data and fit. Residuals from ≈200 fs after the pump pulse (see below) and out to 40-ps delay were padded with zeroes to give an array of 128 points, which was then Fourier transformed to obtain the spectrum of the oscillations. In the resulting discrete FT spectrum, a change corresponding to the frequency difference between subsequent points (8 cm−1) is significant. Fig. 2 illustrates the steps in the data analysis. The kinetics were analyzed in this way throughout the region of stimulated emission from P*, at intervals of 5 nm, and the FT amplitudes were added to obtain an average spectrum of the oscillations for each mutant.
Figure 2.

Example of the data processing. The transient A at 935 nm of the HL168L RC was chosen as sample data. (Upper) The upper curve shows the raw data. Removing uncorrelated noise by singular value decomposition filtering gives the middle curve. To extract the oscillations, the filtered data were fitted to a biexponential decay (not shown). The residuals (data minus fit) are shown in the lower curve; the FT of this curve gives the frequency spectrum shown in the Lower panel.
It should be noted that the procedure of subtracting a “nonoscillating” decay from the signal by using an exponential fit can introduce artifacts into the spectrum of the oscillations. It is our experience that the shape of the frequency spectrum below 30 cm−1 depends strongly on the fit used but that no reasonable fit can completely remove the signals in this region (8, 35).
In addition to the absorption change of excited molecules, the measured signal displays rapid variations around t = 0 due to the optical Stark effect (36). As discussed above, imperfect fitting can give rise to spurious peaks in the spectrum of oscillations, and for rapidly varying features, these artifacts may fall in the spectral region of interest (50–200 cm−1). Hence, it is necessary to begin both fitting and applying the FT procedure well after t = 0, as shown in Fig. 2.
RESULTS
The transient absorption was measured for the wt RC and the five mutant proteins. Fig. 3 shows plots of the signals at selected wavelengths at the red side of the stimulated emission band, showing the decay of P*-stimulated emission and typical oscillatory features that arise from coherent nuclear motion. Both properties of the signal are seen to be affected by the mutations.
Figure 3.

Transient absorption data. The plot shows kinetic data for wt RC and for the single site mutants at selected wavelengths at the red side of the stimulated emission band (cf. ref. 2) that display typical decay times and magnitudes of the oscillatory features.
Overall Decay.
Even in the absence of oscillations, it is not possible to extract a single, overall curve that describes the decay of the P* state, because the spectral evolution of the stimulated emission in the first few ps is quite complex (7, 37–39). The presence of oscillations further complicates the picture. The practical approach is to take the transient signal at the stimulated emission maximum, where the amplitude of the oscillations is lowest (2, 40). The decay thus defined is known to be multiexponential in the wt RC at low temperatures. We found >90% of the amplitude of the decay in a component with a lifetime of 1.2 ps. The HL168F, HL168L, and HL168D mutant proteins displayed significantly faster kinetics of P* decay than the wt RC, but showed qualitatively the same behavior, with a main component (>90%) of 0.8 ps in all three cases. The decay observed with the LL131H and FM197H RCs, on the other hand, was much slower (Fig. 3). Data recorded to a delay time of 500 ps for the FM197H RC showed that the decay was highly multiphasic, with components at all time scales out to several hundred picoseconds (not shown). Fitting such a curve by a sum of exponential terms is highly degenerate, in the sense that many quite different sets of parameters can all describe the data well (41). We have therefore refrained from presenting any fit parameters for the P* decay in these two mutants.
Oscillatory Features.
Fig. 4 shows the averaged vibrational spectrum for each of the RCs studied. The spectrum of the wt RC has main peaks at 62 cm−1, 94 cm−1, 125 cm−1 and 156 cm−1. Taking into account the 8 cm−1 resolution, these compare well with those published previously from kinetic analyses performed at single wavelengths (8, 13, 35). It is evident that the mutations induce large changes in the vibrational spectrum.
Figure 4.

Spectra of vibrational modes coupled to the P→P* transition. The spectra are averages over the wavelengths at which there were visible oscillations in the stimulated emission of P*. The spectra are normalized with the maximum value of the kinetics at the stimulated emission maximum.
Addition of a Hydrogen Bond at L131.
In the LL131H RC, a hydrogen bond is introduced, at the edge of the P dimer, to the 9-keto carbonyl of PL (19, 20). The vibrational spectrum in this mutant was a little different from that in the wt RC. The two most intense peaks in the wt spectrum, at 94 cm−1 and 156 cm−1, were conserved but shifted to slightly lower frequencies. A new peak appeared at 54 cm−1, but the dependence of this feature on the probe wavelength showed that it was due to a vibration in the ground-state (data not shown). Very weak oscillations arising from ground-state vibrational modes have been reported recently for wt RCs (34). The enhancement of such oscillations is consistent with the shift of the P→P* transition absorption maximum from 892 nm in the wt RC to 904 nm in the LL131H RC (cf. ref. 25) because the amplitude of the ground-state vibrations is expected to be proportional to the detuning of the excitation pulse (at 880 nm) relative to the absorption maximum (C.R., M.H.V., and J.-L.M., unpublished results). Data at long wavelengths, where ground-state absorption does not contribute to the signal, suggested that the feature at 62 cm−1 in the spectrum of the wt RC shifts to a lower frequency and merges with the feature arising from the ground-state mode at ≈54 cm−1 in the averaged spectrum of the LL131H RC.
Addition of a Hydrogen Bond at M197.
In the FM197H mutant, a hydrogen bond is introduced at the symmetry-related position to the existing hydrogen bond donated by His-L168 (18). This modification of the protein-cofactor coupling induced very drastic changes in the observed vibrational frequencies. The frequency spectrum consists of a broad feature centered at ≈100 cm−1, with peaks at 86 cm−1 and 102 cm−1 (Fig. 4). The peak at 156 cm−1 in the spectrum of the wt RC completely disappeared, and the peaks at 62 cm−1 and 125 cm−1 at most remained as shoulders on the broad signal in the FM197H RC. The signal at 94 cm−1 in the wt RC may have been shifted to either 86 cm−1 or 102 cm−1 in the mutant, but it was not possible to establish an unambiguous correspondence between the peaks observed for the mutant and for the wt RC.
Removal of a Hydrogen Bond at L168.
In the HL168F mutant RC, the naturally occurring hydrogen bond between His-L168 and the 2-acetyl carbonyl of PL is broken (17). The HL168F mutant showed a rich spectrum of modes, with peaks at 117 cm−1, 133 cm−1, 39 cm−1, 172 cm−1 (in order of decreasing intensity), and several others, all displaying a wavelength dependence that is characteristic of excited state vibrational modes. As with the FM197H mutation, an unambiguous correspondence to the vibrational spectrum seen in the wt RC could not be constructed.
To examine the effect of changing the structure of the protein at the L168 position, over and above the breakage of the hydrogen bond donated by His-L168, mutations to Leu (HL168L) and Asp (HL168D) were constructed, in addition to the HL168F RC described above. For both mutants, the changes in the FT-Raman spectrum and in the P/P+ redox potential (Em) with respect to the wt RC were found to be similar to the changes in the HL68F mutant (D.S., T. A. Mattioli, R. W. Visschers, M.R.J., and B. Robert, unpublished data). These results strongly suggest that, as for the HL168F RC (21), the 2-acetyl carbonyl group is no longer hydrogen bonded to the protein in the HL168L or HL168D mutant.
Although the geometrical shapes of the residues of leucine and phenylalanine are rather different, there are similarities in that both residues are entirely apolar, and they have similar van der Waals volumes, 124 Å3 for Leu and 135 Å3 for Phe (42). It is therefore remarkable that the frequency spectra of the HL168F and HL168L mutants showed clear differences (Fig. 4). The spectrum of the HL168L RC had peaks at 156 cm−1, 125 cm−1, and 94 cm−1, and bore some resemblance to the spectrum of the wt RC, although the relative intensities of the peaks were very different.
Introduction of an aspartic acid at the L168 position represents a more drastic change in local structure. Not only is the van der Waals volume smaller than either Phe or Leu (91 Å3), but the polar or charged carboxyl group probably has a strong influence on the electronic structure of PL. The spectrum of the HL168D RC showed peaks at 62 cm−1, 94 cm−1, and possibly around 117 cm−1, in addition to a strong feature at 23 cm−1, which may have been partly artifactual. In contrast to the other L168 mutants, there was no significant vibrational strength in the spectrum above 125 cm−1. The overall amplitude of the oscillations in the kinetic data was significantly smaller for this mutant (see Fig. 3). At present, the reason for this behavior is not understood. However, the width of the P-band was somewhat larger for the HL168D RC than for the HL168L and HL168F mutants (not shown), possibly indicating that the origin of this effect is a larger inhomogeneity in protein-cofactor structure.
DISCUSSION
The Origin of Frequency Shifts.
Our principal finding is that the spectrum of observed vibrational modes is altered drastically by modification of the pattern of hydrogen bonds between the Bchls of P and the surrounding protein (Fig. 4). Physically, these changes can have two origins: (i) Electronic: If the modification of P and its surroundings changes the properties of the ground (P) and excited (P*) states then, in principle, the coupling between the change in electronic configuration and the bath of vibrational modes (the “electron-phonon” couplings) can be altered. As a consequence, the P → P* transition may set in motion another group of vibrations, with different frequencies. (ii) Vibrational: If the addition or removal of a hydrogen bond modifies the inter-atomic forces, this may affect the geometries of the vibrational modes. This can alter the electron-phonon couplings (band intensities) and frequencies (band position) of the modes. The dependence of the spectral changes on the pattern of hydrogen bonds then gives, in principle, information on the contribution of specific atomic displacements to the composition of the modes.
To evaluate the relative importance of electronic vs. vibrational effects, other experimental results must be used. The change in charge distribution upon P → P* excitation is in principle composed of intrachromophore redistributions as well as charge transfer between the two chromophores (i.e., PL+PM− or PL−PM+). It is, unfortunately, difficult to directly characterize the charge distribution in P* for the different mutants. However, such a characterization is available for the oxidized P+ state, for which the charge also is distributed unequally between PL and PM. Rautter et al. (26) have performed Electron Paramagnetic Resonance (EPR), Electron Nuclear Double Resonance (ENDOR), and Electron Nuclear Triple Resonance (TRIPLE) measurements on the LL131H, FM197H, and HL168F RCs examined in the present study and observed no significant changes in the spin density within either PL+ or PM+. By combining their results with the IR-spectroscopic measurements of Nabedryk et al. (20), they also concluded that the resonance integral βD, which is a measure of the strength of the Bchl–Bchl interaction, is unaffected by the mutations. They observed large shifts in the charge distribution between PL and PM, but these changes do not correlate with the differences we observe. The charge distribution between PL and PM in the LL131H RC is different to that in the wt RC but similar to that in the HL168F RC. In contrast, the vibrational spectrum of the LL131H RC is similar to that of the wt RC and different to that of the HL168 RC. Although these results do not directly relate to the excited state, they strongly suggest that the electronic states are not altered in a way that would greatly change the individual electron-phonon couplings and that the changes of the vibrational spectrum are due to a significant extent to modified inter-atomic force constants.
Origin and Identity of the Vibrational Modes.
The results presented above show that the low frequency vibrations of a protein-embedded cofactor can be profoundly altered by a point mutation in the surrounding protein. It is important to realize, that if a change in the protein-cofactor coupling affects a vibrational mode significantly, then the protein must take part in the vibration. Therefore, our findings imply that chromophore-driven motions of the protein moiety play an important role in these low frequency vibrations, and they do not lend support to proposals that they are essentially cofactor modes (10, 11).
Two observations are particularly striking. First, mutations in residues not bound to P produce distinct changes in the vibrational spectrum. This is seen when comparing, for example, the spectrum of the HL168L and HL168F RCs. This observation shows that even protein–cofactor interactions that do not involve a direct bond are sufficiently strong to alter the character of particular vibrational modes. Generally consistent with this observation, in several mutant RCs of the same type as those studied here, surprisingly large frequency shifts in Fourier-transform IR P/P+ difference spectra compared with that of wt RCs were reported for carbonyl groups that are not in the immediate vicinity of the targeted site of hydrogen bond alteration (20). Second, modification of residues close to the interface of the Bchls in the P dimer (M197F and L168H) induces much larger changes than the modification of a residue more distant from this interface (L131H). This result suggests that at least some of the observed low frequency modes are associated with this interface region and that the mutations affecting the hydrogen bonds to the 2-acetyl carbonyl groups, or the packing of the protein in those regions, may change the steric interaction between PL and PM. The overlap of PL and PM is such that the 2-acetyl carbonyl group of each Bchl is located immediately adjacent to the magnesium atom of the other Bchl. The orientation of this acetyl group may therefore affect the separation, or the freedom of movement relative to one another, of PL and PM. In this context, it has been demonstrated recently in a RC with a Phe to Arg mutation at the M197 position that the 2-acetyl carbonyl group of PM is rotated out of the plane of the Bchl ring relative to the orientation of the substituent group in the wt RC (44). Determination of the crystal structures of the HL168F and FM197H mutant RCs is currently in progress.
Raman spectroscopy, using excitation that is in resonance with the P → P* transition, probes the same vibrational modes that are observed in the present study as coherent oscillations, except that Raman spectroscopy gives the frequency of modes associated with the ground-state. Spectra obtained with the two techniques show good general agreement (10, 45, 46, 47); so it is of interest to compare the results of the present work with those from resonance Raman investigations.
In a study of the low frequency spectrum of the P dimer, the monomeric accessory Bchls (B) of the RC, and isolated Bchl in a film, Palaniappan et al.(10) deduced a one-to-one correspondence between the observed vibrations in all three forms of Bchl and therefore concluded that all of these vibrations were attributable to modes internal to the Bchl macrocycle. In particular, a consideration of physically reasonable force constants led them to rule out the presence of any true intradimer modes in the spectrum of P. Moreover, from investigation of the HM202L RC mutant, in which the Bchl at PM is replaced by a bacteriopheophytin, they inferred that the observed modes are essentially localized on the PL half of the dimer. These conclusions are at odds with the present results because the vibrational spectrum obtained for the wt RC is profoundly altered when a hydrogen bond is introduced to PM in the FM197H mutant (Fig. 4). This observation argues against the idea that the low frequency modes associated with P* are confined to PL. Clearly, a low frequency resonance Raman study of mutants with altered hydrogen bonding would be of great interest in addressing this question further.
Recently, Czarnecki et al. (11) investigated the geometry of the low frequency vibrations by using isotope substitution of the N and Mg atoms of P and by theoretical modeling of a Bchl monomer. The model included an imidazole ligated to the Mg atom, to simulate the histidine axial ligand associated with all of the RC Bchls, but did not include any additional protein matrix. The conclusion from this work was that the modes are localized mainly near the interface of the two halves of the Bchl dimer. This is in general agreement with our finding that the strongest modifications in the vibrational pattern are induced by mutations adjacent to the interface area. However, as stated above, our results indicate that the protein moiety also must be taken into account for a correct description of these vibrational modes.
With the present data set, a clear correlation of the peaks in the frequency spectra of the different RCs is not possible, and we can only say that the mutations induce large changes in the spectrum and, by extension, in the underlying vibrational modes. This ambiguity prevents assignment of the modes at a molecular level. At present, we are attempting to clarify possible correlations by extending this study to multiple mutants. In principle, an alternative pathway which could lead to a more precise assignment of the observed vibrational modes is the use of molecular dynamics simulations. In a recent study (48), spectra were calculated for the wt RC that exhibited similar features to those observed in the experimental spectra. In future work, the inclusion of the chromophore self-energies may enhance the quantitative correspondence between simulation and experiment.
Speed of Electron Transfer.
In the set of L168 mutants examined in this report, electron transfer from P* to P+HL− occurred somewhat more rapidly than in the wt RC, whereas it was much slower and multiphasic in the FM197H and LL131H mutants. This multiphasic behavior prohibits the quantitative use of standard Marcus theory to describe electron transfer (49). Qualitatively, the slowing down of the electron transfer in the mutants with additional hydrogen bonds can be explained by a decrease in the driving force for the reaction. This is expected from the increase in Em P/P+ of 80 mV and 125 mV measured for the wt LL131H and FM197H RC respectively, relative to the wt RC (18). In titrations on membrane-bound RCs, the L168 set of mutant RCs had an Em P/P+ of between 80 and 115 mV lower than that in the wt RC (D.S., R.W.V., and M.R.J., unpublished data). Despite the expected consequent increase in driving force, the rate of electron transfer was found to speed up only modestly at 18 K in the present study and not at all in the case of detergent-solubilized HL168F RCs at ambient temperature (17). Within Marcus theory, a lower sensitivity to an increase in driving force than to a decrease is expected when one approaches the activationless regime, in which the driving force equals the reorganization energy. If the reorganization energy is assumed to be the same for all mutants, it could be tentatively assigned a value that is a little higher than the value of the driving force in the wt RC. The temperature dependence of the electron transfer rate in R. sphaeroides wt RCs is well described by the formula for an activationless reaction (7), using a model in which the modes are thermally populated on the timescale of the reaction (50). The motions coupled to the P → P* transition are in fact described by a superposition of coherent oscillations rather than by thermally activated motions. At low temperatures, this may make the reaction insensitive to small activation barriers, as will be discussed elsewhere.
Functional Relevance of Changes in Vibrational Motion.
The vibrations that we observe are those coupled to the P → P* excitation, and they are not necessarily involved in the transition from P* to the charge-separated state. Indeed, a comparison between the decay times and the vibrational spectra in Fig. 4 gives no indication that the change in vibrations directly influence the time constant for electron transfer. However, the fact that the surrounding protein matrix affects the vibrations coupled to the P → P* transition suggests that the frequencies of any modes coupled to the electron transfer reaction driven by the P* state could be altered in a similar way, by accommodation of the protein. This would correspond to a change of the reorganization energy. A close analogy is provided by the activity of enzymes, which is believed to reside in an ability to lower the free energy barrier between reactant states and product states of bound molecules, by stabilizing the transition state. In general, it is very difficult to demonstrate this mechanism. Our results may provide a means to address this question by directly showing that the protein environment can have a significant influence on the low frequency motions of a molecule bound to the protein.
Acknowledgments
C.R. was supported by a grant from the Carlsberg Foundation. D.S. is supported by the Human Capital and Mobility programme of the European Community. J.P.R. is a Biotechnology and Biological Sciences Research Council Postgraduate student, and M.R.J. is a Biotechnology and Biological Sciences Research Council Advanced Research Fellow. M.H.V. is supported by Centre National de la Recherche Scientifique.
ABBREVIATIONS
- RC
reaction center
- Bchl
bacteriochlorophyll,
- wt
wild type
- FT
Fourier transform
References
- 1. Vos M H, Lambry J-C, Robles S J, Youvan D C, Breton J, Martin J-L. Proc Natl Acad Sci USA. 1991;88:8885–8889. doi: 10.1073/pnas.88.20.8885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Vos M H, Rappaport F, Lambry J-C, Breton J, Martin J-L. Nature (London) 1993;363:320–325. [Google Scholar]
- 3.Wang Q, Schoenlein R W, Peteanu L A, Mathies R A, Shank C V. Science. 1994;266:422–424. doi: 10.1126/science.7939680. [DOI] [PubMed] [Google Scholar]
- 4.Zhu L, Li P, Huang M, Sage J T, Champion P M. Phys Rev Lett. 1994;72:301–304. doi: 10.1103/PhysRevLett.72.301. [DOI] [PubMed] [Google Scholar]
- 5.Owrutsky J C, Raftery D, Hochstrasser R M. Annu Rev Phys Chem. 1994;45:519–555. doi: 10.1146/annurev.pc.45.100194.002511. [DOI] [PubMed] [Google Scholar]
- 6.Hoff A J, Deisenhofer J. Phys Rep. 1997;287:1–247. [Google Scholar]
- 7.Fleming G R, Martin J-L, Breton J. Nature (London) 1988;333:190–192. [Google Scholar]
- 8.Vos M H, Jones M R, Hunter C N, Breton J, Martin J-L. Biochemistry. 1994;33:6750–6757. doi: 10.1021/bi00188a002. [DOI] [PubMed] [Google Scholar]
- 9.Jonas D M, Bradforth S E, Passino S A, Fleming G R. J Phys Chem. 1995;99:2594–2608. [Google Scholar]
- 10.Palaniappan V, Schenck C C, Bocian D F. J Phys Chem. 1995;99:17049–17058. [Google Scholar]
- 11.Czarnecki K, Diers J R, Chynwat V, Erickson J P, Frank H A, Bocian D F. J Am Chem Soc. 1997;119:415–426. [Google Scholar]
- 12.Czarnecki K, Chynwat V, Erickson J P, Frank H A, Bocian D F. J Am Chem Soc. 1997;119:2594–2595. [Google Scholar]
- 13.Vos M H, Jones M R, Breton J, Lambry J-C, Martin J-L. Biochemistry. 1996;35:2687–2692. doi: 10.1021/bi9521708. [DOI] [PubMed] [Google Scholar]
- 14.Yeates T O, Komiya H, Chirino A, Rees D C, Allen J P, Feher G. Proc Natl Acad Sci USA. 1988;85:7993–7997. doi: 10.1073/pnas.85.21.7993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.El-Kabbani O, Chang C-H, Tiede D, Norris J, Schiffer M. Biochemistry. 1991;30:5361–5369. doi: 10.1021/bi00236a006. [DOI] [PubMed] [Google Scholar]
- 16.Ermler U, Michel H, Schiffer M. J Bioenerg Biomembr. 1994;26:5–15. doi: 10.1007/BF00763216. [DOI] [PubMed] [Google Scholar]
- 17.Murchison H A, Alden R G, Allen J P, Peloquin J M, Tagushi A K W, Woodbury N W, Williams J C. Biochemistry. 1993;32:3498–3505. doi: 10.1021/bi00064a038. [DOI] [PubMed] [Google Scholar]
- 18.Lin X, Murchison H A, Nagarajan V, Parson W W, Allen J P, Williams J C. Proc Natl Acad Sci USA. 1994;91:10265–10269. doi: 10.1073/pnas.91.22.10265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Williams J C, Alden R G, Murchison H A, Peloquin J M, Woodbury N W, Allen J P. Biochemistry. 1992;31:11029–11037. doi: 10.1021/bi00160a012. [DOI] [PubMed] [Google Scholar]
- 20.Nabedryk E, Allen J P, Tagushi A K W, Williams J C, Woodbury N W, Breton J. Biochemistry. 1993;32:13879–13885. doi: 10.1021/bi00213a017. [DOI] [PubMed] [Google Scholar]
- 21.Mattioli T A, Williams J C, Allen J P, Robert B. Biochemistry. 1994;33:1636–1643. doi: 10.1021/bi00173a004. [DOI] [PubMed] [Google Scholar]
- 22.Wachtveitl J, Farchaus J W, Das R, Lutz M, Robert B, Mattioli T A. Biochemistry. 1993;32:12875–12886. doi: 10.1021/bi00210a041. [DOI] [PubMed] [Google Scholar]
- 23.Woodbury N W, Peloquin J M, Alden R G, Lin X, Lin S, Tagushi A K W, Williams J C, Allen J P. Biochemistry. 1994;33:8101–8112. doi: 10.1021/bi00192a015. [DOI] [PubMed] [Google Scholar]
- 24.Allen J P, Williams J C. J Bioenerg Biomembr. 1995;27:275–283. doi: 10.1007/BF02110097. [DOI] [PubMed] [Google Scholar]
- 25.Mattioli T A, Lin X, Allen J P, Williams J C. Biochemistry. 1995;34:6142–6152. doi: 10.1021/bi00018a017. [DOI] [PubMed] [Google Scholar]
- 26.Rautter J, Lendzian F, Schulz C, Fetsch A, Kuhn M, Lin X, Williams J C, Allen J P, Lubitz W. Biochemistry. 1995;34:8130–8143. doi: 10.1021/bi00025a020. [DOI] [PubMed] [Google Scholar]
- 27.Woodbury N W, Lin S, Lin X, Peloquin J M, Tagushi A K W, Williams J C, Allen J P. Chem Phys. 1995;197:405–421. [Google Scholar]
- 28.Muegge I, Apostolakis J, Ermler U, Fritzsch G, Lubitz W, Knapp E W. Biochemistry. 1996;35:8359–8370. doi: 10.1021/bi952214c. [DOI] [PubMed] [Google Scholar]
- 29.Jones M R, Fowler G J S, Gibson L C D, Grief G G, Olsen J D, Crielaard W, Hunter C N. Mol Microbiol. 1992;6:1173–1184. doi: 10.1111/j.1365-2958.1992.tb01556.x. [DOI] [PubMed] [Google Scholar]
- 30.Jones M R, Visschers R W, van Grondelle R, Hunter C N. Biochemistry. 1992;31:4458–4465. doi: 10.1021/bi00133a011. [DOI] [PubMed] [Google Scholar]
- 31.Jones M R, Heer-Dawson M, Mattioli T A, Hunter C N, Robert B. FEBS Lett. 1994;339:18–24. doi: 10.1016/0014-5793(94)80376-5. [DOI] [PubMed] [Google Scholar]
- 32.McGlynn P, Hunter C N, Jones M R. FEBS Lett. 1994;349:349–353. doi: 10.1016/0014-5793(94)00701-2. [DOI] [PubMed] [Google Scholar]
- 33.Press W H, Flannery B P, Teukolsky S A, Vetterling W T. Numerical Recipes. Cambridge, U.K.: Cambridge University Press; 1988. [Google Scholar]
- 34.Vos M H, Jones M R, Martin J-L. Chem Phys. 1998;233:179–190. [Google Scholar]
- 35.Stanley R J, Boxer S G. J Phys Chem. 1995;99:859–863. [Google Scholar]
- 36.Becker P C, Fork R L, Brito Cruz C H, Gordon J P, Shank C V. Phys Rev Lett. 1988;60:2462–2464. doi: 10.1103/PhysRevLett.60.2462. [DOI] [PubMed] [Google Scholar]
- 37.Nagarajan V, Parson W W, Gaul D, Schenck C. Proc Natl Acad Sci USA. 1990;87:7888–7892. doi: 10.1073/pnas.87.20.7888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nagarajan V, Parson W W, Davis D, Schenck C. Biochemistry. 1993;32:12324–12336. doi: 10.1021/bi00097a008. [DOI] [PubMed] [Google Scholar]
- 39.Peloquin J M, Lin S, Taguchi A K W, Woodbury N W. J Phys Chem. 1995;99:1349–1356. [Google Scholar]
- 40.Jean J. J Chem Phys. 1994;101:10464–10473. [Google Scholar]
- 41.Provencher S W. Comp Phys Comm. 1982;27:213–227. [Google Scholar]
- 42.Richards F M. J Mol Biol. 1974;82:1–14. doi: 10.1016/0022-2836(74)90570-1. [DOI] [PubMed] [Google Scholar]
- 43.Plato M, Lendzian F, Lubitz W, Möbius K. In: The Photosynthetic Bacterial Reaction Center II: Structure, Spectroscopy and Dynamics. Breton J, Vermeglio A, editors. New York: Plenum; 1992. pp. 109–118. [Google Scholar]
- 44.McAuley-Hecht K M, Fyfe P K, Ridge J P, Prince S, Hunter C N, Isaacs N W, Cogdell R J, Jones M R. Biochemistry. 1998;37:4740–4750. doi: 10.1021/bi971717a. [DOI] [PubMed] [Google Scholar]
- 45.Shreve A P, Cherepy N J, Franzen S, Boxer S G, Mathies R A. Proc Natl Acad Sci USA. 1991;88:11207–11211. doi: 10.1073/pnas.88.24.11207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cherepy N J, Shreve A P, Moore L J, Franzen S, Boxer S G, Mathies R A. J Phys Chem. 1994;98:6023–6029. [Google Scholar]
- 47.Lutz M. Biospectroscopy. 1995;1:313–327. [Google Scholar]
- 48.Souaille M, Marchi M. J Am Chem Soc. 1997;119:3948–3958. [Google Scholar]
- 49.Marcus R A, Sutin N. Biochim Biophys Acta. 1985;811:265–322. [Google Scholar]
- 50. Bixon M, Jortner J. J Phys Chem. 1986;90:3795–3800. [Google Scholar]