

Abstract
A complex interaction between the retroviral envelope glycoproteins and a specific cell surface protein initiates viral entry into cells. The avian leukosis-sarcoma virus (ALV) group of retroviruses provides a useful experimental system for studying the retroviral entry process and the evolution of receptor usage. In this study, we demonstrate that evolutionary pressure on subgroup A ALV [ALV(A)] entry exerted by the presence of a competitive inhibitor, a soluble form of the ALV(A) Tva receptor linked to a mouse immunoglobulin G tag (quail sTva-mIgG), can select different populations of escape variants. This escape population contained three abundant ALV(A) variant viruses, all with mutations in the surface glycoprotein hypervariable regions: a previously identified variant containing the Y142N mutation in the hr1 region; a new variant with two mutations, W141G in hr1 and K261E in vr3; and another new variant with two mutations, W145R in hr1 and K261E. The W141G K261E and W145R K261E viruses escape primarily by lowering their binding affinities for the quail Tva receptor competitive inhibitor while retaining wild-type levels of binding affinity for the chicken Tva receptor. A secondary phenotype of the new variants was an alteration in receptor interference patterns from that of wild-type ALV(A), indicating that the mutant glycoproteins are possibly interacting with other cellular proteins. One result of these altered interactions was that the variants caused a transient period of cytotoxicity. We could also directly demonstrate that the W141G K261E variant glycoproteins bound significant levels of a soluble form of the TvbS3 ALV receptor in a binding assay. Alterations in the normally extreme specificity of the ALV(A) glycoproteins for Tva may represent an evolutionary first step toward expanding viral receptor usage in response to inefficient viral entry.
Retroviruses share a common overall strategy for entry into cells (for recent reviews, see references 26 and 40). The retroviral envelope glycoproteins are initially synthesized as a polyprotein precursor that is subsequently processed into two glycoproteins: the surface glycoprotein (SU), which contains the major domains that interact with the host receptor, and the transmembrane glycoprotein (TM), which anchors SU to the membrane and is directly involved in the fusion of viral and host membranes. The entry process is initiated by a complex interaction between SU and a specific cell surface protein that acts as a receptor, involving multiple, noncontiguous determinants in both proteins that specify receptor choice and binding affinity. Only a proper interaction triggers a conformational change in the structure of the viral glycoproteins which unlocks the fusion peptide located in TM. The exposed fusion peptide interacts with the target cell membrane, initiating a multistep process leading to fusion of the viral and cellular membranes and delivery of a subviral particle into the cell. Despite the complexity of the initial viral SU-cellular receptor interaction, retroviruses have the ability to evolve the structure of their envelope glycoproteins so that they can use a different cellular protein as a receptor (at times a protein that has no obvious homology to the original receptor) and retain efficient entry functions.
The avian leukosis-sarcoma virus (ALV) group of retroviruses provides a useful experimental system for studying the initial interactions of retroviral entry and the evolution of receptor usage. ALV envelope subgroups A through E [ALV(A) through ALV(E)] are highly related, suggesting that these viruses have evolved from a common viral ancestor to use distinct cellular proteins as receptors in order to gain entry into chicken cells, presumably in response to the development of host resistance to viral entry. ALV(A) to ALV(E) SU glycoproteins are almost identical except for five hypervariable regions designated vr1, vr2, hr1, hr2, and vr3 (Fig. 1) (6, 7, 13). Past analyses have suggested that the principal receptor interaction determinants are contained in the hr1 and hr2 domains of ALV SU, with vr3 playing a role in the specificity of receptor recognition but not in receptor binding affinity (14, 36, 37). The vr1 and vr2 hypervariable regions did not appear to be essential for receptor specificity or binding affinity.
FIG. 1.

(A) Schematic representations of the ALV-based RCASBP replication-competent retroviral vector and the major domains of the envelope glycoproteins. The five regions of amino acid sequence variation (vr1, vr2, hr1, hr2, and vr3) identified by comparing the sequences of the surface glycoproteins (SU) of ALV subgroups A to E are also shown. (B) Comparison of the amino acid sequences of three SU hypervariable domains, hr1, hr2, and vr3, of ALV envelope subgroups A to E. The sequences were aligned with the ClustalW Multiple Alignment program of MacVector, version 6.5. Dots, amino acids identical to those in SR-A; dashes, gaps in the alignment. (C) Comparison of the extracellular domains of the quail (Q) and chicken (CK) Tva receptors used in the soluble Tva receptor constructs. Dots, chicken Tva amino acids identical to those in quail Tva. The 40-amino-acid region of Tva related to the human low-density lipoprotein receptor-related motifs is underlined. Brackets indicate the three disulfide bonds.
Five cell surface proteins have been identified as ALV receptors. The two subgroup A receptors, quail Tva and the chicken Tva homologue, are related to the low-density lipoprotein receptor family (4, 5, 41). The three receptors TvbS1 (subgroups B, D, and E) (2), TvbS3 (subgroups B and D) (8), and TvbT (subgroup E) (1) are related to the tumor necrosis factor receptor family. All of these receptors are monomeric, type I membrane glycoproteins that span the cellular membrane only once, and each receptor appears to be necessary and sufficient to confer susceptibility to the specific virus subgroup(s). Soluble forms of the Tva receptors are sufficient to bind the viral glycoproteins and trigger conformational changes in them similar to changes expected to occur during the initiation of the infection process (12, 19-21). Soluble forms of the Tva receptors can specifically block ALV(A) infection of cells in culture and in chickens by binding directly to the virion so as to block access to the membrane-bound Tva receptor (25).
Because of the complexity of the interaction between a retrovirus and its receptor that productively initiates virus entry, the molecular mechanisms of this process are still poorly understood. The ALV system, with highly related envelope subgroups that use relatively simple receptors, offers a simple system in which to genetically define the determinants in both proteins important for retroviral receptor use and entry. However, only a limited number of studies have been conducted to identify residues within the hypervariable regions of ALV SU that are important for receptor interaction. Previous studies have demonstrated that regions and residues in the ALV(A) SU hr1 hypervariable domain that are important for ALV(A) receptor use and virus entry could be identified by using genetic selection strategies that block virus entry based on receptor interference (23) or by using a competitive inhibitor of the Tva receptor (24). Both of these studies identified residues in hr1 that are important for Tva binding affinity and receptor usage. In another genetic approach, an ALV(B) strain was propagated on a mixture of permissive chicken cells (C/E) and nonpermissive quail cells (QT6/BD) (34). A variant virus with two amino acid changes in the hr1 hypervariable region that expanded receptor usage to also include the related quail ALV(E) receptor was selected. These strategies successfully modeled both the evolutionary pressure on ALV encountering host resistance and the ability of a replicating retrovirus to alter receptor usage by mutation and/or recombination in order to counteract resistance to entry in a cell culture system. All three genetic selections identified only amino acids in the hr1 hypervariable domain as important for the interaction of ALV envelope glycoproteins with their receptor. In another study, site-directed mutagenesis of basic amino acids in the ALV(A) SU hr2 hypervariable region resulted in reduced infectivity of murine leukemia virus pseudotyped with the variant envelope glycoproteins in NIH 3T3 cells expressing the quail Tva receptor (31). Three mutant glycoproteins produced in NIH 3T3 cells as glycosyl phosphatidylinositol-linked forms and subsequently cleaved were further characterized and showed altered receptor binding and/or altered efficiency of receptor-triggered conformational changes (11). However, the entry and replication phenotypes of ALV(A) viruses with these mutations were not determined.
In a previous study, the selective pressure of a soluble form of the quail Tva receptor (sTva linked to a mouse immunoglobulin G tag [sTva-mIgG]) on ALV(A) entry into chicken cells produced a population of ALV(A) escape variants with mutations in the subgroup A envelope glycoproteins (24). As was proposed, the mutations were located in a hypervariable region of SU, hr1, and reduced the binding affinities of the mutant glycoproteins for the quail sTva-mIgG, the competitive inhibitor of the membrane-bound chicken Tva receptor. The ALV(A) escape population consisted of two equally abundant variants: variants with the Y142N mutation, which reduced the binding affinity for quail sTva-mIgG ∼100-fold, and variants with the E149K mutation, which reduced the binding affinity for quail sTva-mIgG ∼10-fold. While the Y142N and E149K variant glycoproteins had reduced binding affinities for quail sTva-mIgG, both mutant proteins bound chicken sTva-mIgG with wild-type affinity. This preference in binding for the chicken over the quail sTva receptor was directly related to the efficiency of the variant viruses at infecting cells expressing the chicken or quail Tva receptor. The genetic selection strategy of inhibiting ALV(A) entry into chicken cells with quail sTva as the competitor selected mutant ALV(A) glycoproteins that could exploit the differences in the Tva receptor homologues (Fig. 1C) in order to escape and to improve viral entry efficiency.
This selected variant population represents one escape outcome of ALV(A) replication in chicken cells in the presence of the quail sTva-mIgG competitor. Are there other escape pathways that ALV(A) can take in order to evade this selective pressure on viral entry? If so, will the mutations in the viral glycoproteins identify additional residues that reduce the binding affinity for quail Tva while retaining high affinity for chicken Tva—including residues in the other hypervariable regions, especially hr2? Are there additional ways for ALV(A) to escape the antiviral effect (e.g., by evolving to use a non-Tva receptor)? To answer these questions, ALV(A) was propagated in chicken cells in the presence of the quail sTva-mIgG competitor. The genetic strategy again selected a population of ALV(A) viruses that could replicate efficiently in chicken cells in the presence of quail sTva-mIgG, but this population contained different escape variants. The new escape variant viruses displayed a preference for the chicken Tva receptor and had a significantly reduced binding affinity for quail sTva-mIgG. However, some of the selected variant viruses also appeared to interact with other cellular proteins that resulted in altered receptor interference patterns and periods of cytotoxicity.
MATERIALS AND METHODS
Virus sequence alignment.
The deduced amino acid sequences of the SU regions of ALV(A) through ALV(E) were compared using the ClustalW Multiple Sequence Alignment program of MacVector (version 7.0; Oxford Molecular Ltd., Oxford, England). The Schmidt-Ruppin subgroup A strain of Rous sarcoma virus (SR-A; GenBank accession no. M14901), the Prague subgroup C strain of Rous sarcoma virus (PR-C; GenBank accession no. J02342), Rous associated virus type 2 (RAV-2; GenBank accession no. M14902), the Schmidt-Ruppin subgroup D strain of Rous sarcoma virus (SR-D; GenBank accession no. D10652), and RAV-0 (GenBank accession no. M12171) were used in the sequence alignments.
Vector constructions.
A soluble form of the chicken Tva receptor, encoding the leader and the first 6 amino acids of quail Tva fused to the extracellular region of chicken Tva (residues 7 to 83), has been described previously (9). The construction of the chicken stva-mIgG (ckstva-mIgG) gene in the CLA12NCO plasmid (CLA12NCO/ckstva-mIgG) has been described previously (24). The ckstva-mIgG gene cassette was isolated as a ClaI fragment and subcloned into the ClaI site of the TFANEO expression plasmid (TFANEO/ckstva-mIgG). The expression cassette of TFANEO consists of two long terminal repeats derived from the RCAS vector that provide strong promoter, enhancer, and polyadenylation sites flanking a unique ClaI insertion site (15, 16). The TFANEO plasmid also contains a neo resistance gene expressed under the control of the chicken β-actin promoter and an ampicillin resistance gene for selection in Escherichia coli. Generation of the TF/sTva-4 cell line, a clonal line derived from DF-1 cells expressing quail sTva-mIgG from the TFANEO expression plasmid, has been described previously (24).
Construction of the RCASBP(A)AP retroviral vector (the ALV-based replication-competent RCASBP vector with a subgroup A env gene and the heat-stable human placental alkaline phosphatase [AP] gene [16-18]) and the RCASBP(A)APSal− retroviral vector [the RCASBP(A)AP vector with the SalI sites flanking the AP gene removed (24)] has been described previously. All mutant SU regions were isolated as Asp718-to-SalI fragments from the cloned PCR-amplified env genes in pCR2.1-TOPO (Invitrogen, Carlsbad, Calif.) and were cloned into the unique Asp718 and SalI sites of the RCASBP(A)APSal− vector. The mutations in the env genes of the recombinant RCASBP(A)APSal− clones were verified by nucleotide sequence analysis.
Cell culture and virus propagation.
DF-1 cells (22, 32) and QT6 cells (29) were grown in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum (both from GIBCO/BRL), 100 U of penicillin per ml, and 100 μg of streptomycin per ml (Quality Biological, Inc., Gaithersburg, Md.) at 39°C under 5% CO2. Human 293 cells were grown in the same medium but at 37°C. The TF/sTva-4 cell line (39°C) (24) and the 293tvbS3 cell line (37°C) (a 293 cell line stably expressing the TvbS3 receptor; a gift of John Young) were grown in the above medium but supplemented with 250 μg of G418/ml. NIH 3T3 cells were grown in Dulbecco's modified Eagle's medium supplemented with 10% calf serum (GIBCO/BRL), 100 U of penicillin per ml, and 100 μg of streptomycin per ml (Quality Biological, Inc.) at 37°C under 5% CO2. NIH 3T3pg950 cells were grown similarly to NIH 3T3 cells but supplemented with 250 μg of G418/ml. All cultures were passaged 1:3 when confluent (cells on one plate were split into three plates). Dead DF-1 cells were identified by the trypan blue exclusion method by using the trypan blue solution (0.4%; Sigma) and were visualized with a Nikon Diaphot 300 microscope using 20× Hoffman optics. Photographs were taken with a Nikon CoolPix 950 digital camera.
Virus propagation was initiated either by transfection of plasmid DNA that contained the retroviral vector in proviral form (16) or by direct infection. In standard transfections, 5 μg of purified plasmid DNA was introduced into DF-1 cells by the calcium phosphate precipitation method (27). Viral spread was monitored by assaying culture supernatants for ALV capsid protein (CA) by enzyme-linked immunosorbent assay (ELISA) (33). Virus stocks were generated from cell supernatants cleared of cellular debris by centrifugation at 2,000 × g for 10 min at 4°C and were stored in aliquots at −80°C. DF-1 cells transfected with TFANEO/ckstva-mIgG plasmid DNA were grown in 500 μg of G418 per ml to select for neomycin-resistant cells. Clones were isolated by using cloning cylinders (Bellco Glass Inc., Vineland, N.J.), expanded, and maintained with standard medium supplemented with 250 μg of G418/ml. DF-1 cell cultures chronically infected with RCASBP(A), RCASBP(B), RCASBP(C), or HPRS-103 were produced. The RCASBP viruses with subgroup A, B, and C env genes have been described previously (16). HPRS-103 (GenBank accession no. Z46390) is an ALV with a subgroup J env gene (3) and was obtained from Michael A. Skinner (Institute for Animal Health, Compton, Near Newbury, Berkshire, United Kingdom).
ELISA.
The ALV CA protein was detected in culture supernatants by ELISA as described previously (33). Levels of sTva-mIgG and cksTva-mIgG were quantitated in culture supernatants by ELISA for the mouse IgG tag as described previously (25). The linear range for a standard experiment was 0.5 to 50 ng of ImmunoPure mouse IgG Fc fragment per ml.
ALV AP assay.
For AP assays, DF-1, NIH 3T3, NIH 3T3pg950, 293, or 293tvbS3 cell cultures (∼30% confluent) were incubated with 10-fold serial dilutions of the appropriate RCASBP-AP virus stocks for 36 to 48 h. The assay for AP activity has been described previously (25).
SDS-PAGE and Western immunoblot analysis.
Supernatants from confluent cultures were cleared of cellular debris by centrifugation at 2,000 × g for 10 min at 4°C. Virions (10 ml of culture supernatant) were pelleted through 1 ml of a 20% sucrose pad (20% sucrose, 100 mM NaCl, 20 mM Tris · Cl [pH 7.5], 1 mM EDTA) by ultracentrifugation at 35,000 rpm in a Beckman SW41 rotor for 60 min at 4°C. The viral pellet was resuspended in 100 μl of Laemmli loading buffer (2% sodium dodecyl sulfate [SDS], 10% glycerol, 50 mM Tris · Cl [pH 6.8], 5% β-mercaptoethanol, 0.1% bromophenol blue) and boiled for 5 min. Viral proteins were separated by SDS-polyacrylamide gel electrophoresis (PAGE) (12% polyacrylamide) and transferred to a nitrocellulose membrane.
The Western transfer filters were blocked in phosphate-buffered saline (PBS) with 10% nonfat dry milk (NFDM) for 1 h at 25°C. The filters were then rinsed briefly in rinse buffer (100 mM NaCl, 10 mM Tris · Cl [pH 8], 1 mM EDTA, 0.1% Tween 20) and incubated with either a rabbit anti-ALV p27 antiserum (SPAFAS, Inc., Norwich, Conn.) (1:5,000 dilution) or an anti-ALV(A) SU monoclonal antibody (30) (purified from the mc8C5 hybridoma; a kind gift of Christina Ochsenbauer-Jambor and Eric Hunter, University of Alabama at Birmingham) (1:1,000 dilution) in rinse buffer containing 1% NFDM for 1 h at 25°C. The filters were washed extensively with rinse buffer and then incubated with 50 ng of peroxidase-labeled rabbit anti-goat or goat anti-mouse IgG (heavy plus light chains) (Kirkegaard & Perry, Gaithersburg, Md.)/ml in rinse buffer with 1% NFDM for 1 h at 25°C. After extensive washing with rinse buffer, immunodetection of the protein-antibody-peroxidase complexes was performed with the Western Blot Chemiluminescence reagent (DuPont, NEN, Boston, Mass.). The immunoblots were then exposed to Kodak X-Omat film.
Cloning and nucleotide sequence analysis of integrated viral DNA.
DNA was isolated from infected cells in culture by using the QIAamp Tissue kit (Qiagen). The entire env gene was amplified by PCR using Taq DNA polymerase (Promega, Madison, Wis.) with primers 5′-GGGACGAGGTTATGCCGCTG-3′ (∼50 bp upstream of the Asp718 site) and 5′-TACCACCACCCATGTACTGCC-3′ (just downstream of the env gene). Each Taq PCR mixture contained 1.25 μl of 10× PCR buffer (final concentrations, 50 mm Tris-Cl [pH 8.3], 50 mM KCl, 7 mM MgCl2, and 1.1 mM β-mercaptoethanol), 1.25 μl of 1.7-mg/ml bovine serum albumin, 0.5 μl of each deoxynucleoside triphosphate at 25 mM, 0.5 μl of each primer (A260, 5), 6.0 μl of H2O, and 1.0 μl of DNA (genomic DNA at ∼100 ng/μl; plasmid DNA at ∼2 ng/μl). The reaction mixtures were heated to 90°C for 1 min, and reactions were initiated by addition of 1.5 μl of Taq DNA polymerase diluted 1:10, vol/vol (0.75 U). Thirty cycles of PCR were carried out as follows: 90°C for 40 s, followed by 59°C for 80 s. The amplified products were separated by agarose gel electrophoresis, and the ∼2.0-kb product was purified and cloned into pCR2.1-TOPO by using the TOPO TA Cloning kit (Invitrogen). The nucleotide sequences of the env genes were determined by the Mayo Clinic Molecular Biology Core facility on an ABI PRISM 377 DNA sequencer (with XL upgrade) with an ABI PRISM dRhodamine Terminator Cycle Sequencing Ready Reaction kit and AmpliTaq DNA polymerase (Perkin-Elmer Applied Biosystems, Foster City, Calif.).
Fluorescence-activated cell sorting (FACS) analysis of envelope glycoprotein binding to receptor.
Uninfected DF-1 cells or DF-1 cells infected with either wild-type or mutant ALVs were removed from culture with Trypsin de Larco (Quality Biological, Inc.) and washed with Dulbecco's PBS. The cells were fixed with 4% paraformaldehyde in PBS at room temperature for 15 min and then washed with PBS. Approximately 106 cells in PBS supplemented with 1% calf serum (PBS-CS) were incubated with a supernatant containing either chicken or quail sTva-mIgG, or sTvbS3-mIgG, on ice for 30 min. The stable DF-1 cell lines TF/cksTva-15 (expressing chicken sTva-mIgG) and TF/sTva-4 (expressing quail sTva-mIgG) were the sources of the sTva-mIgG proteins. The sTvbS3-mIgG protein was produced by the stable DF-1 cell line TF/sTvbS3. The cells were then washed with PBS-CS and incubated with 5 μl of goat anti-mouse IgG (heavy plus light chains) linked to phycoerythrin (Kirkegaard & Perry Laboratories) in PBS-CS (total volume, 1 ml) on ice for 30 min. The cell-soluble receptor-mIgG-Ig-phycoerythrin complexes were washed with PBS-CS, resuspended in 0.5 ml of PBS-CS, and analyzed with a Becton Dickinson FACScalibur using CellQuest (version 3.1) software.
Kd calculations.
The maximum possible bound fluorescence and apparent dissociation constant (Kd) for each data set obtained from the FACS binding assays were estimated by fitting the data via nonlinear least squares to a log logistic growth curve function, 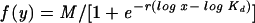 , where y is the mean fluorescence, M is the maximum fluorescence, r is the rate, x is the concentration of sTva-mIgG, and Kd is the dissociation constant, defined as the concentration of sTva-mIgG at half-maximal binding (24). The statistical significance among the estimated Kd values was analyzed by analysis-of-variance (ANOVA) methods. The estimated average Kd for each glycoprotein was obtained along with the associated 95% confidence interval.
, where y is the mean fluorescence, M is the maximum fluorescence, r is the rate, x is the concentration of sTva-mIgG, and Kd is the dissociation constant, defined as the concentration of sTva-mIgG at half-maximal binding (24). The statistical significance among the estimated Kd values was analyzed by analysis-of-variance (ANOVA) methods. The estimated average Kd for each glycoprotein was obtained along with the associated 95% confidence interval.
RESULTS
Experimental design.
To test whether additional ALV(A) variants resistant to the antiviral effect of quail sTva-mIgG could be selected, the experimental protocol developed in the previous study of Holmen et al. (24) was used (Fig. 2A). The ALV(A)-based vector RCASBP(A)AP was propagated in the chicken DF-1 cell line TF/sTva-4, which expresses high levels of quail sTva-mIgG (∼50 nM). Detectable levels of ALV were not observed until 16 days postinfection, as monitored by an ELISA for ALV CA, and reached a peak by day 20; a period of transient cytotoxicity was observed (data not shown). In order to determine if the virus pool contained variants resistant to the quail sTva-mIgG antiviral effect and in order to narrow the mutant population to the most robust variants, the mutant virus pool (0.10 ml) was repassaged in uninfected TF/sTva-4 cells and virus replication was compared to that of wild-type virus. The mutant virus pool had a significant replication advantage over wild-type virus in the TF/sTva-4 cells (data not shown).
FIG. 2.

(A) Schematic representation of the experimental approach. (B) Summary of the SU mutations selected by replicating ALV(A) in chicken cells expressing quail sTva-mIgG.
A different population of ALV(A) variants was selected.
The entire env gene regions (∼2.0 kb) of integrated proviruses were amplified by PCR from genomic DNA isolated from the TF/sTva-4 culture infected with the repassaged mutant virus pool, and the PCR products were cloned. Two separate amplifications with Taq DNA polymerase were performed to control for any changes that might have been introduced by the PCR. The nucleotide sequences of the cloned env genes were compiled, and the deduced amino acid sequences of each clone were compared to those of the subgroup A glycoproteins of the parental RCASBP(A)AP virus. Three different viral variants were present in the population (20 clones sequenced) at >5% frequency (Fig. 2B): 80% of the clones contained a tyrosine (TAT)-to-asparagine (AAT) mutation at codon 142 (Y142N); 10% of the clones contained a tryptophan (TGG)-to-glycine (GGG) mutation at codon 141 and a lysine (AAA)-to-glutamic acid (GAA) mutation at codon 261 (W141G K261E); and 10% of the clones contained a tryptophan (TGG)-to-arginine (CGG) mutation at codon 145 and the same K261E mutation (W145R K261E). In a previous study, 46% of the resistant population contained the Y142N mutation, 50% contained the E149K mutation in hr1, and 4% of the population contained both the Y142N and E149K mutations.
ALV(A) molecular clones containing the putative mutations can replicate in the presence of quail sTva-mIgG.
The previous study had identified and characterized the Y142N mutation phenotype as conferring resistance to the quail sTva-mIgG antiviral effect (24). Therefore, we set out to determine the entry phenotype of the W141G K261E and W145R K261E double mutations and of each mutation singly by introducing the mutations into the env gene of the RCASBP(A)AP molecular clone. Wild-type and mutant viral plasmid DNAs were transfected into both TF/sTva-4 cells and DF-1 cells, and subsequent virus production was monitored. All five mutant ALVs had a replication advantage over wild-type virus in the TF/sTva-4 cells expressing the quail sTva-mIgG antiviral protein (Fig. 3A). The W141G K261E and W145R K261E viruses had a relative growth advantage in TF/sTva-4 cells over the viruses with the W141G, W145R, and K261E single mutations (three independent experiments were conducted). The wild-type and mutant viruses replicated well in DF-1 cells (Fig. 3B); however, the W145R K261E virus replicated at a slightly lower rate (Fig. 3C).
FIG. 3.

Replication of recombinant RCASBP(A)AP vectors containing mutations conferring resistance to the quail sTva-mIgG antiviral effect. Viral growth was monitored by ELISA of the culture supernatants for ALV CA protein. Each panel shows one representative result of three total experiments. (A and B) Plasmids encoding molecular clones of either the wild-type RCASBP(A)AP virus (WT) or a mutant virus containing either the W141G K261E, W145R K261E, W141G, W145R, or K261E mutation were transfected into TF/sTva-4 cells expressing quail sTva-mIgG (A) or into DF-1 cells (B). (C) Virus titers were determined from the virus stocks produced in DF-1 cells (panel B, day 14) by an AP assay. To compare the initial rates of viral replication, fresh DF-1 cells were infected with each virus (multiplicity of infection, 0.01) and the infected cultures were passaged when confluent.
All five mutant envelope glycoproteins were efficiently incorporated into virions at levels similar to those of wild-type glycoproteins: the level of the CA viral structural protein of each virus, used to normalize the amount of protein on the Western immunoblot, is also shown (Fig. 4). The W141G K261E and W145R K261E viruses produced infectious virus in DF-1 cells at 10- to 20-fold lower titers than the wild-type virus (Table 1). The K261E virus produced a slightly lower infectious virus titer in DF-1 cells (∼2- to 5-fold), while the W141G and W145R viruses produced titers similar to those of the wild type. Unexpectedly, DF-1 cultures infected with several of the mutant viruses, the W141G K261E, K261E, and W141G mutants, displayed a transient and variable period of cytotoxicity upon extended passage (Fig. 5). The culture infected with the W141G K261E virus went through an obvious period of cytotoxicity lasting 14 to 16 days, after which it recovered. The level and length of cytotoxicity induced by the W141G K261E mutant were very similar to those of ALV(B)- and ALV(D)-induced cytotoxicity (data not shown). In contrast, the effect of K261E or W145G virus infection was relatively mild, causing a slowing of cell replication but without the obvious cytopathic effect seen with the W141G K261E virus (Fig. 5A).
FIG. 4.

Western immunoblot analysis of the levels of SU glycoprotein in wild-type and mutant virions. Virions from day-16-infected DF-1 culture supernatants were pelleted, and the proteins were denatured, separated by SDS-12% PAGE, and transferred to a nitrocellulose membrane. A Western immunoblot containing the pelleted virus from 5 ml of supernatant was probed with an anti-subgroup A SU monoclonal antibody (SUA), and the bound protein complexes were visualized by chemiluminescence. A Western immunoblot containing the pelleted virus from 1 ml of supernatant was probed with anti-ALV CA sera (CA), and the bound protein complexes were visualized by chemiluminescence. In both immunoblots, proteins were analyzed from uninfected DF-1 cells (lane 1) and from DF-1 cells infected with either wild-type (lane 2), W141G K261E (lane 3), W145R K261E (lane 4), W141G (lane 5), K261E (lane 6), or W145R (lane 7) virus. Molecular sizes (in kilodaltons) are given on the left.
TABLE 1.
Abilities of ALVs with wild-type or mutant subgroup A envelope glycoproteins to infect cells expressing the chicken or quail TVA receptor
| Envelope glycoprotein | Virus titera
|
||
|---|---|---|---|
| In chicken DF-1 cells | In quail QT6 cells | Chicken/ quail ratio | |
| Wild type | (8.9 ± 4.2) × 106 | (9.5 ± 4.7) × 105 | 9.4 |
| W141G K261E | (6.3 ± 3.9) × 105 | (2.6 ± 6.2) × 104 | 23.9 |
| W145R K261E | (5.0 ± 5.4) × 105 | (9.7 ± 5.9) × 103 | 51.7 |
| W141G | (5.7 ± 1.8) × 106 | (3.4 ± 6.5) × 105 | 17.0 |
| W145R | (7.2 ± 3.3) × 106 | (2.1 ± 3.1) × 105 | 35.3 |
| K261E | (1.4 ± 0.9) × 106 | (2.1 ± 4.9) × 105 | 6.7 |
All viruses were derived from the RCASBP(A)AP molecular clone. Virus titers were determined by assaying 10-fold serial dilutions of infected DF-1 cell supernatants for AP. Results shown are averages and standard deviations from four experiments. Boldfaced ratios are statistically different from wild type.
FIG. 5.

(Top) Growth rates of DF-1 cells infected with RCASBP(A) viruses with wild-type or mutant envelope glycoproteins. Plasmids encoding molecular clones of either the wild-type RCASBP(A)AP virus or a mutant virus containing either the W141G K261E, W145R K261E, W141G, W145R, or K261E mutation were transfected into DF-1 cells. Buffer (Mock) was used as a control. The cultures were split 1:3 when confluent (normally every 2 days), which was scored as 1 passage. Transient periods of cytotoxicity and/or slowed cell replication are shaded. (Bottom) Cultures stained with trypan blue solution. (A) Example of the transient cytotoxicity observed in the culture infected with the W141G K261E virus at passage 11. (B) Cells infected with the wild-type virus at passage 12. (C) Mock-infected cells at passage 12.
Variants with envelope glycoprotein mutations in both hr1 and vr3 have a complex phenotype.
Since the soluble Tva receptor inhibits ALV entry by binding directly to the virions and competitively inhibiting the virus's access to the membrane-bound Tva receptor, we have proposed that the phenotype of all escape variant viruses would require a reduced binding affinity for the soluble receptor. To estimate the binding affinities of the W141G K261E and W145R K261E envelope glycoproteins for Tva receptors, infected DF-1 cells expressing the wild-type, W141G K261E, or W145R K261E envelope glycoproteins were assayed for binding to chicken and quail sTva-mIgG by FACS as described previously (24). Both W141G K261E and W145R K261E glycoproteins bound quail sTva-mIgG with significantly lower affinity (>50-fold), but bound chicken sTva-mIgG with similar or slightly lower affinities, than wild-type glycoproteins (Table 2). Viruses with the W141G K261E and W145R K261E glycoproteins also were less efficient at infecting quail QT6 cells (two to fivefold), which express quail Tva, than was the wild-type virus (Table 1).
TABLE 2.
Estimated binding affinities of wild-type and mutant subgroup A envelope glycoproteins for soluble forms of the chicken and quail TVA receptors
| Envelope glycoprotein | Apparent Kd (nM)a
|
|
|---|---|---|
| Chicken sTva-mIgG | Quail sTva-mIgG | |
| Wild type | 0.45 (0.30-0.66) | 0.62 (0.42-0.92) |
| W141G K261E | 0.88 (0.60-1.31) | 30.47 (20.53-45.23) |
| W145R K261E | 1.07 (0.72-1.60) | 29.17 (19.65-43.30) |
| W141G | 0.78 (0.53-1.16) | 5.12 (3.45-7.61) |
| W145R | 0.81 (0.54-1.20) | 4.87 (3.28-7.23) |
| K261E | 0.73 (0.49-1.08) | 1.54 (1.04-2.29) |
Apparent Kd values were estimated by fitting the data via nonlinear least squares to a log logistic growth curve function as described in Materials and Methods. Each result is the estimated mean and 95% confidence interval (given in parentheses) from five experiments. In general, if the confidence intervals do not overlap between values, the values are statistically different. Boldfaced values are statistically different from wild type.
To determine if these envelope glycoprotein mutations also altered the viruses' overall receptor usage, we first performed receptor interference assays. The infectious virus titers of wild-type and mutant virus stocks produced in DF-1 cells were assayed on uninfected DF-1 cells and on DF-1 cells chronically infected with ALV(A), ALV(B), ALV(C), or ALV(J). The susceptibility of this panel of cells to wild-type ALV(A) was demonstrated with a RCASBP(A)AP infection (Fig. 6). RCASBP(A)AP efficiently infected each culture except for cells previously infected with ALV(A), which caused a ∼64,000-fold inhibition. In contrast, the receptor interference patterns of the W141G K261E and W145R K261E ALV(A)s were significantly different from the wild-type ALV(A) pattern. Both mutant viruses infected cells previously infected with ALV(A) ∼6-fold more efficiently than the wild-type virus, and the entry of both mutant viruses into cells previously infected with ALV(B) or ALV(C) was less efficient (5- to 10-fold) than that of the wild-type virus (Fig. 6). While interference with the Tvb and Tvc receptors partially blocked W141G K261E and W145R K261E virus entry, interference with the subgroup J receptor did not alter the infection efficiency of the mutant viruses.
FIG. 6.

Analysis of the receptor interference patterns of wild-type and mutant RCASBP(A)AP viruses produced in DF-1 cells 14 days posttransfection. Uninfected DF-1 cells (DF-1) and DF-1 cells chronically infected with RCASBP(A) (A), RCASBP(B) (B), RCASBP(C) (C), or subgroup J HPRS-103 (J) virus were infected with 10-fold serial dilutions of the culture supernatants, and titers were determined by an AP assay. Results are averages and standard deviations from six experiments. Inhibition of virus entry was calculated by dividing the viral titer determined in uninfected DF-1 cells by the titer determined in DF-1 cells chronically infected with the different ALV subgroups. Standard deviations for the ratios of means were derived by using formulas from Levy and Lemeshow (28). To compare the differences between “treatments” across viruses, an ANOVA model was fit by using individual natural log-transformed ratios (n = 6 per treatment and virus) as the response variable, main model effects for virus and treatment, and an interaction term between virus and treatment. A significant interaction effect would indicate that treatment differences changed with the virus. These tests were conducted overall, and if results were significant, specific comparisons were evaluated to determine which virus-treatment combinations differed. Values that differ significantly from those for the wild-type virus are shaded.
To further analyze the receptor usage of the W141G K261E and W145R K261E mutant viruses, mammalian cells that either do not express ALV receptors (NIH 3T3 and 293), express only quail Tva (NIH 3T3pg950), or express only TvbS3 (293tvbS3) and the chicken DF-1 cell line (expressing chicken Tva, TvbS1, TvbS3, and Tvc) were infected with the mutant virus stocks or the wild-type subgroup A and B viruses RCASBP(A)AP and RCASBP(B)AP (Fig. 7A). The mutations in the W141G K261E and W145R K261E glycoproteins did not significantly alter the viral tropism from that of a normal subgroup A phenotype except for the lack of infection of 3T3pg950 cells expressing the quail Tva receptor. However, the lower binding affinity of the mutant glycoproteins for quail Tva (Table 2), which leads to lower infection efficiency with quail Tva (Table 1), may explain this result. To directly test whether the mutations in the W141G K261E and W145R K261E glycoproteins enabled binding to a Tvb receptor, DF-1 cells infected with the mutant wild-type viruses were incubated with soluble forms of ALV receptors, either chicken sTva-mIgG or chicken sTvbS3-mIgG, and the binding was detected by FACS (Fig. 7B). Of the two mutants, only the W141G K261E mutant glycoproteins could bind significant levels of sTvbS3-mIgG. Despite the ability of W141G K261E to bind a soluble form of TvbS3, viral entry of 293 cells using the TvbS3 receptor could not be detected (Fig. 7A). We conclude from these studies that the W141G K261E and W145R K261E variants interact with other cellular proteins, but we do not have conclusive proof that these interactions can mediate productive infection.
FIG. 7.

Further analysis of receptor usage by the W141G K261E and W145R K261E mutant viruses. (A) Cell lines that do not express ALV receptors (NIH 3T3 and 293), cell lines that express only the quail Tva receptor (3T3pg950) or only the TvbS3 receptor (293tvbS3), or DF-1 cells that express the chicken Tva, TvbS3, TvbS1, and Tvc receptors were infected with 1 ml and 10-fold serial dilutions of W141G K261E, W145R K261E, RCASBP(A)AP [WT(A)], or RCASBP(B)AP [WT(B)] virus stocks, and viral titers were determined by AP assay. Results marked with an asterisk are less than 1 PFU/ml. (B) DF-1 cells chronically infected with a wild-type or mutant virus were removed from culture, fixed with paraformaldehyde, and incubated with 600 ng of chicken sTva-mIgG protein or 2,000 ng of sTvbS3-mIgG protein. The soluble receptor-envelope glycoprotein complexes were bound to a goat anti-mouse Ig antibody linked to phycoerythrin, and the levels of phycoerythrin were measured by FACS. In both graphs, results are averages from three different experiments. Error bars, standard deviations. ifu, infections unit.
Characterization of the W141G, W145R, and K261E single mutations.
The combinations of mutations in the W141G K261E and W145R K261E viruses appear to confer two different phenotypes: a reduced binding affinity for quail Tva and an increase in the interactions of the mutant glycoproteins with other cellular proteins. To determine the contribution of each mutation to these phenotypes, the envelope glycoproteins and viruses with each single mutation were constructed, characterized, and compared to W141G K261E, W145R K261E, and wild-type viruses. There was no significant difference in the binding affinities of W141G, W145R, K261E, and wild-type glycoproteins for chicken sTva-mIgG (Table 2). Both the W141G and W145R mutant glycoproteins had lower binding affinities for quail sTva-mIgG than wild-type glycoproteins (∼8-fold), while the affinity of K261E glycoproteins for quail sTva-mIgG averaged only slightly lower than that of wild-type glycoproteins (Table 2). However, the combination of the K261E mutation with either the W141G or the W145R mutation reduced the binding affinity for quail sTva-mIgG an additional fivefold over the reduction with either of the tryptophan mutations alone. The abilities of viruses with each of these single mutations and with the double mutations to infect quail cells correlated with the binding affinities of the mutant envelope glycoproteins for quail sTva-mIgG (Table 1). The receptor interference patterns of both the W141G and W145R viruses were similar to the pattern of a wild-type subgroup A virus, while the K261E virus was statistically more efficient at infecting ALV(A)-infected cells than was the wild-type virus (Fig. 6). No other significant differences were observed between the receptor interference patterns of mutant viruses with each single mutation and wild-type ALV(A). Therefore, the phenotypes of the W141G K261E and W145R K261E viruses are not a simple sum of the phenotypes of the single mutations. Rather, the K261E mutation in vr3 in combination with either the W141G or the W145R mutation has a synergistic effect both in lowering the binding affinity for quail Tva and in altering the interactions of the viral glycoproteins with other cellular proteins.
DISCUSSION
Initiation of retroviral entry involves multiple, noncontiguous determinants in both the viral SU and receptor proteins, which makes defining these determinants in a meaningful way problematic. Our approach to defining these critical determinants utilizes the ability of retroviruses to escape environmental pressure by mutation and/or recombination to select a viable variant. It has been shown that even a modest level of selective pressure on ALV entry results in the generation of viral variants with altered entry properties (e.g., expanded host range or reduced receptor binding affinity) (23, 24, 34). These genetic approaches do not make assumptions about the location of interaction determinants in SU (in contrast to site-directed mutagenesis), and viral variants with multiple mutations can be selected to identify functional but noncontiguous determinants. For example, in this study, individual ALV(A) variants were selected with mutations in two different hypervariable regions more than 100 amino acid residues apart that resulted in an improved entry phenotype under the selective conditions. We have also approached the study of the evolution of ALV glycoprotein-receptor interactions in cell culture by trying to mimic the selective pressures on ALV replication in birds as closely as possible. Therefore, the processes of ALV infection, replication, and subsequent spread were studied in normal-host-range cells (i.e., avian cells) in which the receptors and viral proteins are synthesized, posttranslationally modified, and expressed at normal levels. We believe that these environmental conditions are especially important in studying the evolution of ALV receptor usage, since the known receptors for ALV are expressed on the avian cell surface at extremely low levels yet are efficiently employed by the viruses for entry.
We have shown that the same evolutionary pressure applied to ALV(A) entry can select different populations of resistant variants and that the variants can have different entry phenotypes. The preferred mechanism by which ALV escapes the block to entry by the quail soluble Tva receptor is the acquisition of mutations in SU that significantly reduce the binding affinity for this competitive inhibitor. The particular selective pressure exerted by expression of the quail sTva-mIgG in chicken cells expressing the membrane-bound chicken Tva receptor drove the evolution of ALV(A) to acquire mutations that enabled it to distinguish between these two Tva receptor homologues, lowering binding affinity for the quail receptor while retaining wild-type affinity for the chicken receptor. In two different studies, five different ALV(A) variants have been selected with a broad range of binding affinities (expressed as apparent Kd values) for quail Tva: 9.7 nM (E149K variant) (24), 29.2 nM (W145R K261E variant), 30.5 nM (W141G K261E variant), 100.4 nM (Y142N variant) (24), and 1,469 nM (Y142N E149K variant) (24). The binding affinity of wild-type RCASBP(A) glycoproteins for quail Tva is 0.6 nM.
In addition to the lower binding affinity for quail Tva, several variants caused a period of transient cytotoxicity in DF-1 cells (Fig. 5), a phenotype very different from that of wild-type subgroup A viruses. ALV(A) to ALV(E) have been classified as noncytopathic (subgroups A, C, and E) or cytopathic (subgroups B and D) (38, 39). Some ALV(C) strains can also cause cytotoxicity in the chicken DF-1 fibroblast cell line (22, 32). Replication of cytopathic ALV subgroups in chicken fibroblasts causes a transient period of cytotoxicity that results in the death of 30 to 40% of the cells. However, not all ALV(B), ALV(D), and ALV(C) strains induce detectable cytotoxicity; this divergence may be related to the expression level of the ALV envelope glycoproteins (32). Central to the proposed mechanism(s) whereby some retroviruses induce cytotoxicity is the specific receptor-envelope glycoprotein interaction, which may result in toxicity due to either (i) accumulation of unintegrated viral DNA from superinfection, (ii) downregulation of the cellular protein used by the virus as a receptor, or (iii) activation of a signaling cascade through the receptor, leading to apoptosis (35). Our results imply that some of the mutant subgroup A glycoproteins have either expanded their interactions to cellular proteins in addition to the Tva receptor, thereby causing cytotoxicity, or altered the interaction with the chicken Tva receptor to retain high binding affinity (with the result that the interaction causes cytotoxicity), or both.
The alterations in the receptor interference patterns of W141G K261E and W145R K261E variants relative to that of wild-type ALV(A) lend further support to the conclusion that some of the mutant glycoproteins are interacting with other cellular proteins and possibly altering the virus's receptor usage (Fig. 6). Not only do the W141G K261E and W145R K261E variants enter ALV(A)-infected cells more efficiently than the wild type, but preinfection of cells with ALV(B) or ALV(C) interferes with infection by W141G K261E and W145R K261E viruses but not by wild-type subgroup A viruses. However, the data clearly show that the most efficient receptor for the W141G K261E and W145R K261E viruses is the chicken Tva receptor. The additional mutant glycoprotein-cellular protein interactions are not sufficient to alter the viral tropism to mammalian cells that do not express a Tva receptor, for example (Fig. 7A), but the W141G K261E glycoproteins could bind a significant but low level of the sTvbS3-mIgG receptor (Fig. 7B). This secondary phenotype, the increased interactions of the variant glycoproteins with other cellular proteins, may represent an evolutionary first step toward altering viral receptor usage in response to inefficient viral entry.
The Y142N virus is the favored escape variant under the conditions of these experiments. The Y142N mutant glycoproteins have the lowest binding affinity for the competitive inhibitor but use the chicken Tva receptor efficiently for entry, and Y142N virus infection does not induce cytotoxicity, resulting in wild-type levels of virus production. The W141G K261E and W145R K261E variants are less favored than Y142N, due to slightly higher binding affinities for the competitive inhibitor and an increase in interactions with other cellular proteins. However, both variant viruses produced lower levels of virus, presumably due to these additional interactions which induced cytotoxicity (e.g., W141G K261E [Fig. 5A]) and/or reduced the rate of replication (e.g., W145R K261E [Fig. 3C]). While these studies sought to model ALV evolution by use of cultured cells, the selective environment in the animal (e.g., immune response) may result in the selection of viral variants with alternate or additional mutations reflecting these selective forces.
Our studies have identified a region of ALV(A) SU hr1 (residues 141 to 149) that appears to be particularly important for the ability of ALV(A) to lower binding affinity for quail Tva while retaining affinity for the chicken receptor homologue (Fig. 8) (23, 24). The three aromatic residues in this region, W141, Y142, and W145, appear to play a central role in determining this receptor preference. In addition, ALV(A) variants were selected with an additional mutation, K261E in the vr3 region, that appears to broaden the interactions of the mutant glycoproteins to other cellular proteins, possibly broadening receptor usage, but does not significantly affect receptor binding affinity by itself. This is the same role proposed for the vr3 region in earlier studies of recombinant viruses containing various portions of the ALV(B), ALV(C), and ALV(E) hypervariable regions (13, 36, 37). What was not necessarily predicted from these earlier experiments was the synergistic effect of a mutation in vr3 combined with a mutation in hr1 in lowering ALV receptor binding affinity (Table 2). An earlier study identified another region of ALV(A) hr1 (Fig. 8, residues 155 to 160) deletion of which broadened the receptor usage of the variant but allowed it to retain wild-type binding affinity for the Tva receptors (23). Despite the reports that residues in ALV(A) hr2 are critical for Tva receptor interactions (11, 31), these genetic selection strategies have not yet identified a variant with mutations in this region.
FIG. 8.

Summary of the amino acid residues in the SR-A ALV SU hypervariable regions identified as important for Tva receptor binding affinity and specificity. The amino acids identified by genetic selection strategies in replicating ALV in avian cells (23, 24; this study) are boldfaced. The basic amino acids in hr2 identified in studies using mammalian cells expressing the ALV glycoprotein mutants as well as murine leukemia virus vectors pseudotyped with the ALV glycoproteins to measure infectivity are underlined (11, 31).
There are now four genetically selected ALV(A) variants that display some level of expanded interactions with cellular proteins in addition to Tva: three variants selected with the soluble quail Tva inhibitor (the E149K [24], W141G K261E, and W145R K261E variants) and one variant selected by receptor interference with a soluble form of ALV(A) SU (the Δ155-160 variant [23]). Compared to wild-type ALV(A), all four variant viruses have an altered receptor interference pattern: an increased entry efficiency in cells previously infected with ALV(A) and a decreased entry efficiency in cells previously infected with ALV(B) or ALV(C). However, only the Δ155-160 variant could infect ALV(A)-infected cells at levels >1,000-fold those of wild-type ALV(A), indicating altered receptor usage, most likely because the genetic selection required the acquisition of mutations that allow the use of a non-Tva receptor. The other variant mutations may induce conformational changes in the ALV(A) envelope glycoproteins similar to those induced by soluble forms of Tva, enabling infection of receptor-deficient cells, albeit with a much lower efficiency than that of receptor-positive cells (10). Therefore, one possible explanation of the broadened cellular protein interactions of the selected variants is that the mutations alter the structure of the envelope glycoprotein trimer such that the glycoproteins are more easily triggered to change conformation and/or fuse more readily with the target cell. However, this mechanism does not readily explain why the variant viruses still efficiently enter cells previously infected with ALV(J), i.e., why only subgroup A, B, and C envelope glycoproteins interfere with variant virus entry.
The data from this and previous studies suggest that ALV(A) through ALV(E) share a common link in the mechanism of virus entry. A link between the Tva, Tvb, and Tvc receptors, which excludes the ALV(J) receptor, could explain why the ALV(A) to ALV(E) envelope glycoproteins are homologous and highly related while the ALV(J) glycoprotein is very different. A possible link between the Tva, Tvb, and Tvc receptors could be a structural motif that is shared by these apparently very different proteins but is not contained in the subgroup J receptor. Another possible link could be a coreceptor or facilitator protein required for efficient virus entry (similar to that required for the entry of human immunodeficiency virus type 1) that is shared by ALV(A) through ALV(E), while ALV(J) would require an unrelated protein. If this model is correct, the coreceptor or facilitator protein must be evolutionarily conserved and expressed in a variety of species to explain the observation that expression of Tva or Tvb in mammalian cells confers susceptibility to ALV infection. While the primary viral escape mechanism from the soluble quail Tva receptor inhibitor was the acquisition of mutations in the viral glycoproteins that lowered binding affinity for quail Tva and retained affinity for chicken Tva, glycoprotein mutations were also selected to increase the interactions of viral glycoproteins with other, non-Tva cellular proteins. The broadening of the viral glycoprotein interactions to other proteins may represent a first step toward evolving receptor usage in response to inefficient viral entry.
Acknowledgments
We thank Ana G. Rosales for help with the statistical analysis and Stephen Hughes, Roberto Cattaneo, Stephen Russell, and the members of the Federspiel laboratory for helpful discussions and critical reading of the manuscript.
This work was supported in part by the USDA NRI Competitive Grants Program (98-35204-6392) and the Mayo Foundation (M.J.F.).
REFERENCES
- 1.Adkins, H. B., J. Brojatsch, J. Naughton, M. M. Rolls, J. M. Pesola, and J. A. T. Young. 1997. Identification of a cellular receptor for subgroup E avian leukosis virus. Proc. Natl. Acad. Sci. USA 94:11617-11622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Adkins, H. B., J. Brojatsch, and J. A. T. Young. 2000. Identification and characterization of a shared TNFR-related receptor for subgroup B, D, and E avain leukosis viruses reveal cysteine residues required specifically for subgroup E viral entry. J. Virol. 74:3572-3578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bai, J., L. N. Payne, and M. A. Skinner. 1995. HPRS-103 (exogenous avian leukosis virus, subgroup J) has an env gene related to those of endogenous elements EAV-0 and E51 and an E element found previously only in sarcoma viruses. J. Virol. 69:779-784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bates, P., L. Rong, H. E. Varmus, J. A. T. Young, and L. B. Crittenden. 1998. Genetic mapping of the cloned subgroup A avian sarcoma and leukosis virus receptor gene to the TVA locus. J. Virol. 72:2505-2508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bates, P., J. A. T. Young, and H. E. Varmus. 1993. A receptor for subgroup A Rous sarcoma virus is related to the low density lipoprotein receptor. Cell 74:1043-1051. [DOI] [PubMed] [Google Scholar]
- 6.Bova, C. A., J. P. Manfredi, and R. Swanstrom. 1986. env genes of avian retroviruses: nucleotide sequence and molecular recombinants define host range determinants. Virology 152:343-354. [DOI] [PubMed] [Google Scholar]
- 7.Bova, C. A., J. C. Olsen, and R. Swanstrom. 1988. The avian retrovirus env gene family: molecular analysis of host range and antigenic variants. J. Virol. 62:75-83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brojatsch, J., J. Naughton, M. M. Rolls, K. Zingler, and J. A. T. Young. 1996. CAR1, a TNFR-related protein, is a cellular receptor for cytopathic avian leukosis-sarcoma viruses and mediates apoptosis. Cell 87:845-855. [DOI] [PubMed] [Google Scholar]
- 9.Connolly, L., K. Zingler, and J. A. T. Young. 1994. A soluble form of a receptor for subgroup A avian leukosis and sarcoma viruses (ALSV-A) blocks infection and binds directly to ALSV-A. J. Virol. 68:2760-2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Damico, R., and P. Bates. 2000. Soluble receptor-induced retroviral infection of receptor-deficient cells. J. Virol. 74:6469-6475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Damico, R., L. Rong, and P. Bates. 1999. Substitutions in the receptor-binding domain of the avian sarcoma and leukosis virus envelope uncouple receptor-triggered structural rearrangements in the surface and transmembrane subunits. J. Virol. 73:3087-3094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Damico, R. L., J. Crane, and P. Bates. 1998. Receptor-triggered membrane association of a model retroviral glycoprotein. Proc. Natl. Acad. Sci. USA 95:2580-2585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dorner, A. J., and J. M. Coffin. 1986. Determinants for receptor interaction and cell killing on the avian retrovirus glycoprotein gp85. Cell 45:365-374. [DOI] [PubMed] [Google Scholar]
- 14.Dorner, A. J., J. P. Stoye, and J. M. Coffin. 1985. Molecular basis of host range variation in avian retroviruses. J. Virol. 53:32-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Federspiel, M. J., L. B. Crittenden, and S. H. Hughes. 1989. Expression of avian reticuloendotheliosis virus envelope confers host resistance. Virology 173:167-177. [DOI] [PubMed] [Google Scholar]
- 16.Federspiel, M. J., and S. H. Hughes. 1997. Retroviral gene delivery. Methods Cell Biol. 52:179-214. [PubMed] [Google Scholar]
- 17.Fekete, D. M., and C. L. Cepko. 1993. Retroviral infection coupled with tissue transplantation limits gene transfer in the chicken embryo. Proc. Natl. Acad. Sci. USA 90:2350-2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fields-Berry, S. C., A. L. Halliday, and C. L. Cepko. 1992. A recombinant retrovirus encoding alkaline phosphatase confirms clonal boundary assignment in lineage analysis of murine retina. Proc. Natl. Acad. Sci. USA 89:693-697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gilbert, J. M., P. Bates, H. E. Varmus, and J. M. White. 1994. The receptor for the subgroup A avian leukosis-sarcoma viruses binds to subgroup A but not to subgroup C envelope glycoprotein. J. Virol. 68:5623-5628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gilbert, J. M., L. D. Hernandez, J. W. Balliet, P. Bates, and J. M. White. 1995. Receptor-induced conformational changes in the subgroup A avian leukosis and sarcoma virus envelope glycoprotein. J. Virol. 69:7410-7415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hernandez, L. D., R. J. Peters, S. E. Delos, J. A. T. Young, D. A. Agard, and J. M. White. 1997. Activation of a retroviral membrane fusion protein: soluble receptor-induced liposome binding of the ALSV envelope glycoprotein. J. Cell Biol. 139:1455-1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Himly, M., D. N. Foster, I. Bottoli, J. S. Iacovoni, and P. K. Vogt. 1998. The DF-1 chicken fibroblast cell line: transformation induced by diverse oncogenes and cell death resulting from infection by avian leukosis viruses. Virology 248:295-304. [DOI] [PubMed] [Google Scholar]
- 23.Holmen, S. L., and M. J. Federspiel. 2000. Selection of a subgroup A avian leukosis virus [ALV(A)] envelope resistant to soluble ALV(A) surface glycoprotein. Virology 273:364-373. [DOI] [PubMed] [Google Scholar]
- 24.Holmen, S. L., D. C. Melder, and M. J. Federspiel. 2001. Identification of key residues in subgroup A avian leukosis virus envelope determining receptor binding affinity and infectivity of cells expressing chicken or quail Tva receptor. J. Virol. 75:726-737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Holmen, S. L., D. W. Salter, W. S. Payne, J. B. Dodgson, S. H. Hughes, and M. J. Federspiel. 1999. Soluble forms of the subgroup A avian leukosis virus [ALV(A)] receptor Tva significantly inhibit ALV(A) infection in vitro and in vivo. J. Virol. 73:10051-10060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hunter, E. 1997. Viral entry and receptors, p. 71-120. In J. M. Coffin, S. H. Hughes, and H. E. Varmus (ed.), Retroviruses. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y. [PubMed]
- 27.Kingston, R. E., C. A. Chen, and H. Okayama. 1989. Introduction of DNA into eukaryotic cells, p. 911-919. In F. M. Ausubel, R. Brent, R. E. Kingston, D. D. Moore, J. G. Seidman, J. A. Smith, and K. Struhl (ed.), Current protocols in molecular biology, vol. 1. John Wiley & Sons, Inc., New York, N.Y.
- 28.Levy, P. S., and S. Lemeshow. 1980. Sampling for health professionals. Lifetime Learning Publications, Belmont, Calif.
- 29.Moscovici, C., M. Moscovici, H. Jimenez, M. M. C. Lai, M. J. Hayman, and P. K. Vogt. 1977. Continuous tissue culture cell lines derived from chemically induced tumors of Japanese quail. Cell 11:95-103. [DOI] [PubMed] [Google Scholar]
- 30.Ochsenbauer-Jambor, C., S. E. Delos, M. A. Accavitti, J. M. White, and E. Hunter. 2002. Novel monoclonal antibody directed at the receptor binding site on the avian sarcoma and leukosis virus Env complex. J. Virol. 76:7518-7527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rong, L., A. Edinger, and P. Bates. 1997. Role of basic residues in the subgroup-determining region of the subgroup A avian sarcoma and leukosis virus envelope in receptor binding and infection. J. Virol. 71:3458-3465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schaefer-Klein, J., I. Givol, E. V. Barsov, J. M. Whitcomb, M. VanBrocklin, D. N. Foster, M. J. Federspiel, and S. H. Hughes. 1998. The EV-0-derived cell line DF-1 supports efficient replication of avian leukosis-sarcoma viruses and vectors. Virology 248:305-311. [DOI] [PubMed] [Google Scholar]
- 33.Smith, E. J., A. M. Fadly, and W. Okazaki. 1979. An enzyme-linked immunosorbent assay for detecting avian leukosis-sarcoma viruses. Avian Dis. 23:698-707. [PubMed] [Google Scholar]
- 34.Taplitz, R. A., and J. M. Coffin. 1997. Selection of an avian retrovirus mutant with extended receptor usage. J. Virol. 71:7814-7819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Temin, H. M. 1988. Mechanisms of cell killing/cytopathic effect by nonhuman retroviruses. Rev. Infect. Dis. 10:399-405. [DOI] [PubMed] [Google Scholar]
- 36.Tsichlis, P. N., and J. M. Coffin. 1980. Recombinants between endogenous and exogenous avian tumor viruses: role of the C region and other portions of the genome in the control of replication and transformation. J. Virol. 33:238-249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tsichlis, P. N., K. F. Conklin, and J. M. Coffin. 1980. Mutant and recombinant avian retroviruses with extended host range. Proc. Natl. Acad. Sci. USA 77:536-540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Weller, S. K., A. E. Joy, and H. M. Temin. 1980. Correlation between cell killing and massive second-round superinfection by members of some subgroups of avian leukosis viruses. J. Virol. 33:494-506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Weller, S. K., and H. M. Temin. 1981. Cell killing by avian leukosis viruses. J. Virol. 39:713-721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Young, J. A. T. 2001. Virus entry and uncoating., p. 87-103. In D. M. Knipe, P. M. Howley, D. E. Griffin, R. A. Lamb, M. A. Martin, B. Roizman, and S. E. Straus (ed.), Fields virology, 4th ed., vol. 2. Lippincott Williams & Wilkins, Philadelphia, Pa.
- 41.Young, J. A. T., P. Bates, and H. E. Varmus. 1993. Isolation of a chicken gene that confers susceptibility to infection by subgroup A avian leukosis and sarcoma viruses. J. Virol. 67:1811-1816. [DOI] [PMC free article] [PubMed] [Google Scholar]