

Abstract
The genus Streptomyces produces two-thirds of microbially derived antibiotics. Polyketides form the largest and most diverse group of these natural products. Antibiotic diversity of polyketides is generated during their biosynthesis by several means, including postpolyketide modification performed by oxidoreductases, a broad group of enzymes including cytochrome P450 monooxygenases (CYPs). CYPs catalyze site-specific oxidation of macrolide antibiotic precursors significantly affecting antibiotic activity. Efficient manipulation of Streptomyces CYPs in generating new antibiotics will require identification and/or engineering of monooxygenases with activities toward a diverse array of chemical substrates. To begin to link structure to function of CYPs involved in secondary metabolic pathways of industrially important species, we determined the X-ray structure of Streptomyces coelicolor A3(2) CYP154A1 at 1.85 Å and analyzed it in the context of the closely related CYP154C1 and more distant CYPs from polyketide synthase (EryF) and nonribosomal peptide synthetase (OxyB) biosynthetic pathways. In contrast to CYP154C1, CYP154A1 reveals an active site inaccessible from the molecular surface, and an absence of catalytic activities observed for CYP154C1. Systematic variations in the amino acid patterns and length of the surface HI loop correlate with degree of rotation of the F and G helices relative to the active site in CYP154A1-related CYPs, presumably regulating the degree of active site accessibility and its dimensions. Heme in CYP154A1 is in a 180° flipped orientation compared with most other structurally determined CYPs.
Keywords: Streptomyces, P450, CYP154A1, antibiotic biosynthesis, heme orientation, X-ray structure
The genus Streptomyces produces a large number of secondary metabolites, including numerous medicinal antibacterial (Haydock et al. 1991; Inouye et al. 1994; Arisawa et al. 1995), antifungal (Dairi et al. 1997), and anticancer (Mao et al. 1999; Molnar et al. 2000; Cheng et al. 2002) compounds, as well as immunosuppressant agents (Aparicio et al. 1996; Molnar et al. 1996). Polyketides form the largest and most diverse group of these natural products. Although diverse in their structures, these molecules share common features in their biosynthetic pathways. Polyketides are synthesized by polyketide synthases (PKS). Antibiotic diversity of polyketides is generated by several means, including postpolyketide modification performed mainly by group transferases and oxidoreductases, the latter being a very broad group of enzymes consisting of oxygenases, oxidases, peroxidases, reductases, and dehydrogenases. Cytochrome P450 monooxygenases (CYPs) are among the most common enzymes in PKS gene clusters and introduce oxygen-containing functionalities into polyketide natural products. Another group of biologically important secondary metabolites consists of low molecular weight bioactive peptides, such as vancomycin (Solenberg et al. 1997), complestatin (Chiu et al. 2001), and teicoplanin (Pootoolal et al. 2002), synthesized by nonribosomal peptide synthetases and further tailored by CYPs catalyzing oxidative phenolic coupling yielding a rigid cross-linked architecture.
CYPs constitute a superfamily of NADPH/NADH- and O2-dependent heme-thiolate enzymes (Guengerich and MacDonald 1990) that play important roles in the biosynthesis of sterols, fatty acids, prostaglandins in animals, antibiotics and other biologically active molecules in bacteria, fungi, and plants, as well as in the metabolism of xenobiotic drugs and toxic chemicals in most organisms (Guengerich 2002). Efficient manipulation of Streptomyces in generating new antibiotics by a combinatorial biosynthesis approach could be achieved through modification of postpolyketide synthase tailoring steps (Rix et al. 2002), including those catalyzed by P450 monooxygenases. Identification and/or engineering of the monooxygenases with activities toward a diverse array of chemical substrates would be beneficial in the context of combinatorial biosynthesis to generate new “unnatural” or “hybrid” natural products with potentially greater or altered biological activities.
The ability to predict the 3D structure for a CYP with chosen substrate specificity once the X-ray structure of one family member is determined would undoubtedly be beneficial for such a combinatorial biosynthesis approach, as well as for building 3D models for physiologically important mammalian CYPs whose structures have not yet been determined. However, this goal is far from being achieved, even though 12 CYP structures are available. CYP structures have demonstrated that on the one hand, a P450 structural fold is preserved during evolution and is almost certainly repeated in all of the more than 2500 members of this superfamily currently available in genomic databases (http://drnelson.utmem.edu/CytochromeP450.html). On the other hand, they also demonstrate that fold similarity coexists in P450 enzymes with notable local deviations in spatial arrangements of helices and loops, which, together with substitutions of residues directly interacting with a substrate in the binding site, confers CYPs with an ability to metabolize incredibly structurally diverse substrates. To better understand the relation between structure and function of P450 monooxygenases from secondary metabolic pathways of industrially important Streptomyces and related species, we have initiated systematic structural evaluation of the CYP complement from Streptomyces coelicolor A3(2).
S. coelicolor A3(2) has been investigated extensively as a model system for the study of morphological and physiological development of Streptomyces and for investigation of the genetic control of antibiotic production (Hopwood 1999). Among about 8000 genes encoded by the ~8.0-Mb S. coelicolor A3(2) genome (Bentley et al. 2002) are 18 CYPs (Bentley et al. 2002; Lamb et al. 2002) with high sequence similarity to CYPs from antibiotic-producing pathways in industrially important Streptomyces species. However, the roles of CYPs in various metabolic functions of S. coelicolor A3(2) remain largely unknown. Recently, we have determined the structure of CYP154C1 from S. coelicolor A3(2) (Protein Data Bank code 1GWI) as having multiple hydroxylation functions toward 12- and 14-membered ring macrolactones in vitro (Podust et al. 2003). Here we report the 1.85 Å crystal structure of another member of this P450 gene family, CYP154A1 (Protein Data Bank code 1ODO), which, however, catalyzes none of the same reactions of CYP154C1. Comparison of these two structures with those of other CYPs indicates regions located outside of the substrate binding site that are predicted to influence the catalytic specificity.
Results and Discussion
Comparison of overall structures for CYP154A1 and CYP154C1
This is the first time that 3D structures are reported for CYPs assigned, based on sequence homology, to the same CYP family. Protein identity of ~40% is chosen as criteria for family assignment for P450 monooxygenases. However, assignment to the same CYP family does not always reflect a similarity in function (Andersen and Hutchinson 1992), which in the case of most of CYPs is simply unknown. Although both CYP154A1 and CYP154C1 are assigned to the same CYP family (42% sequence identity), their 3D appearance reveals notable differences (Fig. 1A ▶). Most pronounced is closed access to the active site in CYP154A1 in contrast to the open conformation of CYP154C1 owing to a repositioning of the F and G helices. A superimposition of just the F and G helices gives excellent alignment with a root mean square (rms) deviation of 1.07 Å. This indicates that the F/G helix region is repositioned as a single unit, similar to what has been seen in the substrate-free and -bound P450BM-3 structures (Li and Poulos 1997). The CYP154A1 structure is obtained in the presence of inhibitor 4-phenylimidazole bound to the heme iron in the substrate binding site, whereas CYP154C1 is ligand free, a difference that by itself might significantly influence the conformation of the F/G region, as previously was observed for CYP119 (Yano et al. 2000; Park et al. 2002). Additionally, analysis of aligned se- quences shows that there are only two gaps longer than one amino acid in the CYP154C1 sequence compared with CYP154A1, which could have a potential impact on the spatial arrangement of the F and G helices. They are located in the HI loop (three-residue gap) and in the DE′ loop (two-residue gap; Fig. 1B ▶). CYP154A1 does not contain gaps longer than one residue, usually Gly, in its sequence compared with CYP154C1.
Figure 1.
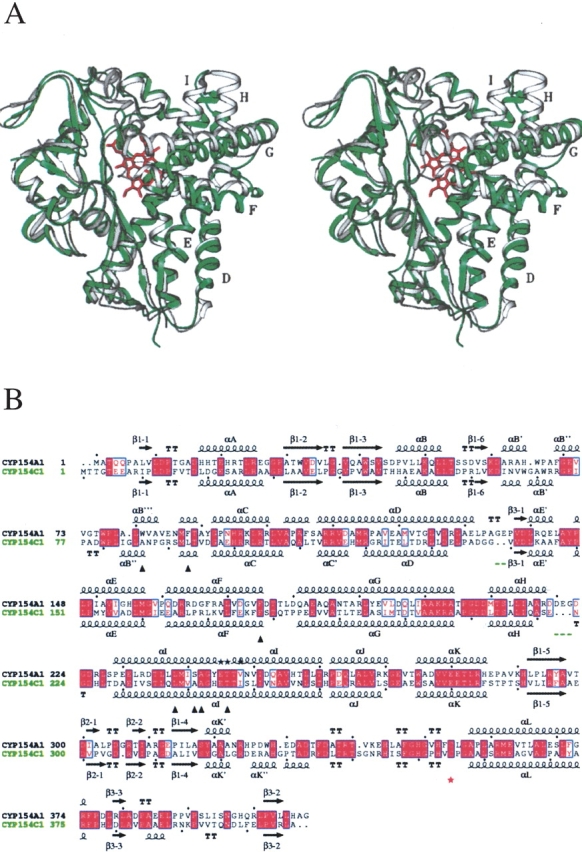
Comparison of CYP154A1 and CYP154C1 structures. (A) Stereo view of superimposition between two structures, CYP154A1 (in gray) and CYP154C1 (in green). Heme is in rose. Images here and in the following figures are generated using SETOR (Evans 1993). Helices mentioned in the text are labeled with letters. (B) Sequence alignment between CYP154A1 and CYP154C1. Residues interacting with 4-phenylimidazole in CYP154A1 are labeled with black triangles. The heme thiolate ligand is indicated by a red star. Black stars in place of secondary structure elements indicate residues in alternate conformations. Gaps in CYP154C1 larger than one residue are underlined in green. Black dots point at every tenth residue in CYP154A1 and CYP154C1 sequences. The alignment was performed using BCM Search Launcher (Smith et al. 1996). The image was generated using the program ESPript (Gouet et al. 1999) with secondary structure elements assigned based on 1GWI for CYP154C1 and 1ODO for CYP154A1.
Significant structural deviations between CYP154A1 and CYP154C1 are also observed in the region of the BC loop despite obvious conservation of residues in both amino acid sequences (Fig. 2 ▶). The CYP154A1 BC loop is overall more structured, owing to an extra 310 helix and an additional pair of short α-helices compared with CYP154C1. The positions and number of the glycines and prolines in this region, however, are not conserved, which we believe affects local helical propensities in the BC loop region and results in displacement of those residues that are conserved and well aligned between the two sequences. CYP154C1 has four glycine and three proline residues in the BC loop compared with two glycines and two prolines in CYP154A1. Residues in positions corresponding Gly69 and Gly84 in CYP154C1 are missing in CYP154A1 and are represented by gaps in the sequence alignment (Fig. 2 ▶). Gly and Pro residues in the BC loop are distributed differently along the sequence, except for G70 (G74 in CYP154C1) and P77 (P81 in CYP154C1), which align well in both proteins. However, in the structures, G70 and its counterpart in CYP154C1 are separated in space by a distance of 7.4 Å, whereas P77 and P81 in both proteins occupy closer positions with a distance between α carbons of 1.5 Å. We assume that G69, G74, P77, and P81 in CYP154C1 are responsible for the unstructured stretch of residues, which in CYP154A1 accommodates the extra αB″′ helix.
Figure 2.

Superimposition and alignment of the BC loops in CYP154A1 and CYP154C1. CYP154A1 is in gray; CYP154C1, in green. Positions of Pro residues and of Gly α-carbons are shown in gray for CYP154A1 and in red for CYP154C1. The residues noted in the superimposition are highlighted in alignment with yellow boxes.
Substrate binding site of CYP154A1
The presence of 4-phenylimidazole in the CYP154A1 substrate binding site allows us to locate amino acid residues that are likely to participate in the binding of endogenous substrates. Seven CYP154A1 residues are localized within 4 Å of 4-phenylimidazole, including four residing in the middle portion of the I helix (Fig. 1B ▶, labeled with black triangles), two coming from the BC loop, and one from the C terminus of the F helix. These same regions provide residues for interactions with the endogenous substrate 6-dioxyerythronolide B in EryF (Cupp-Vickery and Poulos 1995; Fig. 6 ▶, labeled with green triangles). Thus, the phenyl ring of 4-phenylimidazole in CYP154A1 is gripped by three aromatic residues, W81, F88, F175, and one nonpolar residue, L238 (Fig. 3A ▶), which build a hydrophobic cavity of about 340 Å3. The size of the cavity is larger than required to accommodate 4-phenylimidazole, which occupies a volume of 125 Å3. Four residues interacting with 4-phenylimidazole, F175, L238, A242, and T246, are conserved between CYP154A1 and CYP154C1.
Figure 6.
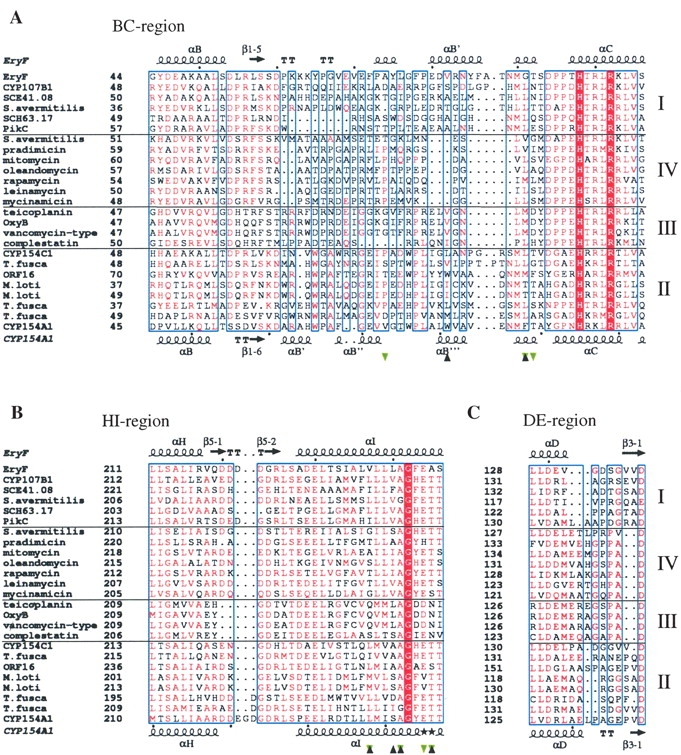
Fragments of CYP sequence alignment. (A) BC region, (B) HI region, and (C) DE region. The alignment was performed using MULTALIN (Corpet 1988; Gouet et al. 1999) with a similarity global score of 0.3 and a differentiation score of 0.3. Residue colors are according to the following rules: red box/white character, strict identity; red character, similarity in the group; blue frames, similarity across groups. Groups are defined based on similarity scores calculated using the algorithm implemented in the program. Branches of sequences are separated by solid horizontal lines and numbered on the right. Sequences are labeled at the left with the protein or gene name if available, and otherwise according to the biosynthetic pathway or organism of origin if anything else is unknown. Residues interacting with 4-phenylimidazole in CYP154A1 are labeled with black triangles; those interacting with 6-deoxyerythronolide B in Eryf are labeled with green triangles. Secondary structure elements are assigned based on 1OXA for Eryf and 1ODO for CYP154A1. Black stars in place of secondary structure element symbols label residues with alternate conformations. Black dots point at every tenth residue in the CYP154A1 and EryF sequences. The image was generated using the program ESPript (Gouet et al. 1999) The full sequence alignment is represented in the Supplemental Material.
Figure 3.
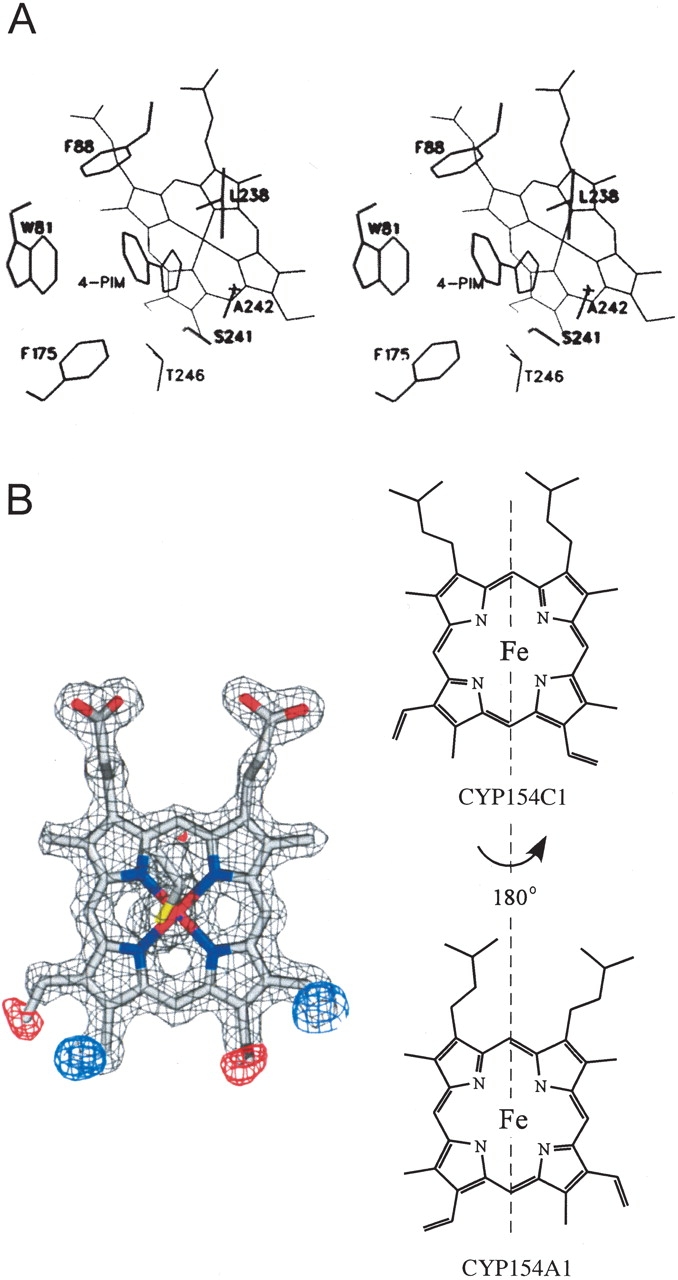
Substrate binding site and heme orientation in CYP154A1. (A) Stereo view of 4-phenylimidazole binding in the active site of CYP154A1. (B) Heme orientation acquired from molecular replacement model CYP154C1. Fragments of Fo-Fc map (red and blue) indicate the requirement of a 180° heme rotation along the axis defined by the α- and γ-meso carbons, as schematically illustrated in the right part of the figure. Fragments of electron density from 2Fo-Fc composite omit map are contoured at 1.6σ (gray), from Fo-Fc map at −3.4σ (red), and 3.4σ (blue). The colors of the atoms are as follows: oxygen and iron, red; nitrogen, blue; sulfur, yellow; carbon, gray.
Despite the stabilizing role of the inhibitor in the active site, residues in the middle portion of the I helix are not stabilized in a single conformation. Alternative conformations were assigned to the residues E245, T246, and V248 (Fig. 1B ▶, alternative conformations labeled with black stars). The presence of alternative conformations in this region is unusual and was not observed previously for other CYPs. This region is one of the most conserved parts of the protein core and reveals almost no deviations from one CYP structure to another.
Heme orientation in CYP154A1
In nearly all reported P450 structures, the heme has been found in a single orientation with respect to orientation around the axis defined by the heme α- and γ-meso carbons. However, in the recently determined atomic resolution structure of CYP121 from Mycobacterium tuberculosis, heme was found in two distinct orientations in a ratio of 7 : 3 (Leys et al. 2003). CYP154A1 is the second example of a CYP in which heme is in the flipped 180° orientation (Fig. 3B ▶). However, in contrast to CYP121, no mixture of conformers was observed at the available resolution. The only difference between the two orientations is the positioning of two of the vinyl moieties in the heme binding pocket, which certainly makes the issue of how these two orientations are differentiated during biosynthesis quite intriguing.
Although only one specific heme orientation has so far been observed in enzymes like peroxidases, and before structures of CYP121 and CYP154C1 have been determined, the opposite of that in P450s, the phenomenon of the mixture of two heme conformations was previously described for the electron-transfer mediators cytochromes b5 (Keller and Wuthrich 1980; McLachlan et al. 1986; Banci et al. 2000) and for the oxygen reservoir myoglobin (La Mar et al. 1983; Aojula et al. 1986). The two forms of cytochrome b5 have slightly different redox potentials, which is thought not to be functionally relevant (Walker et al. 1988). The two populations of myoglobin, which differ in the orientation of the heme by a 180° rotation, have identical absorption spectra and equilibrium-thermodynamic stability, although their unfolding and refolding rate constants differ by a factor of 10 (Moczygemba et al. 2000). The question arises as to whether two different heme orientations might exist in cytochromes P450 in equilibrium, or whether once incorporated, heme remains in the original orientation. Further, what guides heme in one or another orientation in CYP proteins? Analysis of the heme amino acid environment in CYP154A1 and CYP121, and its comparison with other available CYP structures, does not provide a clue to understanding the reason for heme flipping in CYPs. These questions may be answered with the accumulation of more high-resolution CYP structures with clearly resolved heme orientations and using techniques other than X-ray crystallography (i.e., NMR).
Sequence homology of CYP154A1 and CYP154C1 with selected monooxygenases
Most currently identified antibiotics are produced by genes clustered on Streptomyces chromosomes, which has been helpful in identification of CYPs involved in antibiotic biosynthesis (Haydock et al. 1991; Arisawa et al. 1995; Rodriguez et al. 1995; Molnar et al. 1996, 2000; Xue et al. 1998b; Mao et al. 1999; Chiu et al. 2001; Cheng et al. 2002). The closest homologs of CYP154A1 identified using BLAST (Altschul et al. 1997) clearly point toward macrolide monooxygenases as the most related enzymes (Figs. 4 ▶, 5 ▶). The exact function of the majority of related CYPs whose sequences are available in different databases remains unknown. Hierarchical clustering of BLAST-selected sequences performed using the program MULTALIN (Corpet 1988) results in a sequence-based phylogenetic tree (Fig. 4 ▶), which, to a certain degree, predicts the evolution of this group of enzymes and permits correlation of sequence similarity with similarity of function, if the function is known. For convenience of discussion, numbers were assigned to the branches of sequences coming from the common root primarily in those cases in which something is known about function of the CYPs that constitute the branch, branch II being an exception because no information on function is available. Yet, it is of interest to present this branch because the structures of two members, CYP154A1 and CYP154C1, have been determined. CYPs constituting branch II exhibit 40%–50% identity with CYP154A1. They include CYP154C1 from S. coelicolor A3(2) (42% identity to CYP154A1) and the orf16 product from the tylosin producer Streptomyces fradiae (CYP154B1; 50% identity). Although known to be part of the tylosin biosynthetic gene cluster in S. fradiae, CYP154B1 has an elusive role because tylosin ring hydroxylations are catalyzed by other enzymes (Bate et al. 1999; Fouces et al. 1999). CYP154C1 possesses multiple catalytic activities toward 12- and 14-membered ring macrolactones in vitro (Podust et al. 2003). All three, CYP154A1, CYP154B1, and CYP154C1, have notably high sequence similarity to the product of MycG (CYP107E1) from the mycinamicin producer Micromonospora griseorubida (Branch IV). MycG possesses two separate activities, 12, 13-epoxidation and 14-hydroxylation on the same polyketide ring (Inouye et al. 1994), which place it in the same row with polyketide oxygenases catalyzing multiple hydroxylations, such as PikC (CYP107L1) of Streptomyces venezuelae (Xue et al. 1998a) and CYP154C1 of S. coelicolor A3(2) (Podust et al. 2003). Other members of branch II come from the nitrogen-fixing symbiotic bacterium Mesorhizobium loti (Sullivan et al. 2002), and cellulose-degrading actinomycete Thermobifida fusca, where their biological roles also are unknown.
Figure 4.
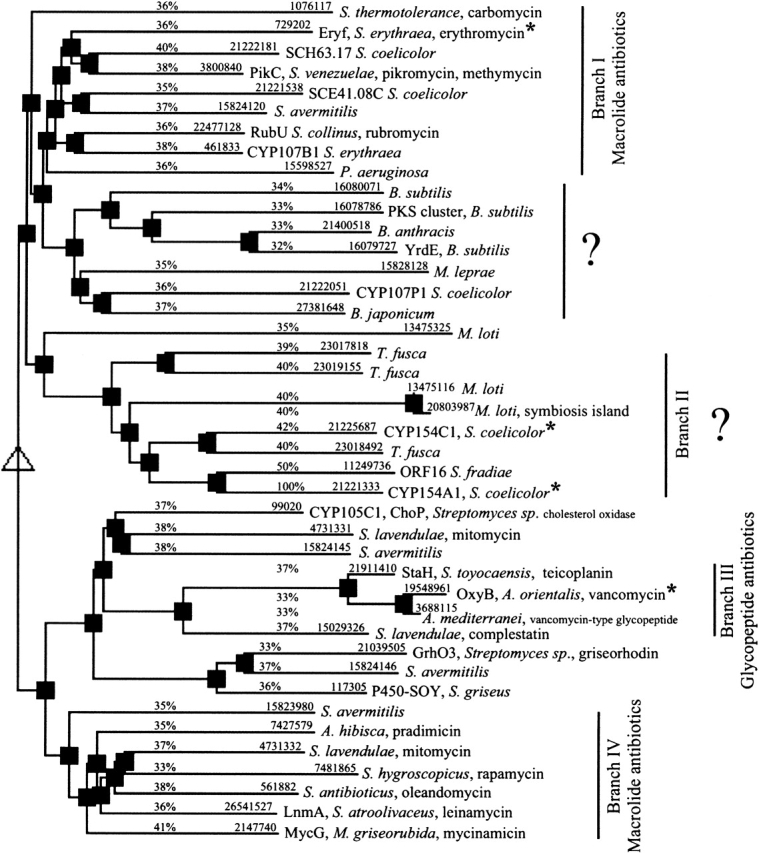
Hierarchical clustering of CYPs having highest homology with CYP154A1 and CYP154C1. Hierarchical clustering was performed using the program MULTALIN (Corpet 1988; http://prodes.toulouse.inra.fr/multalin/multalin.html) for CYP154A1-related sequences identified by BLAST (Altschul et al. 1997). An identity of 32% has been arbitrarily chosen as a threshold. The numbers above the lines are protein GI codes in the NCBI database and percents indicating sequence identity with CYP154A1. CYPs with available 3D structures are labeled with an asterisk. Proteins are also labeled with the protein or gene name, whichever is available, as well as with the organism of origin and the biosynthetic pathway if available.
Figure 5.
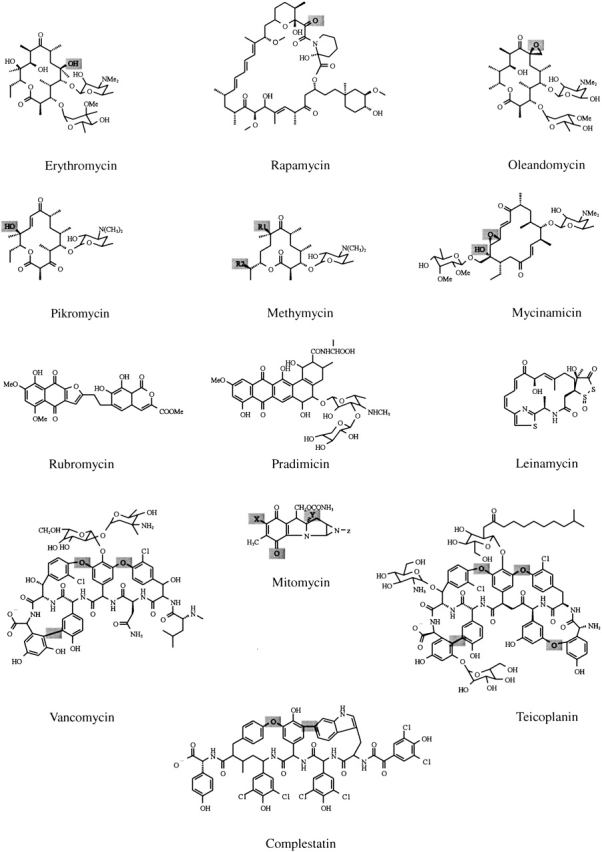
Chemical structures of selected antibiotics functionalized by CYPs. Examples of chemical structures of selected antibiotics functionalized by CYPs represented in the alignment. Modification sites, if established, are highlighted in gray.
CYP154A1 and CYP154C1 share 35%–41% identity with macrolide monooxygenases, which are divided in two branches (Fig. 4 ▶, branches I and IV) based solely on the algorithm of the alignment implemented in MULTALIN (Corpet 1988). CYPs that catalyze oxidative phenol coupling reactions in glycopeptide antibiotics produced by nonribosomal peptide synthetases form a separate branch III. Despite the striking structural difference of products derived from nonribosomal peptide synthetase and PKS gene clusters (Fig. 5 ▶), the similarity of CYPs functionalizing glycopeptides fits within 33%–37% identity with macrolide monooxygenases (Fig. 4 ▶).
At the level of sequence alignment, the differences between the branches are most prominent in the regions of the BC and HI loops (Fig. 6A,B ▶). The BC loop is known to be involved in interactions with substrate and is defined as a substrate recognition site (SRS1) in cytochromes P450 (Gotoh 1992). Accordingly, the BC loop lacks extensive conservation of residues between branches. The length of the BC loop also varies even within a branch, as for branch I. As we have discussed elsewhere (Podust et al. 2003), the length of the BC loop might be directly related to the size of substrate and selectivity of the enzyme. The most prominent reduction of the BC loop is observed for a number of macrolide monooxygenases (Fig. 6A ▶), including those that are known to perform multiple hydroxylation reactions in vivo, the PikC from S.venezuelae and MycG from M.griseorubida. CYPs catalyzing reactions of oxidative phenol coupling in glycopeptide antibiotics of the vancomycin-type (Fig. 5 ▶) also have a relatively short BC loop (Fig. 6A ▶), which is consistent with the large size of the presumed heptapeptide substrate (Zerbe et al. 2002).
In contrast to the BC loop, the HI loop that in some CYPs accommodates the 2-stranded β-sheet 5, and the DE loop are unlikely to be involved in direct interactions with substrate, because of their remote locations in respect to the substrate-binding site. However, the length of both loops varies between proteins represented in the alignment (Fig. 6B,C ▶), with residues in the HI loop demonstrating clear conservation patterns within branches, which are notably different from one branch to another. This fact indicates that the HI loop is one of the signature regions for this group of CYPs and might be one of the determinants for substrate specificity of these enzymes. This is opposite to the DE loop, where very similar residue patterns are observed for macrolide monooxygenases from branch IV and vancomycin-type monooxygenases from branch III (Fig. 7C ▶). We speculate that although the regions directly contacting substrate mediate precise enzyme–substrate fit, the HI loop might play a role as a course regulator, which roughly dictates the size and shape of incoming substrate by positioning the G and F helices relative to the substrate binding site.
Figure 7.
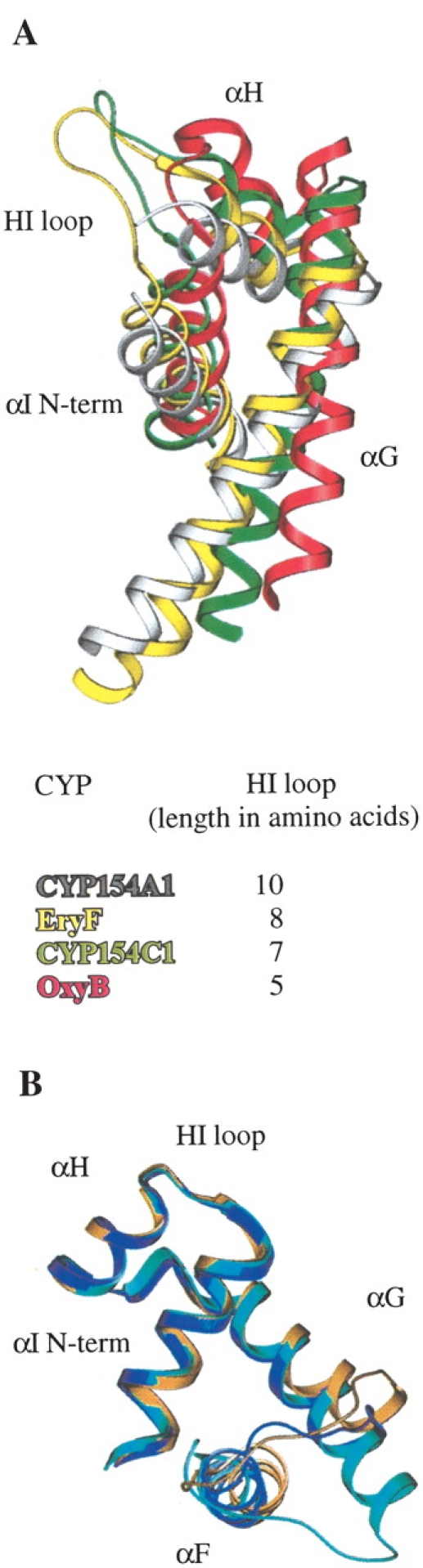
Superimposition between structures. (A) Region comprising G, H, and I helices and loops in between as a result of superimposition of CYP154C1 (1GWI), OxyB (1LFK), and EryF (1OXA) against CYP154A1 (1ODO). The superimposition was performed using SWISS-PDB VIEWER (Guex and Peitsch 1997). Five residues within the HI loop in CYP154A1, E221, G222, D223, G224, and D225 lack electron density and are omitted from the final model of CYP154A1 and absent in the figure. Colors are as follows: gray for CYP154A1, yellow for EryF, green for CYP154C1, and red for OxyB. (B) Region comprising F, G, H, and I helices as a result of superimposition between CYP119 structures in inhibitor free (1IO7), cyan; imidazole bound (1F4U), gold; and 4-phenylimidazole bound (1F4T), blue; viewed along the F helix.
Presumed role of the HI loop in positioning of G and F helices in CYP154A1, CYP154C1, EryF, and OxyB
Among the CYPs selected by BLAST based on 32% identity as a threshold (Fig. 4 ▶), four CYPs have known structures, CYP154A1, CYP154C1 (Podust et al. 2003), OxyB (CYP165B3; Zerbe et al. 2002), and EryF (CYP107A1; Cupp-Vickery and Poulos 1995). As noted earlier, the HI loop sequence motifs are clearly distinguished between branches, with the loop length largely preserved within branches. The four structurally defined CYPs mentioned earlier have different lengths of the HI loop, which is indicated in the table inserted in Figure 7 ▶. CYP154A1 has the longest HI loop, with EryF, CYP154C1, and OxyB following. Superimposition of the four structures (Fig. 7A ▶) shows that the degree of the G helix rotation directly correlates with the length of the HI loop, resulting in a remarkable difference between CYP154A1 and OxyB, which have a twofold difference in the length of the HI loop, 10 versus 5 amino acids, respectively. Rotation of the G and F helices out of the active site in OxyB results in a more open active site compared with the other three CYPs (Fig. 8 ▶). As a result of the G helix displacement, the F helix in all four structures is also gradually displaced, reaching its extreme position in OxyB, where the C terminus of the F helix is situated between the N terminus of the G helix and the middle part of the I helix (Fig. 8 ▶). In contrast to the HI loop, the variations in the length of the DE loop result in repositioning of the loop itself, but otherwise the D and the E helices and the β3–1 strand superimpose well in all four proteins.
Figure 8.
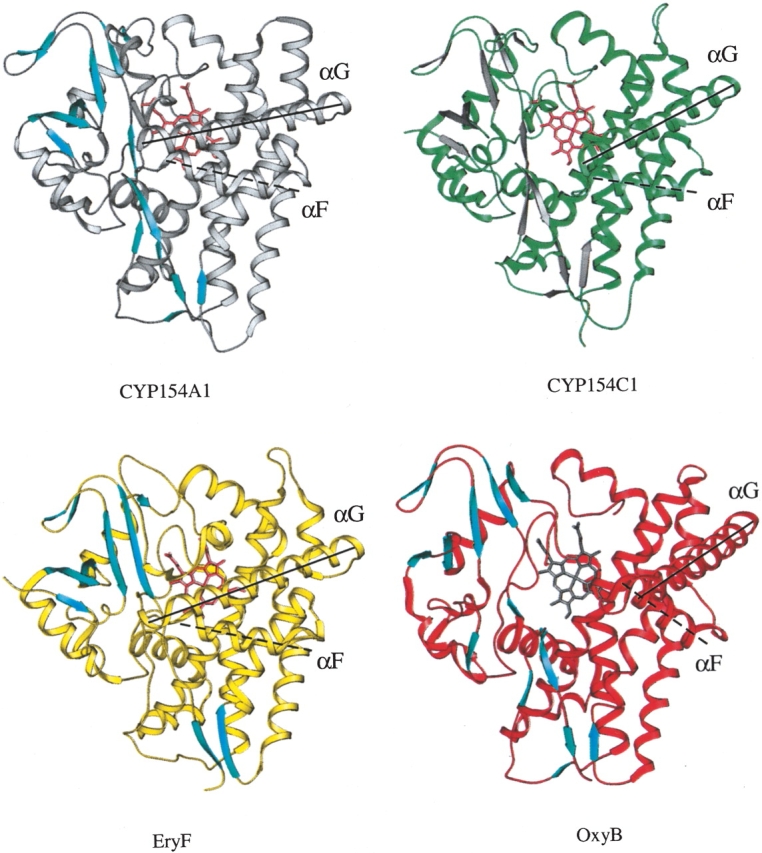
Ribbon representation of CYP154A1, CYP154C1, EryF, and OxyB structures. Positions of the G and F helices are emphasized by solid (G) and dashed (F) lines running along the helical axis. All four structures are in the same orientation. β-Strands are shown in colors contrasting with the rest of the structure.
As noted earlier, the presence of an inhibitor or substrate in the substrate binding site may affect the conformation of the F/G region. The fact that the structures of CYP154A1 and EryF were determined in the presence of an inhibitor and a substrate, respectively, whereas the structures of CYP154C1 and OxyB have the substrate binding site empty, complicates analysis of conformational differences between these proteins because structures of none of them are available in both ligand-free and ligand-bound states. As for CYP154A1, crystals have never been obtained in the absence of an inhibitor, which is quite opposite to CYP154C1, which so far has been crystallized only with the empty active site. For those CYPs whose structures are obtained in both ligand-free and ligand-bound states, conformational changes on inhibitor or substrate binding vary from small to large movements (Li and Poulos 1997; Schlichting et al. 2000; Yano et al. 2000; Park et al. 2002). The most significant difference was observed in the structure of the F/G region for inhibitor-free and inhibitor-bound forms of CYP119 (Yano et al. 2000; Park et al. 2002), where the largest displacement was up to 13 Å in the F/G loop. However, binding of the inhibitor causes only slight rotation of the F and G helices themselves and does not involve repositioning of either the H helix or the N terminus of the I helix (Fig. 7B ▶). Although binding of an inhibitor is certainly one of the external determinants of conformational changes in the substrate binding site, on its own it results in less substantial conformational changes for CYP119 than the differences observed between CYP154A1 and CYP154C1. Additionally, pair-wise comparison of ligand-bound CYP154A1 and EryF, and ligand-free CYP154C1 and OxyB indicates that there must be internal protein interactions that control the degree of the G-helix rotation relative to the active site in CYPs. We assume that the length of the HI loop is one of the factors contributing to this phenomenon.
Functional analysis of CYP154A1
Although CYP154A1 and CYP154C1 (branch II) are clearly diverged from the monooxygenases from branches I and IV, CYP154C1 has in vitro catalytic activity toward precursors of macrolide antibiotics produced by S. venezuelae (Podust et al. 2003), which are endogenous substrates for PikC (branch I). In contrast, CYP154A1 shows binding only toward the 14-membered ring macrolide narbomycin (Fig. 9 ▶) with a Kd of over 1 mM, compared with 24 μM for PikC and 400 μM for CYP154C1 (data not shown). No visible spectral changes were obtained when the 12-membered ring macrolide YC-17 was added to CYP154A1, although they are readily observed with PikC and CYP154C1. Further, no CYP154A1 catalysis of narbomycin was observed (D. Sherman, unpubl.). Poor binding of both macrolactones by CYP154A1 is not a surprise, if one takes into account the relatively small size of the substrate binding pocket (340 Å3 versus 480 Å3 and 430 Å3 required for binding of narbomycin and YC-17, respectively). Thus, despite a relatively high sequence identity with CYP154C1, CYP154A1 has a substrate specificity that is different from that of CYP154C1. However, endogenous substrate and biological function are unknown for both proteins.
Figure 9.

Substrate binding and catalytic activity of CYP154A1. (A) Type I binding spectra resulting from CYP154A1 titration with increasing concentration of narbomycin ranging from 0.3 to 0.6 mM and linearization of titration data in the form of S0/ΔA versus S0 plot.
Materials and methods
CYP154A1 purification
The DNA sequence corresponding to CYP154A1 with four histidine codons inserted at the 3′ end was generated by PCR and cloned into the Escherichia coli expression vector pET17b as described elsewhere (Lamb et al. 2002). The protein was expressed in HMS174 (DE3) cells (Novagen) and purified to homogeneity by Ni-NTA (Qiagen) and Q-Sepharose (Amersham Pharmacia Biotech) chromatography.
CYP154A1 crystallization and data collection
Crystals grew in hanging drops from 0.3 mM CYP154A1 in 10 mM Tris-HCl (pH 7.5), 200 mM NaCl, and 0.5 mM EDTA mixed with an equal volume of 0.7 M sodium citrate, 100 mM HEPES (pH 7.5), and 5 mM 4-phenylimidazole at 22°C. Native diffraction data and multiple anomalous dispersion (MAD) data at three wavelengths (Table 1) were collected at 100°K at Southeast Regional Collaborative Access Team (SER-CAT) 22-ID beamline at the Advanced Photon Source, Argonne National Laboratory, USA. Cryoprotectant contained 20% (v/v) glycerol plus mother liquor. Crystals belong in space group P212121, with unit cell dimensions a = 47.52, b = 103.71, c = 104.31, α = β = γ = 90°. There is one molecule per asymmetric unit with 56% solvent content in the crystal.
Table 1.
Crystallographic data and statistics
| Data | λ1 (edge) | λ2 (peak) | λ3 (remote) | Native |
| Data collection statistics | ||||
| Wavelength, Å | 1.74100 | 1.73839 | 1.73284 | 0.9000 |
| Resolution, Å | 2.65 (2.74–2.65)a | 2.3 (2.38–2.30) | 2.7 (2.80–2.70) | 1.85 (1.92–1.85) |
| Unique reflections | 29,694 | 44,999 | 28,285 | 44,967 |
| Redundancy | 7.1 (6.3) | 6.9 (6.2) | 7.1 (6.3) | 7.0 (6.4) |
| Coverage, % | 99.8 (98.9) | 99.6 (99.3) | 99.8 (99.2) | 99.9 (99.7) |
| Rsym,b %a | 12.6 (47.6) | 13.1 (47.1) | 12.2 (43.7) | 8.0 (46.8) |
| I/σ | 23.3 (4.5) | 26.0 (5.2) | 26.3 (4.9) | 17.6 (3.0) |
| MAD phasing statistics | ||||
| Phasing powerc (anomalous) | 1.77 | 1.65 | 1.29 | — |
| Phasing power (isomorphous) | 1.06 | 1.22 | Reference | — |
| FOM (mean figure of merit) after phasing | 0.64 | — | ||
| FOM after solvent flattening and phase extension | 0.87 | — | ||
| Refinement statistics | ||||
| Resolution, Å | 1.85 | |||
| Protein atoms | 3060 | |||
| Ligand atomsd | 54 | |||
| Water/ions atoms | 454 | |||
| R/Rfree,e % | 17.7/20.8 | |||
| rms deviation | ||||
| Bonds, Å | 0.005 | |||
| Bond angles, ° | 1.2 | |||
| Dihedral angles, ° | 20.8 | |||
| Improper angles, ° | 0.81 | |||
| Ramachandran,f % | 91.2/8.8 | |||
a Data for high-resolution bins are listed in parentheses.
b Rsym = ∑|Ii − 〈I〉|/∑Ii, where Ii is the intensity of the ith observation and 〈I〉 is the mean intensity of reflection.
c Phasing power = [∑(FH)2]1/2/[∑(|FPHo|−FPHc|2], where FPHo and FPHc are the observed and calculated structure factors for the anomalous scatterer.
d Ligand atoms include heme and 4-methylimidazole.
e R = ∑| |Fo| − |Fc| |/∑| Fo|, where Fo and Fc are the observed and calculated structure factor amplitudes, Rfree is the same as R but calculated using 10% of reflections omitted from the refinement.
f Program PROCHECK (Laskowski et al. 1993).
CYP154A1 phasing and refinement
The images were integrated and the intensities merged using HKL2000 (Otwinowski and Minor 1997). The position of the Fe site was determined with CNS (Brunger et al. 1998). Phases were calculated by CNS (Brunger et al. 1998), but failed to yield a traceable electron density map. Alternatively, molecular replacement (MR) in CNS (Brunger et al. 1998) was used with CYP154C1 coordinates as a search model. MR resulted in an unambiguous placement of the model in the unit cell. The Fe position found by molecular replacement exactly coincided with the position calculated from the MAD data. Crystallographic refinement was carried out with CNS (Brunger et al. 1998) using native data to a resolution of 1.85 Å against a composite omit map calculated with CNS (Brunger et al. 1998) at each cycle of the refinement. The final atomic model (Table 1) with an R factor of 17.7% (20.8%) was obtained after iterations of refinement (CNS [Brunger et al. 1998]), evaluation (PROCHECK [Laskowski et al. 1993]) and manual building (O [Jones et al. 1991]). The final model consists of residues 7–406, along with the heme group, one molecule of 4-phenylimidazole, and 454 water molecules. Six residues are substituted for Ala in the PDB entry because of insufficient electron density for the side chains. The quality of the final structure (Table 1) was assessed with the program PROCHECK (Laskowski et al. 1993). It was found that 91.2% of the residues are in most-favored regions of the Ramachandran plot, 8.8% in allowed regions.
Substrate binding assay
YC-17- (Djerassi and Zderic 1956) and narbomycin- (Djerassi and Halpern 1958) induced spectral shifts were monitored at 23.5°C using a Shimadzu UV-2401 spectrophotometer. Samples contained 1 mL of 5 μM CYP154A1 in 10 mM Tris-HCl (pH 7.5) and 10% glycerol. YC-17 and narbomycin were dissolved in ethanol at a stock concentration of 100 mM. Kd values were estimated using spectrophotometric titrations by adding 2-μL aliquots of substrate to the sample and of ethanol to the reference cuvette, followed by recording the difference spectra. Data were linearized in the form of an S0/ΔA versus S0 plot, where S0 is a total concentration of substrate in the reaction mixture. The difference in absorbance between 384 nm (peak) and 418 nm (trough) for each spectrum was taken as a ΔA of the reaction. Kd was estimated from the intercept of the linear plot on the S0-axis.
Electronic supplemental material
“Electronic supplement alignment.pdf” represents the results of multiple sequence alignment of CYPs constituting branches I, II, III, and IV (Fig. 4 ▶). The alignment was performed and the figure generated as described in the legend to Figure 6 ▶.
Data deposition
The atomic coordinates have been deposited in the Protein Data Bank with PDB ID code 1ODO.
Acknowledgments
We thank Jarrod A. Smith and the Vanderbilt University Center for Structural Biology computing facilities; Emmanuel Courcelle and the ESPript program development team at CNRS; personnel at SER-CAT 22-ID beamline at APS; Argonne National Laboratory for expert technical assistance; and Thomas L. Poulos at the University of California, Irvine, for critical reading of the manuscript. This work was supported by NIH grants GM37942 and ES00267 (to M.R.W.), P30 ES00267 (to L.M.P.), GM48562 (to D.H.S.), as well as by Biotechnology and Biological Sciences Research Council (to S.L.K. and D.C.L.) and a Wellcome Trust grant (to D.C.L. and M.R.W.).
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Supplemental material: See www.proteinscience.org
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.03384804.
References
- Altschul, S.F., Madden, T.L., Schaffer, A.A., Zhang, J., Zhang, Z., Miller, W., and Lipman, D.J. 1997. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 25 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen, J.F. and Hutchinson, C.R. 1992. Characterization of Saccharopolyspora erythraea cytochrome P-450 genes and enzymes, including 6-deoxyerythronolide B hydroxylase. J. Bacteriol. 174 725–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aojula, H.S., Wilson, M.T., and Drake, A. 1986. Characterization of haem disorder by circular dichroism. Biochem. J. 237 613–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aparicio, J.F., Molnar, I., Schwecke, T., Konig, A., Haydock, S.F., Khaw, L.E., Staunton, J., and Leadlay, P.F. 1996. Organization of the biosynthetic gene cluster for rapamycin in Streptomyces hygroscopicus: Analysis of the enzymatic domains in the modular polyketide synthase. Gene 169 9–16. [DOI] [PubMed] [Google Scholar]
- Arisawa, A., Tsunekawa, H., Okamura, K., and Okamoto, R. 1995. Nucleotide sequence analysis of the carbomycin biosynthetic genes including the 3-O-acyltransferase gene from Streptomyces thermotolerans. Biosci. Biotechnol. Biochem. 59 582–588. [DOI] [PubMed] [Google Scholar]
- Banci, L., Bertini, I., Rosato, A., and Scacchieri, S. 2000. Solution structure of oxidized microsomal rabbit cytochrome b5. Factors determining the heterogeneous binding of the heme. Eur. J. Biochem. 267 755–766. [DOI] [PubMed] [Google Scholar]
- Bate, N., Butler, A.R., Gandecha, A.R., and Cundliffe, E. 1999. Multiple regulatory genes in the tylosin biosynthetic cluster of Streptomyces fradiae. Chem. Biol. 6 617–624. [DOI] [PubMed] [Google Scholar]
- Bentley, S.D., Chater, K.F., Cerdeno-Tarraga, A.M., Challis, G.L., Thomson, N.R., James, K.D., Harris, D.E., Quail, M.A., Kieser, H., Harper, D., et al. 2002. Complete genome sequence of the model actinomycete Streptomyces coelicolor A3(2). Nature 417 141–147. [DOI] [PubMed] [Google Scholar]
- Brunger, A.T., Adams, P.D., Clore, G.M., Delano, W.L., Gros, P., Grosse-Kunstleve, R.W., Jiang, J.-S., Kuszewski, J., Nilges, M., and Pannu, N.S. 1998. Crystallography and NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr. D 54 905–921. [DOI] [PubMed] [Google Scholar]
- Cheng, Y.Q., Tang, G.L., and Shen, B. 2002. Identification and localization of the gene cluster encoding biosynthesis of the antitumor macrolactam leinamycin in Streptomyces atroolivaceus S-140. J. Bacteriol. 184 7013–7024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu, H.T., Hubbard, B.K., Shah, A.N., Eide, J., Fredenburg, R.A., Walsh, C.T., and Khosla, C. 2001. Molecular cloning and sequence analysis of the complestatin biosynthetic gene cluster. Proc. Natl. Acad. Sci. 98 8548–8553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corpet, F. 1988. Multiple sequence alignment with hierarchical clustering. Nucleic Acids Res. 16 10881–10890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cupp-Vickery, J.R. and Poulos, T.L. 1995. Structure of cytochrome P450eryF involved in erythromycin biosynthesis. Nat. Struct. Biol. 2 144–153. [DOI] [PubMed] [Google Scholar]
- Dairi, T., Hamano, Y., Igarashi, Y., Furumai, T., and Oki, T. 1997. Cloning and nucleotide sequence of the putative polyketide synthase genes for pradimicin biosynthesis from Actinomadura hibisca. Biosci. Biotechnol. Biochem. 61 1445–1453. [DOI] [PubMed] [Google Scholar]
- Djerassi, C. and Halpern, O. 1958. Macrolide antibiotics–-VII: The structure of neomethymycin. Tetrahedron 3 255–268. [Google Scholar]
- Djerassi, C. and Zderic, J.A. 1956. The structure of the antibiotic methymycin. J. Am. Chem. Soc. 78 6390–6395. [Google Scholar]
- Evans, S.V. 1993. SETOR: Hardware-lighted three-dimensional solid model representations of macromolecules. J. Mol. Graph. 11 134–138. [DOI] [PubMed] [Google Scholar]
- Fouces, R., Mellado, E., Diez, B., and Barredo, J.L. 1999. The tylosin biosynthetic cluster from Streptomyces fradiae: Genetic organization of the left region. Microbiology 145 855–868. [DOI] [PubMed] [Google Scholar]
- Gotoh, O. 1992. Substrate recognition sites in cytochrome P450 family 2 (CYP2) proteins inferred from comparative analyses of amino acid and coding nucleotide sequences. J. Biol. Chem. 267 83–90. [PubMed] [Google Scholar]
- Gouet, P., Courcelle, E., Stuart, D.I., and Metoz, F. 1999. ESPript: Multiple sequence alignments in PostScript. Bioinformatics 15 305–308. [DOI] [PubMed] [Google Scholar]
- Guengerich, F.P. 2002. Cytochrome P450 enzymes in the generation of commercial products. Nat. Rev. Drug Discov. 1 359–366. [DOI] [PubMed] [Google Scholar]
- Guengerich, F.P. and MacDonald, T.L. 1990. Mechanisms of cytochrome P-450 catalysis. FASEB J. 4 2453–2459. [DOI] [PubMed] [Google Scholar]
- Guex, N. and Peitsch, M.C. 1997. SWISS-MODEL and the Swiss-Pdb Viewer: An environment for comparative protein modeling. Electrophoresis 18 2714–2723. [DOI] [PubMed] [Google Scholar]
- Haydock, S.F., Dowson, J.A., Dhillon, N., Roberts, G.A., Cortes, J., and Leadlay, P.F. 1991. Cloning and sequence analysis of genes involved in erythromycin biosynthesis in Saccharopolyspora erythraea: Sequence similarities between EryG and a family of S-adenosylmethionine-dependent methyltransferases. Mol. Gen. Genet. 230 120–128. [DOI] [PubMed] [Google Scholar]
- Hopwood, D.A. 1999. Forty years of genetics with Streptomyces: From in vivo through in vitro to in silico. Microbiology 145 2183–2202. [DOI] [PubMed] [Google Scholar]
- Inouye, M., Takada, Y., Muto, N., Beppu, T., and Horinouchi, S. 1994. Characterization and expression of a P-450-like mycinamicin biosynthesis gene using a novel Micromonospora-Escherichia coli shuttle cosmid vector. Mol. Gen. Genet. 245 456–464. [DOI] [PubMed] [Google Scholar]
- Jones, T.A., Zou, J.Y., Cowan, S.W., and Kjeldgaard, M. 1991. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr. A 47 110–119. [DOI] [PubMed] [Google Scholar]
- Keller, R.M. and Wuthrich, K. 1980. Structural study of the heme crevice in cytochrome b5 based on individual assignments of the 1H-NMR lines of the heme group and selected amino acid residues. Biochim. Biophys. Acta 621 204–217. [DOI] [PubMed] [Google Scholar]
- La Mar, G.N., Davis, N.L., Parish, D.W., and Smith, K.M. 1983. Heme orientational disorder in reconstituted and native sperm whale myoglobin. Proton nuclear magnetic resonance characterizations by heme methyl deuterium labeling in the Met-cyano protein. J. Mol. Biol. 168 887–896. [DOI] [PubMed] [Google Scholar]
- Lamb, D.C., Skaug, T., Song, H.-L., Jackson, C.J., Podust, L.M., Waterman, M.R., Kell, D.B., Kelly, D.E., and Kelly, S.L. 2002. The cytochrome P450 complement (CYPome) of Streptomyces coelicolor A3(2). J. Biol. Chem. 277 24000–24005. [DOI] [PubMed] [Google Scholar]
- Laskowski, R.A., MacArthur, M.W., Moss, D.S., and Thornton, J.M. 1993. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 26 283–291. [Google Scholar]
- Leys, D., Mowat, C.G., McLean, K.J., Richmond, A., Chapman, S.K., Walkinshaw, M.D., and Munro, A.W. 2003. Atomic structure of Mycobacterium tuberculosis CYP121 to 1.06 A reveals novel features of cytochrome P450. J. Biol. Chem. 278 5141–5147. [DOI] [PubMed] [Google Scholar]
- Li, H. and Poulos, T.L. 1997. The structure of the cytochrome p450BM-3 haem domain complexed with the fatty acid substrate, palmitoleic acid. Nat. Struct. Biol. 4 140–146. [DOI] [PubMed] [Google Scholar]
- Mao, Y., Varoglu, M., and Sherman, D.H. 1999. Molecular characterization and analysis of the biosynthetic gene cluster for the antitumor antibiotic mitomycin C from Streptomyces lavendulae NRRL 2564. Chem. Biol. 6 251–263. [DOI] [PubMed] [Google Scholar]
- McLachlan, S.J., La Mar, G.N., Burns, P.D., Smith, K.M., and Langry, K.C. 1986. 1H-NMR assignments and the dynamics of interconversion of the isomeric forms of cytochrome b5 in solution. Biochim. Biophys. Acta 874 274–284. [DOI] [PubMed] [Google Scholar]
- Moczygemba, C., Guidry, J., and Wittung-Stafshede, P. 2000. Heme orientation affects holo-myoglobin folding and unfolding kinetics. FEBS Lett. 470 203–206. [DOI] [PubMed] [Google Scholar]
- Molnar, I., Aparicio, J.F., Haydock, S.F., Khaw, L.E., Schwecke, T., Konig, A., Staunton, J., and Leadlay, P.F. 1996. Organisation of the biosynthetic gene cluster for rapamycin in Streptomyces hygroscopicus: Analysis of genes flanking the polyketide synthase. Gene 169 1–7. [DOI] [PubMed] [Google Scholar]
- Molnar, I., Schupp, T., Ono, M., Zirkle, R., Milnamow, M., Nowak-Thompson, B., Engel, N., Toupet, C., Stratmann, A., Cyr, D.D., et al. 2000. The biosynthetic gene cluster for the microtubule-stabilizing agents epothilones A and B from Sorangium cellulosum So ce90. Chem. Biol. 7 97–109. [DOI] [PubMed] [Google Scholar]
- Otwinowski, Z. and Minor, W. 1997. Processing of x-ray diffraction data collected in oscillation mode. Methods Enzymol. 276 307–326. [DOI] [PubMed] [Google Scholar]
- Park, S.Y., Yamane, K., Adachi, S., Shiro, Y., Weiss, K.E., Maves, S.A., and Sligar, S.G. 2002. Thermophilic cytochrome P450 (CYP119) from Sulfolobus solfataricus: High resolution structure and functional properties. J. Inorg. Biochem. 91 491–501. [DOI] [PubMed] [Google Scholar]
- Podust, L.M., Kim, Y., Arase, M., Neely, B.A., Beck, B.J., Bach, H., Sherman, D.H., Lamb, D.C., Kelly, S.L., and Waterman, M.R. 2003. The 1.92-Å structure of Streptomyces coelicolor A3(2) CYP154C1. A new monooxygenase that functionalizes macrolide ring systems. J. Biol. Chem. 278 12214–12221. [DOI] [PubMed] [Google Scholar]
- Pootoolal, J., Thomas, M.G., Marshall, C.G., Neu, J.M., Hubbard, B.K., Walsh, C.T., and Wright, G.D. 2002. Assembling the glycopeptide antibiotic scaffold: The biosynthesis of A47934 from Streptomyces toyocaensis NRRL15009. Proc. Natl. Acad. Sci. 99 8962–8967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rix, U., Fischer, C., Remsing, L.L., and Rohr, J. 2002. Modification of post-PKS tailoring steps through combinatorial biosynthesis. Nat. Prod. Rep. 19 542–580. [DOI] [PubMed] [Google Scholar]
- Rodriguez, A.M., Olano, C., Mendez, C., Hutchinson, C.R., and Salas, J.A. 1995. A cytochrome P450-like gene possibly involved in oleandomycin biosynthesis by Streptomyces antibioticus. FEMS Microbiol. Lett. 127 117–120. [DOI] [PubMed] [Google Scholar]
- Schlichting, I., Berendzen, J., Chu, K., Stock, A.M., Maves, S.A., Benson, D.E., Sweet, R.M., Ringe, D., Petsko, G.A., and Sligar, S.G. 2000. The catalytic pathway of cytochrome P450cam at atomic resolution. Science 287 1615–1622. [DOI] [PubMed] [Google Scholar]
- Smith, R.F., Wiese, B.A., Wojzynski, M.K., Davison, D.B., and Worley, K.C. 1996. BCM Search Launcher—An integrated interface to molecular biology data base search and analysis services available on the World Wide Web. Genome Res. 6 454–462. [DOI] [PubMed] [Google Scholar]
- Solenberg, P.J., Matsushima, P., Stack, D.R., Wilkie, S.C., Thompson, R.C., and Baltz, R.H. 1997. Production of hybrid glycopeptide antibiotics in vitro and in Streptomyces toyocaensis. Chem. Biol. 4 195–202. [DOI] [PubMed] [Google Scholar]
- Sullivan, J.T., Trzebiatowski, J.R., Cruickshank, R.W., Gouzy, J., Brown, S.D., Elliot, R.M., Fleetwood, D.J., McCallum, N.G., Rossbach, U., Stuart, G.S., et al. 2002. Comparative sequence analysis of the symbiosis island of Mesorhizobium loti strain R7A. J. Bacteriol. 184 3086–3095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker, F.A., Emrick, D., Rivera, J.E., Hanquet, B.J., and Buttlaire, D.H. 1988. Effect of heme orientation on the reduction potential of cytochrome b5. J. Am. Chem. Soc. 110 6234–6240. [DOI] [PubMed] [Google Scholar]
- Xue, Y., Wilson, D., Zhao, L., Liu, H., and Sherman, D.H. 1998a. Hydroxylation of macrolactones YC-17 and narbomycin is mediated by the pikC-encoded cytochrome P450 in Streptomyces venezuelae. Chem. Biol. 5 661–667. [DOI] [PubMed] [Google Scholar]
- Xue, Y., Zhao, L., Liu, H.W., and Sherman, D.H. 1998b. A gene cluster for macrolide antibiotic biosynthesis in Streptomyces venezuelae: Architecture of metabolic diversity. Proc. Natl. Acad. Sci. 95 12111–12116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yano, J.K., Koo, L.S., Schuller, D.J., Li, H., Ortiz de Montellano, P.R., and Poulos, T.L. 2000. Crystal structure of a thermophilic cytochrome P450 from the archaeon Sulfolobus solfataricus. J. Biol. Chem. 275 31086–31092. [DOI] [PubMed] [Google Scholar]
- Zerbe, K., Pylypenko, O., Vitali, F., Zhang, W., Rouset, S., Heck, M., Vrijbloed, J.W., Bischoff, D., Bister, B., Sussmuth, R.D., et al. 2002. Crystal structure of OxyB, a cytochrome P450 implicated in an oxidative phenol coupling reaction during vancomycin biosynthesis. J. Biol. Chem. 277 47476–47485. [DOI] [PubMed] [Google Scholar]