

Abstract
Analysis of circular dichroism spectra of proteins provides information about protein secondary structure. Analytical methods developed for such an analysis use structures and spectra of a set of reference proteins. The reference protein sets currently in use include soluble proteins with a wide range of secondary structures, and perform quite well in analyzing CD spectra of soluble proteins. The utility of soluble protein reference sets in analyzing membrane protein CD spectra, however, has been questioned in a recent study that found current reference protein sets to be inadequate for analyzing membrane proteins. We have examined the performance of reference protein sets available in the CDPro software package for analyzing CD spectra of 13 membrane proteins with available crystal structures. Our results indicate that the reference protein sets currently available for CD analysis perform reasonably well in analyzing membrane protein CD spectra, with performance indices comparable to those for soluble proteins. Soluble + membrane protein reference sets, which were constructed by combining membrane proteins with soluble protein reference sets, gave improved performance in both soluble and membrane protein CD analysis.
Keywords: protein secondary structure, reference protein set, membrane proteins, protein CD, CDPro
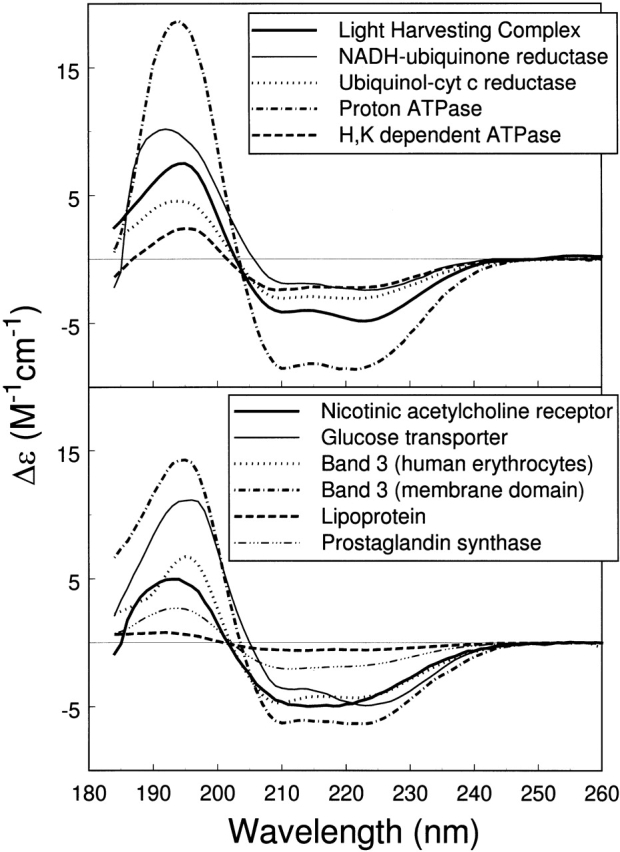
Circular dichroism (CD) spectroscopy is a widely used technique for obtaining information about protein structure and conformation. The sensitivity of far-UV protein CD spectra to protein secondary structure is used in one of the most successful applications of CD, the determination of secondary structure composition of a protein (Yang et al. 1986; Johnson Jr. 1988; Greenfield 1996; Venyaminov and Yang 1996; Sreerama and Woody 2000a, 2004). The approximation that a given protein CD spectrum (Cλ) can be expressed as a linear combination of secondary structure component spectra, Bkλ, given as
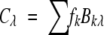 |
where fk is the fraction of secondary structure k, forms the basis for such an analysis. Earlier methods used Bkλ obtained from polypeptides in specific conformations (Greenfield and Fasman 1969; Brahms and Brahms 1980). Most current methods (Hennessey Jr. and Johnson Jr. 1981; Provencher and Glöckner 1981; Pancoska and Keiderling 1991; Böhm et al. 1992; Andrade et al. 1993; Sreerama and Woody 1993) use Bkλ derived from a set of CD spectra of proteins with known secondary structure, that is, a reference protein set. Reference protein sets that include a large number of proteins, belonging to different tertiary structure classes and with varying secondary structure contents, have been constructed (Hennessey Jr. and Johnson Jr. 1981; Yang et al. 1986; Pancoska et al. 1995; Sreerama and Woody 2000b; Sreerama et al. 2000, 2001) and are expected to provide a good representation of the spectral and structural variability in proteins. However, such reference protein sets currently available for protein CD analysis include only soluble proteins due to the paucity of membrane protein structures.
Wallace et al. (2003) have recently examined the performance of soluble protein reference sets in analyzing membrane protein CD spectra, using CDPro software (Sreerama and Woody 2000b). Their analysis was performed for eight membrane proteins, and the results for two representative proteins were presented. They concluded that the soluble protein reference sets give inaccurate results for membrane protein CD analysis, which they attributed to differences in spectral characteristics of membrane and soluble proteins, thus necessitating the development of a membrane protein reference set.
A reference set of membrane protein CD spectra, but without any secondary structure information, was developed by Park et al. (1992). This set of CD spectra was used to estimate the transmembrane and peripheral helical content in the corresponding membrane proteins with the convex constraint analysis (CCA; Perczel et al. 1991). Such an analysis of membrane protein CD spectra without the knowledge of secondary structures was possible because CCA extracts the so-called pure component spectra in a data set without requiring any structural information (Perczel et al. 1991). In the CCA method, secondary structure content is estimated by assigning the extracted pure component spectra to specific structures and determining the fractions of each component spectrum in a given protein CD spectrum. Park et al. (1992) were partially successful in the analysis of CD spectra of three membrane proteins for which structures were available.
The methods for protein CD analysis and the availability of membrane protein structures have improved since the publication of Park et al. (1992). The improved CD analysis methods, however, require both the spectra and secondary structures for the reference proteins (Sreerama and Woody 2000b). By using a subset of the Park et al. (1992) membrane protein data set for which crystal structures are available (13 membrane proteins), we have examined the performance of three popular methods for protein CD analysis and the soluble protein reference sets available in CDPro software (Sreerama and Woody 2000b). Our conclusions differ from those of Park et al. (1992) and Wallace et al. (2003). Both Park et al. (1992) and Wallace et al. (2003) concluded that the soluble protein reference sets are inadequate for the analysis of membrane proteins because of bias effect of the reference proteins, optical artifacts, different spectral characteristics, etc. We did not find any systematic differences in spectral characteristics of soluble and membrane proteins. We also found that the CD analysis results, using soluble protein reference sets for membrane proteins, are only slightly inferior to those obtained for soluble proteins. We constructed a membrane protein reference set with this limited set of 13 membrane proteins and examined its performance, both separately and in combination with soluble protein reference sets, by using the CD analysis programs available in CDPro. The performance of the membrane protein reference set was poor, probably due to the limited number of reference proteins. However, the inclusion of membrane proteins in the soluble protein reference sets resulted in improvements for both membrane and soluble proteins.
Results
We have performed the analysis of CD spectra of a set of 13 membrane proteins by using the CDPro software package (Sreerama and Woody 2000b), which includes three methods (SELCON3, CONTIN/LL, and CDSSTR) for CD analysis, and eight reference protein sets. Five of the eight reference protein sets were from the CDPro software package and included only soluble proteins. CDPro provides seven reference sets, all constructed with soluble proteins, but two of those have definitions of secondary structures different from those used in this study and were not used. Three additional reference protein sets were created by constructing a 13-membrane protein reference set and by combining the membrane proteins with two soluble protein reference sets.
The secondary structures used in CDPro are from the DSSP assignments (Kabsch and Sander 1983) of crystal structures as adapted by Sreerama et al. (1999). The six secondary structures estimated in CDPro are regular α-helix (αR), distorted α-helix (αD), regular β-sheet (βR), distorted β-sheet (βD), turns (T), and unordered (U). For simplicity and comparison with literature data, we have summarized the results from the CD analysis for four secondary structures: α-helix (α), β-sheet (β), turns, and unordered. (Results for individual membrane proteins are provided in Supplemental Material.) The fractions of α and β were obtained by adding the corresponding regular and distorted fractions, for example, α = αR + αD. The performance of the analysis is measured by performance indices: root mean square (RMS) deviations (δ) and correlation coefficients (r) between the crystal structure and the CD predicted values. The performance indices are given for each secondary structure separately (e.g., α-helix, δα, and rα) and all four secondary structures collectively (δ and r, representing overall performance).
Analysis of membrane proteins with soluble protein reference sets
The CD spectra of 13 membrane proteins included in this study are shown in Figure 1 ▶. These spectra were taken (with permission from The Protein Society) from a larger set of 30 CD spectra of membrane proteins measured in the laboratory of Dr. G.D. Fasman (Brandeis University, Waltham, MA), using samples provided by the leading laboratories working on these membrane proteins (Park et al. 1992). We selected these 13 CD spectra because of the availability of the corresponding membrane protein crystal structures in the Protein Data Bank (PDB; Berman et al. 2000). The spectra are identified in the figure by the PDB code of the crystal structure of the membrane protein. The secondary structure fractions for the 13 membrane proteins, assigned by DSSP (Kabsch and Sander 1983), are given in Table 1. Of the 13 membrane proteins, nine have moderate to high α-helical content (αR + αD), and the other four have high β-sheet content (βR + βD).
Figure 1.
CD spectra of 13 membrane proteins drawn using data from Park et al. (1992; with permission from The Protein Society). The proteins are identified by the PDB code for the structure used in this study and the corresponding names of proteins are given in Materials and Methods and in Table 1.
Table 1.
Secondary structure fractions of 13 membrane proteins obtained from crystal structures
| Membrane protein | PBD code | Resolution (Å) | αR | αD | βR | βD | T | U |
| Reaction center (R. viridis) | 1prc | 2.30 | 0.291 | 0.186 | 0.024 | 0.042 | 0.194 | 0.263 |
| Photosystem I | 1jb0 | 2.50 | 0.363 | 0.193 | 0.025 | 0.029 | 0.167 | 0.222 |
| Reaction center (R. sphaeroides) | 1qov | 2.10 | 0.341 | 0.185 | 0.035 | 0.035 | 0.138 | 0.263 |
| Antenna complex (R. acidophila) | 1nkz | 2.00 | 0.569 | 0.161 | 0.000 | 0.000 | 0.086 | 0.183 |
| Ubiquinol-cytochrome c reductase (bovine) | 1bgy | 3.00 | 0.355 | 0.163 | 0.056 | 0.034 | 0.165 | 0.228 |
| Cytochrome oxidase (bovine) | 1occ | 2.80 | 0.434 | 0.146 | 0.031 | 0.022 | 0.141 | 0.226 |
| Rhodopsin (bovine) | 1f88 | 2.80 | 0.482 | 0.153 | 0.012 | 0.025 | 0.160 | 0.166 |
| Bacteriorhodopsin (H. halobium) | 1qhj | 1.90 | 0.605 | 0.154 | 0.035 | 0.017 | 0.109 | 0.079 |
| Ca2+ ATPase (rabbit muscle) | 1eu1 | 2.60 | 0.286 | 0.154 | 0.087 | 0.058 | 0.203 | 0.211 |
| Porin (OmpF, E. coli) | 2omf | 2.40 | 0.010 | 0.035 | 0.462 | 0.118 | 0.223 | 0.153 |
| Porin (R. capsulatus) | 2por | 1.80 | 0.027 | 0.040 | 0.462 | 0.106 | 0.193 | 0.172 |
| Maltoporin (LamB) | 1af6 | 2.40 | 0.000 | 0.028 | 0.482 | 0.114 | 0.159 | 0.216 |
| Phosphoporin (PhoE) | 1pho | 3.00 | 0.000 | 0.020 | 0.433 | 0.115 | 0.236 | 0.194 |
The assignments of secondary structure are from the DSSP method (Kabsch and Sander 1983). The secondary structure fractions are regular α-helix (αR), distorted α-helix (αD), regular β-sheet (βR), distorted β-sheet (βD), turns (T), and unordered (U), as defined by Sreerama et al. (1999). The references for the crystal structures are provided in Electronic Supplemental Material (Table S1).
The results from the analysis of membrane proteins with five soluble protein reference sets from CDPro (Sreerama and Woody 2000b) are summarized in Table 2. These five reference protein sets differ in the number of reference proteins and the wavelength range of CD spectra used in the analysis, and are identified as SP (which stands for soluble protein) followed by the number of reference proteins (NREF) as SPxx (xx = 29, 37, 43, 42, and 48). Addition of five denatured proteins to SP37 and SP43 creates the SP42 and SP48 reference sets (Sreerama et al. 2000). Results from all three CD analysis programs showed similar trends. The performance indices for α and β fractions showed marked improvements with the increase in the NREF from 29 to 43, although results from SP29 were not obtained with the full wavelength range available in the reference set because of the smaller range of membrane protein CD data. When we considered reference sets with NREF from 37 to 48, the performance indices for T and U fractions were comparable to those of soluble proteins and showed, in general, smaller variations with the choice of reference set. The performance indices for α and β fractions were poorer and showed slightly larger variations. Overall performance indices obtained from SP43 and SP48 were similar. These performance indices obtained for membrane proteins compare favorably with those obtained for soluble proteins.
Table 2.
Analysis of membrane protein CD spectra using soluble protein reference sets available in CDPro
| α | β | T | U | |||||||||
| Method | Reference set | λ (nm) | δα | rα | δβ | rβ | δT | rT | δU | rU | δ | r |
| SELCON3 | SP29 | 184–245a | 0.12 | 0.93 | 0.17 | 0.88 | 0.05 | 0.65 | 0.08 | 0.17 | 0.11 | 0.85 |
| SP37 | 185–240 | 0.10 | 0.96 | 0.13 | 0.93 | 0.04 | 0.70 | 0.07 | 0.25 | 0.09 | 0.92 | |
| SP43 | 190–240 | 0.09 | 0.97 | 0.13 | 0.97 | 0.04 | 0.64 | 0.08 | 0.26 | 0.09 | 0.92 | |
| SP42b | 185–240 | 0.10 | 0.96 | 0.12 | 0.94 | 0.04 | 0.82 | 0.06 | 0.35 | 0.09 | 0.93 | |
| SP48b | 190–240 | 0.08 | 0.97 | 0.12 | 0.98 | 0.04 | 0.67 | 0.07 | 0.37 | 0.08 | 0.93 | |
| CONTIL/LL | SP29 | 184–245a | 0.17 | 0.84 | 0.17 | 0.82 | 0.05 | 0.39 | 0.07 | 0.14 | 0.13 | 0.82 |
| SP37 | 185–240 | 0.10 | 0.94 | 0.15 | 0.81 | 0.07 | 0.47 | 0.10 | −0.06 | 0.11 | 0.85 | |
| SP43 | 190–240 | 0.10 | 0.93 | 0.10 | 0.96 | 0.05 | 0.52 | 0.08 | 0.25 | 0.09 | 0.91 | |
| SP42b | 185–240 | 0.12 | 0.92 | 0.15 | 0.86 | 0.05 | 0.47 | 0.08 | 0.17 | 0.11 | 0.87 | |
| SP48b | 190–240 | 0.10 | 0.93 | 0.10 | 0.97 | 0.05 | 0.44 | 0.07 | 0.30 | 0.08 | 0.91 | |
| CDSSTR | SP29 | 184–245a | 0.12 | 0.90 | 0.16 | 0.87 | 0.04 | 0.69 | 0.08 | 0.10 | 0.11 | 0.85 |
| SP37 | 185–240 | 0.09 | 0.96 | 0.14 | 0.96 | 0.04 | 0.76 | 0.08 | 0.09 | 0.09 | 0.90 | |
| SP43 | 190–240 | 0.08 | 0.96 | 0.10 | 0.97 | 0.04 | 0.64 | 0.08 | 0.18 | 0.08 | 0.93 | |
| SP42b | 185–240 | 0.10 | 0.95 | 0.13 | 0.98 | 0.06 | 0.64 | 0.08 | 0.18 | 0.09 | 0.91 | |
| SP48b | 190–240 | 0.08 | 0.96 | 0.12 | 0.99 | 0.05 | 0.53 | 0.08 | 0.20 | 0.08 | 0.92 | |
The results for 13 membrane proteins are summarized in this Table. CDPro software package has three programs for CD analysis (SELCON3, CONTIN/LL, and CDSSTR) and seven reference protein sets. Two of the seven reference protein sets in CDPro correspond to secondary structure assignments that include poly(Pro)II type conformation and were not used in this study. The other five reference protein sets are included in this study (number of reference proteins, NREF, 29 to 48 proteins). The performance indices for each of the secondary structures are given as the RMS deviations and correlation coefficients between the X-ray and the CD predicted fractions (δα, rα, . . .). The fractions of regular and distorted α and β structures from CDPro were combined to obtain α- and β-fractions. Overall, performance indices were calculated as the RMS deviations and correlation coefficients (δ and r) for all four secondary structure fractions collectively.
a Even though SP29 can analyze CD data in the wavelength range 178 to 260 nm, the analysis was performed using the available data in the range 184 to 245 nm.
b SP42 and SP48 were constructed by adding five denatured proteins to SP37 and SP43, respectively (Sreerama et al. 2000).
We find significant improvements in membrane protein CD analysis by increasing the number of reference proteins from 37 to 43 (or from 42 to 48, both involving the same six additional proteins), even with the decrease in the wavelength range of the analyzed CD spectrum from 185 to 240 nm to 190 to 240 nm. It is generally believed that increasing the wavelength range of far-UV CD improves the analysis by including more spectral information in the analysis (Hennessey Jr. and Johnson Jr. 1981). It has also been shown that the increased representation of spectral and structural variations in the reference set leads to improved analysis (Sreerama and Woody 2000b). Both the expanded wavelength range and the expanded reference set increase the information content of a given reference protein set and positively influence the CD analysis. For the set of 13 membrane proteins, we find that the benefits of the expanded reference set (SP43) outweigh those of the expanded wavelength range (SP37), particularly for the β fraction. This is due to the improved analysis of β-rich membrane proteins with the larger reference set. We do not, however, see similar improvements in the analysis with the increase in the number of reference proteins from SP43 to SP48 (or from SP37 to SP42). This is not unexpected because this increase was effected by the addition of denatured proteins to the reference set, and the 13 membrane proteins analyzed are dominated by contributions from either α- or β-structures.
Analysis of membrane proteins with reference sets, including membrane proteins
With the relative success of soluble protein reference sets in analyzing membrane proteins and the availability of both CD spectra and crystal structures for a reasonable number of membrane proteins, we took the next logical step of including membrane proteins in CD analysis. We constructed a membrane protein reference set that includes the 13 membrane protein spectra and the corresponding secondary structures given in Figure 1 ▶ and Table 1, respectively. This reference set is referred to as MP13. We also combined the membrane protein data with those of soluble proteins and constructed soluble + membrane protein reference sets. The wavelength range of the membrane protein CD spectra allowed us to choose SP37 and SP43 for combining with membrane proteins, and the combined soluble + membrane protein reference sets are referred to as SMP followed by the number of proteins in the reference set. The expansion of reference sets SP37 and SP43 by including five denatured CD spectra had mixed effects on the performance of membrane protein CD analysis by different methods (performance worsened with CDSSTR, improved with SELCON3, remained the same with CONTIN/LL), and combining SP42 and SP48 with MP13 was not pursued. The two soluble + membrane protein reference sets constructed represent the effects of the expanded wavelength range (190 to 240 nm to 185 to 240 nm) and increased number of proteins (50 to 56) on the analysis.
The results from the analysis of membrane proteins with three reference protein sets that include membrane proteins, and three programs from CDPro, are summarized in Table 3. The three reference protein sets are identified as MP13, SMP50, and SMP56. The results are obtained from cross-validation analysis, in which the membrane protein analyzed was removed from the reference set and was analyzed with the remaining reference proteins. Our results are compared with those from the CCA method obtained with a 30-membrane protein reference set (MP30; Park et al. 1992), by extracting results for these 13 membrane proteins and obtaining the performance indices.
Table 3.
Analysis of membrane protein CD spectra with the membrane protein and combined soluble + membrane protein reference sets
| α | β | T | U | ||||||||
| Method | Reference set | δα | rα | δβ | rβ | δT | rT | δU | rU | δ | r |
| SELCON3 | MP13a | 0.09 | 0.94 | 0.07 | 0.96 | 0.04 | 0.51 | 0.04 | 0.51 | 0.06 | 0.95 |
| SMP50 | 0.07 | 0.97 | 0.08 | 0.96 | 0.03 | 0.68 | 0.06 | 0.25 | 0.06 | 0.95 | |
| SMP56 | 0.09 | 0.94 | 0.08 | 0.97 | 0.04 | 0.57 | 0.06 | 0.29 | 0.07 | 0.95 | |
| CONTIN/LL | MP13a | 0.13 | 0.89 | 0.07 | 0.96 | 0.04 | 0.48 | 0.08 | 0.02 | 0.09 | 0.92 |
| SMP50 | 0.09 | 0.95 | 0.07 | 0.96 | 0.04 | 0.62 | 0.08 | −0.01 | 0.07 | 0.94 | |
| SMP56 | 0.13 | 0.88 | 0.07 | 0.96 | 0.05 | 0.47 | 0.08 | 0.12 | 0.09 | 0.91 | |
| CDSSTR | MP13a,b | 0.08 | 0.95 | 0.06 | 0.97 | 0.04 | 0.54 | 0.05 | 0.57 | 0.06 | 0.96 |
| SMP50 | 0.08 | 0.96 | 0.08 | 0.99 | 0.05 | 0.75 | 0.08 | 0.42 | 0.08 | 0.93 | |
| SMP56 | 0.09 | 0.94 | 0.08 | 0.99 | 0.04 | 0.64 | 0.07 | 0.22 | 0.07 | 0.93 | |
| Averagec | 0.08 | 0.96 | 0.09 | 0.97 | 0.03 | 0.71 | 0.07 | 0.17 | 0.07 | 0.94 | |
| CCAd | MP30 | 0.13 | 0.92 | 0.17 | 0.72 | 0.10 | 0.50 | 0.14 | 0.56 | 0.14 | 0.75 |
The results for 13 membrane proteins are summarized in this table. The membrane protein reference set was constructed with the 13 membrane protein CD spectra (Fig. 1 ▶) and the corresponding secondary structures (Table 1). The combined soluble + membrane protein reference sets were constructed by adding CD spectra and the corresponding secondary structure fractions of 13 membrane proteins to 37- and 43-protein reference sets provided in CDPro forming SMP50 and SMP56, respectively. The wavelength ranges of CD spectra were 185 to 240 nm for MP13 and SMP50 and 190 to 240 for SMP56.
a Each membrane protein in the 13-membrane protein reference set is analyzed by using the other 12 membrane proteins (wavelength range of CD spectra: 185 to 240 nm), and the results are summarized as performance indices.
b The summary for CDSSTR from MP13 includes results for only nine membrane proteins because solutions were not obtained for four membrane proteins (photosystem I, antenna complex, porin [R. capsulatus] and maltoporin).
c Twelve solutions, from three programs (SELCON3, CONTIN/LL, and CDSSTR) and four reference sets (SP37, SP43, SMP50, and SMP56), were averaged.
d CCA results for the 13 membrane protein CD spectra in the wavelength range 184 to 260 nm were taken from Park et al. (1992), obtained with a 30 membrane protein reference set.
For the reference sets comprised of only membrane proteins, the CDPro results (from MP13) are clearly superior to those from the CCA method (from MP30). Although the correlation coefficients from CCA were comparable to those from SELCON3, CDSSTR, and CONTIN/LL methods, the RMS deviations between the CD predicted and crystal structure values were larger. Although both δ and r are important in determining the performance of a given method, with low δ values and high r values indicating a good performance, the value of r can be skewed by a consistent over- or underprediction of a structure. This is clearly the case here, where high correlation coefficients (>0.85) coupled with large values of δ (~0.13) are observed as a result of consistent under-prediction of the predominant secondary structure (Supplemental Material). In such situations, the smaller value of δ gives a better measure of the performance.
Among the three programs of CDPro, performance of the MP13 reference set decreased in the order, SELCON3 (δ = 0.06), CONTIN/LL (δ = 0.09), and CDSSTR (δ = 0.06, with results for only nine membrane proteins). A careful comparison of results from the individual methods (provided in Supplemental Material) indicated the source of differences in the performance of the three methods, which has origins in both the number of reference proteins and the algorithms followed in these methods. CONTIN/LL (Provencher and Glöckner 1981) uses variable weighting of reference spectra and constrains the sum of secondary structures to unity in fitting the analyzed CD spectrum. In contrast, SELCON3 (Sreerama and Woody 1993) and CDSSTR (Johnson Jr. 1999) do not use any constraints but differ in the implementation of variable selection (Manavalan and Johnson Jr. 1987). SELCON3 uses a locally linearized version (van Stokkum et al. 1990) of variable selection, whereas CDSSTR uses a randomly selected minimal basis (Dalmas and Bannister 1995). The small number of proteins in the membrane protein reference set, 13, gave only 1287 combinations of eight reference proteins in the CDSSTR method (Johnson Jr. 1999), which was not enough to obtain any solution for four spectra (1jb0, 1nkz, 2por, and 1af6). The low information content of the membrane protein reference set was also responsible for poor solutions for three membrane proteins (1jb0, 1qhj, and 2por) from CONTIN/LL (Supplemental Material); a solution was considered poor if the RMS deviation between the CD predicted and crystal structure values of secondary structures for a given membrane protein (δf) was >0.10. The fact that the performance indices obtained with soluble reference proteins were better than those obtained with membrane proteins alone indicates the lack of sufficient information for CD analysis in MP13.
The increased information content provided by combining soluble and membrane proteins leads to improvements in membrane protein CD analysis. In general, the soluble + membrane protein reference sets performed better than either the soluble or the membrane protein reference sets alone. The overall RMS error was reduced from ~7% to 10% with the soluble or membrane protein reference sets alone to ~7% with combined reference sets. In general, the performance indices for α and β fractions improved with combined reference protein sets. The only exception was the performance for the β fraction from SELCON3, which showed an increase in δβ from 0.07 (MP13) to 0.08 (SMP50 and SMP56).
With the soluble + membrane protein reference sets, increasing the wavelength range of the analyzed CD spectra improved the performance of the analysis. In general, performance indices for all secondary structures from SMP50 (wavelength range, 185 to 240 nm) were either better than or comparable to those from SMP56 (wavelength range, 190 to 240 nm). The β-structure was an exception, in which the larger reference set improved the performance slightly. This is in contrast to the results obtained from soluble protein reference sets, in which increasing the number of reference proteins resulted in better performance. The spectral information content of SMP50 is increased by the inclusion of MP13 membrane proteins in SP37. Further addition of soluble proteins with reduction in wavelength range of the reference set (SMP56) leads to poorer analysis, which indicates a decrease in information content. The benefits of the increased spectral information from 185 to 190 nm, in SMP50, outweigh the benefits of additional proteins (SMP56) for the analysis of membrane proteins. The slightly poorer performance of the β-structure with SMP50, in comparison with that from SMP56, indicates an under-representation of β-structures in membrane proteins.
Examination of results for specific membrane proteins of the MP13 reference set (provided in Supplemental Material) indicates that a majority of them are analyzed well, with similar solutions from the three methods. Three proteins, photosystem I, phosphoporin, and porin (R. capsulatus), posed some problems. Photosystem I was analyzed well by the SELCON3 method, but not with the other two. The CD spectrum of photosystem I (Fig. 1 ▶) has a strong inflection at ~225 nm, which affected its analysis with both CONTIN/LL and CDSSTR, as both methods use the similarity between the back-calculated and experimental spectra. CONTIN/LL uses it as a constraint and CDSSTR uses it as a selection rule, whereas in SELCON3 this selection rule is relaxed. Porin (R. capsulatus) was analyzed poorly with all methods, and good analysis of phosphoporin required the larger wavelength range of 185 to 240 nm (SMP50). As Park et al. (1992) observed, the β-strands in membrane proteins are generally longer than those in soluble proteins, and the problems in the analyses of porins are probably due to under-representation of β-rich membrane proteins in the soluble + membrane protein reference sets.
The three methods generally gave similar solutions for a given membrane protein (Supplemental Material), which indicates a reliable analysis. We did not obtain the best solution for all proteins, judging by the RMS difference with the crystal structure, from a single reference protein set. The best solutions for the 13 membrane proteins were spread among different methods and different reference sets. We averaged solutions from SP37, SP43, SMP50, and SMP56, and the performance indices for the averaged solution are also given in Table 3. In the absence of structural information, the average solution obtained by averaging solutions from different methods and different reference sets provides a reliable estimate.
Park et al. (1992) also provided the CD spectrum of F0F1 ATPase, which is shown in Figure 2 ▶. We have performed the analysis of this CD spectrum by using both soluble and soluble + membrane protein reference sets, and the results are presented in Table 4. The results from three programs and four reference sets were averaged to obtain the CD prediction of 59% α-helix and 8% β-sheet for F0F1 ATPase. The relative uncertainty in the β-sheet fraction, given by the standard deviation, was larger than that for α-helix fraction. F0F1 ATPase is a large multimeric protein of Mr ~540 kD (Boyer 1997) and has both soluble (F1) and membrane-bound (F0) components. The soluble component, F1 ATPase (Mr ~ 379 kD), consists of three α-chains, three β-chains, and one chain each of γ, δ, and ɛ. The membrane-bound component, F0 ATPase (Mr ~ 160 kD), consists of one a-chain, two b-chains, and 10 to 12 c-chains. The crystal structure of F1 ATPase (PDB code, 1bmf; subunits α3, β3, and γ; Mr ~ 346 kD; Abrahams et al. 1994) is available, and it has 42% α-helix and 17% β-sheet. By using the CD estimate of α-helix fraction for F0F1 ATPase (fαCD = 0.59) with the crystal structure of F1 ATPase (fαEXP = 0.42), we estimate the α-helix content of the remaining subunits of F0F1 ATPase to be ~ 0.90. This estimate compares quite well with the NMR structures of the c subunit (Girvin et al. 1998) and ac12 complex of F0 ATPase (Rastogi and Girvin 1999), which indicate a very high α-helix content (fαEXP = 0.85 to 0.90). A similar exercise with the β-sheet fraction, however, failed to give meaningful results because the average β-sheet content for F0F1 ATPase (fβCD = 0.08; Table 4) as estimated by CD analysis is quite low in comparison with the crystal structure of α3β3γ portion of F1 ATPase (fβEXP = 0.17). This is probably due to the underestimation of fβ by CD and the larger uncertainty in the value of fβCD (fβCD = 0.08 ± 0.02; range, 0.11 to 0.05). By using the value of 0.11 for fβCD of F0F1 ATPase and 0.17 for fβEXP of F1 ATPase, we estimate the β-sheet fraction in F0 ATPase to be zero.
Figure 2.

CD spectrum of F0F1 ATPase drawn using the data of Park et al. (1992; with permission from The Protein Society).
Table 4.
Analysis of F0F1 ATPase CD spectrum
| Method | Reference set | fα | fβ | fT | fU |
| SELCON3 | SMP50 | 0.59 | 0.06 | 0.14 | 0.21 |
| SMP56 | 0.60 | 0.07 | 0.13 | 0.22 | |
| SP37 | 0.54 | 0.10 | 0.16 | 0.22 | |
| SP43 | 0.58 | 0.10 | 0.13 | 0.21 | |
| CONTIN/LL | SMP50 | 0.59 | 0.06 | 0.14 | 0.21 |
| SMP56 | 0.59 | 0.06 | 0.13 | 0.22 | |
| SP37 | 0.51 | 0.10 | 0.17 | 0.22 | |
| SP43 | 0.57 | 0.11 | 0.12 | 0.19 | |
| CDSSTR | SMP50 | 0.63 | 0.06 | 0.13 | 0.19 |
| SMP56 | 0.64 | 0.05 | 0.12 | 0.20 | |
| SP37 | 0.62 | 0.06 | 0.13 | 0.20 | |
| SP43 | 0.61 | 0.09 | 0.12 | 0.18 | |
| Average ± SD | 0.59 ± 0.04 | 0.08 ± 0.02 | 0.14 ± 0.02 | 0.21 ± 0.01 |
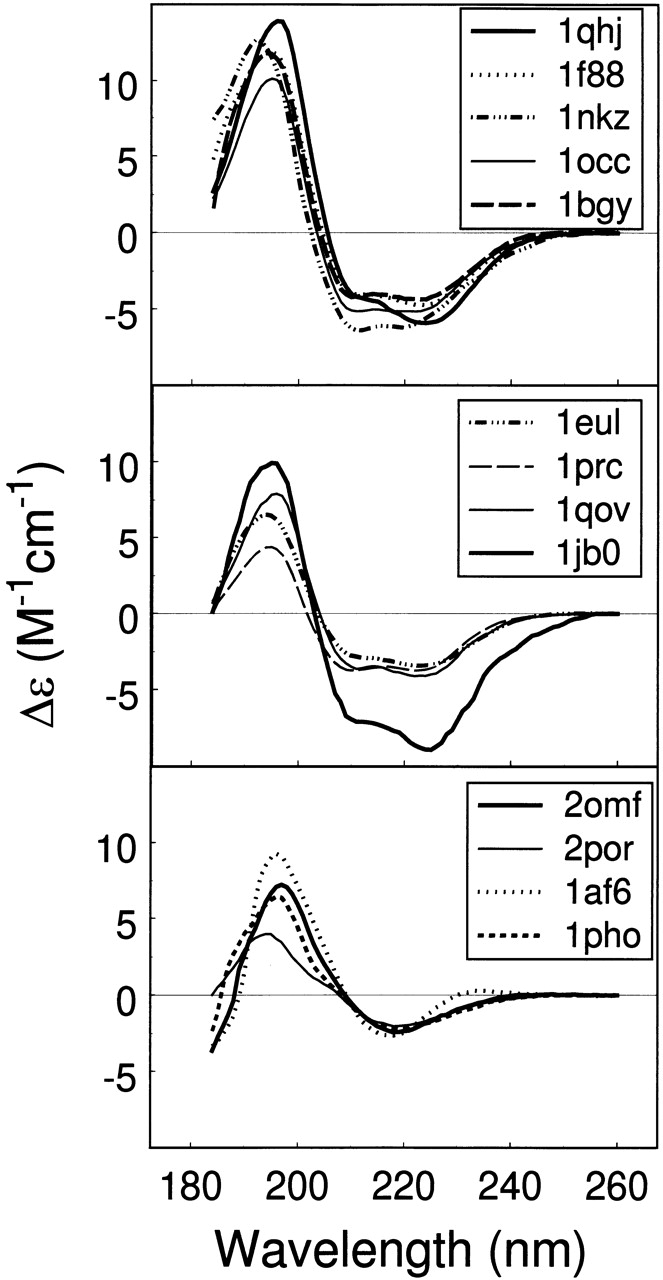
Park et al. (1992) provide a set of 30 membrane protein CD spectra, of which 13 were used in forming the MP13 reference set, and that of F0F1 ATPase is discussed above. By excluding four membrane proteins that were represented twice and colicin A, which is present in our reference set, we are left with 11 additional membrane protein CD spectra, which are shown in Figure 3 ▶. We have performed the analysis of these 11 membrane protein CD spectra, and the results are given in Table 5. The analysis of each CD spectrum was performed by using three methods and four reference protein sets, as in the case of F0F1 ATPase, and the averages and standard deviations are presented. The results from Park et al. (1992), obtained from the CCA method (Perczel et al. 1991) and their 30-membrane protein reference set, are also given for comparison. As indicated in the footnotes to Table 6, a few membrane protein CD spectra were not analyzed well. We either failed to obtain a solution or obtained a poor solution from at least one method; a solution was considered poor if the sum of fractions was not in the range 0.90 to 1.10 or if it differed greatly from solutions from other methods/reference sets. These are, generally, a consequence of the uniqueness of the CD spectrum. The CD spectrum of proton ATPase has a very large amplitude, and consequently, solutions from different methods of CD analysis or different reference sets differ, giving a large uncertainty in the determined secondary structure fractions. Lipoprotein is another example, having a very small CD amplitude that results in a failed analysis from SELCON3 and CDSSTR. Errors in concentration may have contributed to such large or small CD amplitudes. Dissimilar results from different methods, or reference sets, result in a larger uncertainty in the secondary structures determined, affecting the reliability. A majority of membrane proteins, however, gave satisfactory results.
Figure 3.
CD spectra of 11 additional membrane proteins from Park et al. (1992; with permission from The Protein Society), which were analyzed by using the methods developed in this study.
Table 5.
Analysis of CD spectra of 11 additional membrane proteins
| Protein | Method | fα | fβ | fT | fU |
| Light harvesting complex | CDPro | 0.49 ± 0.02 | 0.09 ± 0.03 | 0.17 ± 0.01 | 0.25 ± 0.02 |
| CCAa | 0.44 | 0.14 | 0.03 | 0.39 | |
| NADH-ubiquinone reductase (N. crassa)a,b | CDPro | 0.28 ± 0.17 | 0.32 ± 0.14 | 0.18 ± 0.07 | 0.22 ± 0.07 |
| CCA | 0.25 | 0.27 | 0.49 | 0.00 | |
| Ubiquinol-cytochrome c reductase (N. crassa) | CDPro | 0.32 ± 0.02 | 0.19 ± 0.01 | 0.21 ± 0.01 | 0.28 ± 0.01 |
| CCA | 0.33 | 0.16 | 0.06 | 0.45 | |
| Proton ATPase (yeast plasma membrane)c | CDPro | 0.79 ± 0.11 | 0.03 ± 0.04 | 0.10 ± 0.14 | 0.07 ± 0.06 |
| CCA | 0.44 | 0.01 | 0.56 | 0.00 | |
| H,K-dependent ATPase | CDPro | 0.22 ± 0.02 | 0.26 ± 0.03 | 0.22 ± 0.01 | 0.29 ± 0.01 |
| CCA | 0.20 | 0.15 | 0.12 | 0.53 | |
| Nicotinic acetylcholine receptor | CDPro | 0.37 ± 0.07 | 0.19 ± 0.05 | 0.19 ± 0.04 | 0.26 ± 0.04 |
| CCA | 0.27 | 0.00 | 0.20 | 0.52 | |
| Glucose transporter (human erythrocytes) | CDPro | 0.57 ± 0.02 | 0.11 ± 0.02 | 0.14 ± 0.02 | 0.18 ± 0.01 |
| CCA | 0.50 | 0.32 | 0.07 | 0.11 | |
| Band 3 (human erythrocytes; anion transporter) | CDPro | 0.52 ± 0.04 | 0.07 ± 0.02 | 0.20 ± 0.04 | 0.22 ± 0.07 |
| CCA | 0.40 | 0.10 | 0.02 | 0.48 | |
| Band 3 (human erythrocytes, membrane domain) | CDPro | 0.76 ± 0.06 | 0.02 ± 0.04 | 0.08 ± 0.02 | 0.13 ± 0.03 |
| CCA | 0.69 | 0.18 | 0.01 | 0.13 | |
| Lipoprotein (E. coli, outer membrane)d | CDPro | 0.08 ± 0.05 | 0.40 ± 0.04 | 0.21 ± 0.01 | 0.31 ± 0.01 |
| CCA | 0.25 | 0.24 | 0.00 | 0.51 | |
| Prostaglandin synthasee | CDPro | 0.18 ± 0.02 | 0.31 ± 0.02 | 0.21 ± 0.00 | 0.29 ± 0.01 |
| CCA | 0.27 | 0.18 | 0.06 | 0.49 |
The CD spectra of these 11 membrane proteins, taken from Park et al. (1992), are given in Fig. 3 ▶. CD estimates of secondary structure fractions from CDPro are given as averages (and standard deviations) of solutions from three programs (SELCON3, CONTIN/LL, CDSSTR) and four reference protein sets (SP37, SP43, SMP50, and SMP56).
a Secondary structure fractions from Park et al. (1992), obtained with their 30-membrane protein reference set and the CCA method.
b Poor solutions were obtained from CONTIN/LL and CDSSTR, which resulted in larger uncertainties. The secondary structure fractions obtained by excluding poor solutions are fα, 0.38; fβ, 0.22; fT, 0.17; and fU, 0.23.
c Three poor solutions, obtained from SELCON3, were excluded in determining the averages.
d SELCON3 and CDSSTR failed to give solutions. The averages obtained from four solutions (CONTIN/LL with four reference sets) are reported.
e Poor solutions were obtained from SELCON3 which were excluded in determining the averages.
Table 6.
Analysis of soluble protein CD spectra with combined soluble +membrane protein reference sets
| α | β | T | U | ||||||||
| Method | Reference set | δα | rα | δβ | rβ | δT | rT | δU | rU | δ | r |
| SELCON3 | SMP50 | 0.09 | 0.92 | 0.11 | 0.72 | 0.06 | 0.50 | 0.11 | 0.18 | 0.09 | 0.79 |
| SP37 | 0.08 | 0.94 | 0.11 | 0.71 | 0.06 | 0.57 | 0.11 | 0.15 | 0.09 | 0.80 | |
| SMP56 | 0.07 | 0.93 | 0.10 | 0.77 | 0.07 | 0.44 | 0.10 | 0.22 | 0.09 | 0.82 | |
| SP43 | 0.08 | 0.93 | 0.11 | 0.71 | 0.03 | 0.37 | 0.10 | 0.22 | 0.09 | 0.80 | |
| CONTIN/LL | SMP50 | 0.08 | 0.94 | 0.11 | 0.75 | 0.05 | 0.58 | 0.09 | 0.28 | 0.09 | 0.83 |
| SP37 | 0.08 | 0.93 | 0.12 | 0.65 | 0.07 | 0.42 | 0.09 | 0.28 | 0.09 | 0.81 | |
| SMP56 | 0.07 | 0.94 | 0.10 | 0.80 | 0.07 | 0.40 | 0.09 | 0.27 | 0.08 | 0.84 | |
| SP43 | 0.08 | 0.93 | 0.11 | 0.71 | 0.08 | 0.33 | 0.09 | 0.25 | 0.09 | 0.80 | |
| CDSSTR | SMP50 | 0.09 | 0.94 | 0.11 | 0.75 | 0.06 | 0.59 | 0.09 | 0.44 | 0.09 | 0.84 |
| SP37 | 0.08 | 0.94 | 0.12 | 0.69 | 0.07 | 0.45 | 0.10 | 0.32 | 0.09 | 0.81 | |
| SMP56 | 0.09 | 0.94 | 0.10 | 0.76 | 0.07 | 0.50 | 0.09 | 0.43 | 0.09 | 0.84 | |
| SP43 | 0.09 | 0.92 | 0.11 | 0.71 | 0.07 | 0.46 | 0.09 | 0.37 | 0.09 | 0.80 | |
The secondary structures (αR to U), performance indices (δ, r), and reference sets are defined in footnotes to Tables 1, 2, and 3, respectively. The results for the corresponding soluble protein reference sets (SP37 and SP43) were obtained by cross-validation analysis and are similar to those of Sreerama and Woody (2000b). The wavelength range of CD spectra was 185 to 240 nm for SP37 and SMP50, and 190 to 240 nm for SP43 and SMP56.
Analysis of soluble proteins with soluble + membrane protein reference sets
We have also examined the performance of soluble + membrane protein reference sets for analyzing soluble protein CD spectra. The results of a cross-validation analysis of soluble proteins in SMP50 and SMP56, in which each soluble protein was removed from the reference set and analyzed by using the remaining reference proteins, are compared with those from the corresponding soluble protein reference sets, SP37 and SP43, in Table 6. These represent the effects of including membrane proteins in the analysis of soluble proteins.
In general, the inclusion of membrane proteins led to slightly improved analysis of soluble proteins. The performance indices for α, T, and U fractions showed smaller improvements, and those for β showed slightly larger improvements, with a few exceptions. The extent of improvements, however, depended on the method of analysis. Overall, SMP56 performed the best, with δα, 0.07 to 0.09; δβ, ~0.10; and δ, 0.08 to 0.09. It showed improvements over the corresponding soluble protein reference set, SP43, for all three methods of analysis. SMP50 also showed improvements over the corresponding soluble protein reference set, SP37, for CONTIN/LL and CDSSTR. For SELCON3, the results from SMP50 were slightly worse than those from SP37.
The performance indices for soluble proteins from the two soluble + membrane protein reference sets, SMP50 and SMP56, were comparable. The larger set showed slightly larger improvement for the β fraction than that for the α fraction, which were offset by a slight worsening of the T fraction. The overall performance was similar for CONTIN/LL and CDSSTR, whereas SELCON3 showed slight improvement, which may be a correction for the poorer performance of SMP50. Improvements in the soluble protein analysis obtained by the addition of MP13 to both SP37 and SP43 indicate an increase in the information content of both SMP50 and SMP56 due to membrane proteins.
Discussion
The analysis of membrane protein CD spectra has been considered problematic (Park et al. 1992; Wallace et al. 2003). Among the many reasons listed for the difficulties involved in membrane protein CD analysis are the following: suitability and inadequacy of existing soluble protein reference sets, lack of membrane protein structural information, difficulties in obtaining membrane protein CD spectra, different spectral characteristics of soluble and membrane proteins, and biasing effects of a given reference protein set. The use of a membrane protein reference set specifically for the analysis of membrane proteins has been suggested as an alternative (Park et al. 1992; Wallace et al. 2003). Such a reference set has been developed (Park et al. 1992), albeit without the corresponding structural information.
By using the available spectral and structural data for 13 membrane proteins, we have examined the performance of existing soluble protein reference sets, a newly constructed membrane protein reference set, and combined soluble + membrane protein reference sets for analyzing membrane protein CD spectra. We have also examined the performance of combined souluble + membrane protein reference sets for the analysis of soluble protein CD spectra. Although the existing soluble protein reference sets performed reasonably well in analyzing membrane proteins, the membrane protein reference set performed poorly. The poor performance of the membrane protein reference set was probably due to the low information content, because the number of reference proteins was small. The inclusion of membrane proteins in the soluble protein reference sets increased the spectral information content and improved the performance for both membrane and soluble proteins.
Our results for the analysis of membrane protein CD spectra with both membrane and soluble protein reference sets are better than those of Park et al. (1992). Park et al. used different CD analysis methods for soluble and membrane protein reference sets because of the lack of structural information for membrane proteins. They used the CCA method (Perczel et al. 1991) with the membrane protein reference set without any secondary structure information, and the method of Chang et al. (1978) and the variable selection method (Manavalan and Johnson Jr. 1987) with soluble protein reference sets with secondary structure information. We obtain improvements in the performance of the membrane protein reference set because we use both secondary structure fractions and variable selection of reference proteins in our analysis, which are not included in the CCA method (Perczel et al. 1991). The improvements in the analysis of membrane proteins with soluble protein reference sets over that of Park et al. (1992) are due to the advances in protein CD analyses. We use the latest CD analysis methods, which have increased information content made possible by the inclusion of a large number of soluble proteins and better algorithms.
Our conclusions are different from those of Wallace et al. (2003), who also used CDPro software in their analysis of eight membrane proteins and reported results for two representative CD spectra. Wallace et al. concluded that the existing soluble protein reference sets give inaccurate results for membrane protein CD analysis. They attributed the poor performance to the spectral differences, such as wavelength shifts and intensity differences, between soluble and membrane proteins. They used two parameters in reaching their conclusions: normalized RMS deviation (NRMSD) calculated as
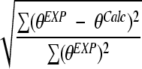 |
(Brahms and Brahms 1980), between the back-calculated (θCalc) and experimental CD spectra (θEXP), and the absolute difference between the secondary structure fractions estimated by CD (fCD) and from crystal structures (fEXP) given as R. Two measures of R were used by Wallace et al. (2003): Rav = (Σ | fEXP − fCD|),1 which gives the total error in the CD-predicted values, and RP = | fEXP − fCD |, which gives the error in the prediction of the predominant secondary structure (either α or β). However, their reliance on NRMSD as a measure of accuracy and the manner in which they determined the secondary structure fractions from crystal structures for comparison with CD estimates may lead to errors.
CDPro provides seven reference protein sets that differ either in the number or in the wavelength range of CD spectra of reference proteins (Sreerama and Woody 2000b), all of which were used by Wallace et al. (2003). CDPro also uses three methods for assigning secondary structure fractions to crystal structures (Kabsch and Sander 1983; Sreerama and Woody 1994a; King and Johnson Jr. 1999), of which the former method is used in five reference sets and does not determine the poly(Pro)II type structure fraction. Wallace et al. (2003) use an average from five different assignments of secondary structures as fEXP in calculating Rav and RP. The CD estimates of secondary structure fractions correspond to a particular definition followed in constructing the reference set used in a given analysis. The CD estimates should be compared with the secondary structure fractions obtained by using the same assignment method used in the construction of the reference protein set. Luckily, the average α and β fractions given by Wallace et al. (2003) are similar to the DSSP values, which are used in six of the seven reference protein sets in CDPro. This allows the comparison of the CD estimate for the predominant secondary structure. Wallace et al. (2003), however, give detailed results for only two proteins. When we consider the results from the reference sets SP37 and SP43 (db = 3 or 4; Table 2b of Wallace et al. [2003]) for the predominantly β-sheet membrane protein ferric enterobactin receptor, the β fraction is determined very accurately (RP = 0.0; SELCON3, SP37). For the predominantly α-helical membrane protein mechanosensitive channel from M. tuberculosis, CD analyses predict a higher α-helical content (RP = 0.28 to 0.38), which is consistent with the intensities of the CD bands (Fig. 1 ▶ of Wallace et al. [2003]); the CD spectrum is comparable to that of bacteriorhodopsin (1qhj, Fig. 1 ▶), which has ~75% α-helical content. This apparent discrepancy between the α-helical contents from CD and the crystal structure may be due to difference between the solution and solid-state structures.
We have previously used the RMS difference between the CD estimates and the crystal structure values of secondary structure fractions,
 |
(Sreerama and Woody 1994b) as a measure of error in the results for specific proteins. Both δf and Rav give a measure of collective error for specific proteins, but Rav seems to accentuate the error. We do not see any advantage of using Rav over δf. RP, on the other hand, could be useful in testing the performance of a given method, although it is of questionable value for many soluble proteins that have no dominant secondary structure. Further, one needs to be careful in drawing conclusions based on just RP. The RP values reported by Wallace et al. (2003) for the α-rich membrane protein appear to be too large for some reference protein sets. For example, Wallace et al. (2003) report the RP value of 0.48 with SELCON3 (Table 2b, db = 2), and the average value obtained from the crystal structure is 0.52 (Table 1b of Wallace et al. [2003]). This indicates a CD estimate (fαCD = fαEXP ± RP) of either 1.00 or 0.04, both of which are improbable. Wallace et al. obtained poor results for db = 2 with the other two programs also, which indicates a failed analysis. We have provided the δf values for the MP13 set in Electronic Supplemental Material.
The back-calculated spectra from the three CD analysis methods provided in CDPro differ qualitatively because they follow different algorithms. CONTIN/LL always gives the best agreement with the experimental spectrum because the algorithm minimizes the error between the fitted and experimental spectra (Provencher and Glöckner 1981) and is expected to have the lowest NRMSD. Both SELCON3 and CDSSTR use the singular value decomposition algorithm (Forsythe et al. 1977) and ignore singular values that correspond to the noise in the CD data set (Hennessey Jr. and Johnson Jr. 1981). The number of singular values included is varied in SELCON3 (Sreerama and Woody 1993), whereas that in CDSSTR is always five (Johnson Jr. 1999), thus affecting overall noise excluded from the analysis. The minimal basis (Dalmas and Bannister 1995) and the locally linearized (van Stokkum et al. 1990) versions of variable selection, respectively, are implemented in CDSSTR and SELCON3. Generally, errors between the experimental and back-calculated spectra from CDSSTR are smaller than those from SELCON3 because of these differences in their algorithms.
For the 13 membrane protein CD spectra analyzed with SMP50, we obtained NRMSD values in the range (and averaged NRMSD) 0.08 to 0.51 (0.22), 0.04 to 0.12 (0.07), and 0.02 to 0.08 (0.04), respectively, from SELCON3, CDSSTR, and CONTIN/LL programs; the corresponding values for soluble proteins from SMP50 were 0.13 to 1.0 (0.24), 0.01 to 0.24 (0.08), and 0.01 to 0.14 (0.03). Moreover, the NRMSD values were uncorrelated with the error in the secondary structure prediction. Wallace et al. (2003) obtained NRMSD values of 0.002 to 0.050 and 0.019 to 0.193, respectively, for the predominantly α-helical and predominantly β-sheet membrane proteins with SP37 and SP43 reference protein sets (Table 2b, Wallace et al. 2003). Given the differences between the three methods in the nature of back-calculated spectra, it is difficult to use them to draw conclusions as to the accuracy of the analysis from NRMSD values.
We did not find evidence to support the suggested (Wallace et al. 2003) wavelength shifts between soluble and membrane protein CD spectra. The variation in the spectral peaks observed in the 30 membrane proteins of Park et al. (1992) was similar to that observed in the soluble protein reference set, SP43. The position of the positive ππ* band varied between 192 and 196 nm in the CD spectra of predominantly α-rich membrane proteins, whereas the corresponding range for α-rich soluble proteins was 192–195 nm. The β-rich soluble proteins showed the largest variation in the position of the positive ππ* band (185 to 197 nm), that in αβ were intermediate (188 to 195 nm). The number of β-rich membrane proteins was too small to compare with the soluble proteins.
It is important to have a good representation of spectral and structural variation of proteins in the reference set. The success of membrane protein CD analysis with soluble protein reference sets indicates the presence of good spectral and structural variation, which is lacking in the small membrane protein reference set. The improvements obtained in the analysis of both membrane and soluble proteins with the addition of a small number of membrane proteins indicate an increase in the information content in the soluble + membrane protein reference sets. Although the existing soluble protein reference sets perform quite well, the inclusion of membrane proteins should further improve protein CD analysis. The MP13 reference set is dominated by α-rich membrane proteins and, by any measure, is not optimal. There is scope for expanding the membrane protein reference set as new and higher resolution structures and CD spectra become available for membrane proteins.
Materials and methods
CD spectra
The following 13 membrane proteins2 were used in this study: reaction center from Rhodopseudomonas viridis (1prc); photosystem I (1jb0); reaction center from Rhodobacter sphaeroides (1qov); B 800–850 antenna complex from Rhodopseudomonas acidophila 10050 (1nkz); ubiquinol-cytochrome c reductase from bovine heart (1bgy); cytochrome c oxidase from bovine heart (1occ)3; rhodopsin from bovine retina (1f88); purple membrane from Halobacterium halobium (1qhj); Ca2+ ATPase from rabbit muscle sarcoplasmic reticulum (1eul); porin (gene OmpF product) from E. coli, batch 2 (2omf)4; porin from Rhodobacter capsulatus (2por); maltoporin (LamB) (1af6); and phosphoporin (PhoE) (1pho). Of these proteins, the first nine (1prc-1eul) are α-rich membrane proteins, and the last four (2omf-1pho) are β-rich membrane proteins. The CD spectra of these proteins (Fig. 1 ▶) were obtained from Park et al. (1992) and are identified following the information given in the supplement to the publication. The PDB (Berman et al. 2000) codes of the crystal structures used to determine secondary structures are given in parenthesis and the citations for crystal structures are provided in the Electronic Supplemental Material (Table S1).
Secondary structure
The secondary structure fractions of the membrane proteins were obtained from the program DSSP (Kabsch and Sander 1983), which uses hydrogen bonding patterns to identify secondary structure elements. The α-helix and β-strand structures were split into regular and distorted classes, considering four residues per α-helix and two residues per β-strand distorted (Sreerama et al. 1999). For proteins with more than one polypeptide chain in the structure, all chains were considered for secondary structure assignment. Our grouping of DSSP assignments gave us six secondary structural classes: regular α-helix, αR; distorted α-helix, αD; regular β-strand, βR; distorted β-strand, βD; turns, T; and unordered, U. The secondary structure fractions are given in Table 1. The secondary structure fractions used in the reference protein sets were also determined in an identical manner (Sreerama and Woody 2000b).
CD analysis
The analysis of CD spectra was performed by using CDPro software (Sreerama and Woody 2000b), which includes three different methods for analyzing protein CD spectra implemented in computer programs CDSSTR (Johnson Jr. 1999), SELCON3 (Sreerama and Woody 1993; Sreerama et al. 1999), and CONTIN/LL (Provencher and Glöckner 1981; Sreerama and Woody 2000b). These methods differ either in the mathematical procedure or in the implementation of variable selection (Manavalan and Johnson Jr. 1987) or both, and they have been described elsewhere (Sreerama and Woody 2000b). Similar results from all three methods provide a measure of the reliability of the analysis. Several reference protein sets with varying number of proteins, inversely related to the wavelength range, are also provided in CDPro and were used in our analysis.
The performance of the analysis was characterized by RMS deviations (δ) and correlation coefficients (r) between the x-ray and CD estimates of secondary structure fractions for different secondary structures. These are denoted by δk and rk, where k is one of the secondary structural types considered. The results from CD analysis for six secondary structures were converted to four secondary structures (α, β, T, and U) by combining the fractions of regular and distorted fractions of α and β. Overall, performance of the analysis for a given set of secondary structure fractions was determined by considering all secondary structure fractions collectively, and these are given by δ and r.
The RMS deviations and correlation coefficients were calculated by using the following equations:
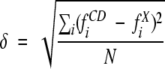 |
and
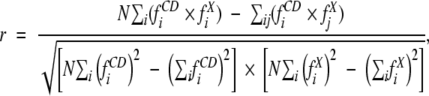 |
where 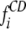 and
and 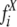 are CD and X-ray estimates of secondary structure types of N reference samples, respectively.
are CD and X-ray estimates of secondary structure types of N reference samples, respectively.
Electronic supplemental material
Citations for the crystal structures used in this study, and detailed results obtained from the analysis of CD spectra of 13 membrane proteins from CDPro software using the existing and newly constructed reference protein sets are provided. Table S1 contains the citations, and three additional tables (Tables S2, S3, and S4) contain results from CD analysis programs SELCON3, CONTIN/LL, and CDSSTR.
Acknowledgments
We thank Dr. W.C. Johnson Jr., Dr. G.D. Fasman, Dr. T.A Keiderling, Dr. S. Yu. Venyaminov, and the late Dr. J.T. Yang, as well as their coworkers, for making the CD data of soluble/membrane proteins available. We thank Dr. A-Y.M. Woody for a critical reading of the manuscript and helpful discussions. This work was supported by NIH research grant EB02803 (formerly GM22994).
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Abbreviations
CD, circular dichroism
CCA, the convex constraint method for protein CD analysis
CDSSTR, Johnson’s minimal basis-random selection method for protein CD analysis
CONTIN/LL, the ridge-regression method for protein CD analysis combined with the locally linearized method for variable selection
DSSP, a computer program for defining secondary structure of proteins
PDB, Protein Data Bank
SELCON3, the self-consistent method for protein CD analysis, version 3
MP13, reference set of 13 membrane proteins
MP30, reference set of 30 membrane proteins
SP29, reference set of 29 soluble proteins
SP37, reference set of 37 soluble proteins
SP42, reference set of 42 soluble proteins
SP43, reference set of 43 soluble proteins
SP48, reference set of 48 soluble proteins
SMP50, reference set of 50 soluble + membrane proteins
SMP56, reference set of 56 soluble + membrane proteins
RMS, root mean square
NRMSD, normalized RMS deviation
δ, RMS deviation
r, correlation coefficient
αR, regular α-helix
αD, distorted α-helix
α, total α-helix
βR, regular β-strand
βD, distorted β-strand
β, total β-sheet
T, turns
U, unordered
fX, fractional content of secondary structure X, X = α, β, T and U
δX, RMS deviation between the CD-estimated and the X-ray values of the secondary structure X for a set of proteins, X = α, β, T and U
rX, correlation between the CD-estimated and the X-ray values of the secondary structure X for a set of proteins, X = α, β, T and U
δf, RMS deviation between the CD estimates and the crystal structure values of secondary structure fractions for a given protein
Supplemental material: See www.proteinscience.org
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.03258404.
Footnotes
Wallace et al. (2003) did not specify absolute values in their definition of R. However, the absence of negative values of R and Rav reported in their article indicates that they have used absolute differences in calculating R values, as it should be. Also, the Rav values they reported included the differences between the CD estimates and crystal structure values of the α, β and turns fractions only.
The bacterial source of the protein is listed as it appears in the corresponding PDB file. The source as listed in Park et al. (1992) may be different because of changes in microbial taxonomy.
Bovine cytochrome oxidase and porin (gene OmpF product) were represented twice in the Park et al. (1992) set of membrane proteins. The cytochrome c oxidase, the source of which was identified as human erythrocytes, was actually bovine cytochrome oxidase; the two CD spectra of bovine cytochrome oxidase were almost identical.
Of the two porin (gene OmpF product) spectra, we selected the one from Dr. A. Tucker and Dr. J.H. Lakey (batch 2). Dr. Tucker also provided the spectra of two other porins.
References
- Abrahams, J.P., Leslie, A.G., Lutter, R., and Walker, J.E. 1994. Structure at 2.8 Å resolution of F1-ATPase from bovine heart mitochondria. Nature 370 621–628. [DOI] [PubMed] [Google Scholar]
- Andrade, M.A., Chacán, P., Merolo, J.J., and Morán, F. 1993. Evaluation of secondary structure of protein from UV circular dichroism spectra using unsupervised learning neural network. Protein Eng. 6 383–390. [DOI] [PubMed] [Google Scholar]
- Berman, H.M., Westbrook, J., Feng, Z., Gilliand, G., Bhat, T.N., Weissig, H., Shindyalov, I.N., and Bourne, P.E. 2000. The Protein Data Bank. Nucleic Acids Res. 28 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Böhm, G., Muhr, R., and Jaenicke, R. 1992. Quantitative analysis of protein far UV circular dichroism spectra by neural networks. Protein Eng. 5 191–195. [DOI] [PubMed] [Google Scholar]
- Boyer, P.D. 1997. The ATP synthase: A splendid molecular machine. Annu. Rev. Biochem. 66 717–749. [DOI] [PubMed] [Google Scholar]
- Brahms, S. and Brahms, J. 1980. Determination of protein secondary structure in solution by vacuum ultraviolet circular dichroism. J. Mol. Biol. 138 149–178. [DOI] [PubMed] [Google Scholar]
- Chang, C.T., Wu, C.-S.C., and Yang, J.T. 1978. Circular dichroism analysis of protein conformation: Inclusion of β-turns. Anal. Biochem. 91 13–31. [DOI] [PubMed] [Google Scholar]
- Dalmas, B. and Bannister, W.H. 1995. Prediction of protein secondary structure from circular dichroism spectra: An attempt to solve the problem of the best-fitting reference protein subsets. Anal. Biochem. 225 39–48. [DOI] [PubMed] [Google Scholar]
- Forsythe, G.E., Malcolm, M.A., and Moler, C.B. 1977. Computer methods for mathematical computations. Prentice-Hall, Englewood Cliffs, NJ.
- Girvin, M.E., Rastogi, V.K., Abildgaard, F., Markley, J.L., and Fillingame, R.H. 1998. Solution structure of the transmembrane H+-transporting subunit c of the F1F0 ATP synthase. Biochemistry 37 8817–8824. [DOI] [PubMed] [Google Scholar]
- Greenfield, N. and Fasman, G.D. 1969. Computed circular dichroism spectra for the evaluation of protein conformation. Biochemistry 8 4108–4116. [DOI] [PubMed] [Google Scholar]
- Greenfield, N.J. 1996. Methods to estimate the conformation of proteins and polypeptides from circular dichroism data. Anal. Biochem. 235 1–10. [DOI] [PubMed] [Google Scholar]
- Hennessey Jr., J.P. and Johnson Jr., W.C. 1981. Information content in the circular dichroism of proteins. Biochemistry 20 1085–1094. [DOI] [PubMed] [Google Scholar]
- Johnson Jr., W.C. 1988. Secondary structure of proteins through circular dichroism spectroscopy. Annu. Rev. Biophys. Biophys. Chem. 17 145–166. [DOI] [PubMed] [Google Scholar]
- ———. 1999. Analyzing protein circular dichroism spectra for accurate secondary structures. Proteins 35 307–312. [PubMed] [Google Scholar]
- Kabsch, W. and Sander, C. 1983. Dictionary of protein secondary structure: Pattern recognition of hydrogen bonded and geometric features. Biopolymers 22 2577–2637. [DOI] [PubMed] [Google Scholar]
- King, S.M. and Johnson Jr., W.C. 1999. Assigning secondary structure from protein coordinate data. Proteins 35 313–320. [PubMed] [Google Scholar]
- Pancoska, P. and Keiderling, T.A. 1991. Systematic comparison of statistical analysis of electronic and vibrational circular dichroism for secondary structure prediction of selected proteins. Biochemistry 30 6885–6895. [DOI] [PubMed] [Google Scholar]
- Pancoska, P., Bitto, E., Janota, V., Urbanova, M., Gupta, V.P., and Keiderling, T.A. 1995. Comparison of and limits of accuracy for statistical analyses of vibrational and electronic circular dichroism spectra in terms of correlations to and predictions of protein secondary structure. Protein Sci. 4 1384–1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park, K., Perczel, A., and Fasman, G.D. 1992. Differentiation between transmembrane and peripheral helices by the deconvolution of circular dichroism spectra of membrane proteins. Protein Sci. 1 1032–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perczel, A., Hollosi, M., Tusnady, G., and Fasman, G.D. 1991. Convex constraint analysis: A natural deconvolution of circular dichroism curves of proteins. Protein Eng. 4 669–679. [DOI] [PubMed] [Google Scholar]
- Provencher, S.W. and Glöckner, J. 1981. Estimation of protein secondary structure from circular dichroism. Biochemistry 20 33–37. [DOI] [PubMed] [Google Scholar]
- Rastogi, V.K. and Girvin, M.E. 1999. Structural changes linked to proton translocation by subunit c of the ATP synthase. Nature 402 263–268. [DOI] [PubMed] [Google Scholar]
- Sreerama, N. and Woody, R.W. 1993. A self-consistent method for the analysis of protein secondary structure from circular dichroism. Anal. Biochem. 209 32–44. [DOI] [PubMed] [Google Scholar]
- ———. 1994a. Poly(Pro)II helices in globular proteins: Identification and circular dichroic analysis. Biochemistry 33 10022–10025. [DOI] [PubMed] [Google Scholar]
- ———. 1994b. Protein secondary structure from circular dichroism spectroscopy: Combining variable selection principle and cluster analysis with neural network, ridge regression and self-consistent methods. J. Mol. Biol. 242 497–507. [DOI] [PubMed] [Google Scholar]
- ———. 2000a. Circular dichroism of peptides and proteins. In Circular dichroism: Principles and applications, 2nd ed. (eds. N. Berova et al.), pp. 601–620. Wiley, New York.
- ———. 2000b. Estimation of protein secondary structure from CD spectra: Comparison of CONTIN, SELCON and CDSSTR methods with an expanded reference set. Anal. Biochem. 287 252–260. [DOI] [PubMed] [Google Scholar]
- ———. 2004. Computation and analysis of protein circular dichroism spectra. Methods Enzymol. (in press). [DOI] [PubMed]
- Sreerama, N., Venyaminov, S.Y., and Woody, R.W. 1999. Estimation of the number of α-helical and β-strand segments in proteins using circular dichroism spectroscopy Protein Sci. 8 370–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ———. 2000. Estimation of protein secondary structure from CD spectra: Inclusion of denatured proteins with native proteins in the analysis. Anal. Biochem. 287 243–251. [DOI] [PubMed] [Google Scholar]
- ———. 2001. Analysis of protein circular dichroism spectra based on tertiary structure classification. Anal. Biochem. 299 271–274. [DOI] [PubMed] [Google Scholar]
- van Stokkum, I.H.M., Spoelder, H.J.W., Bloemendal, M., van Grondelle, R., and Groen, F.C.A. 1990. Estimation of protein secondary structure and error analysis from circular dichroism spectra. Anal. Biochem. 191 110–118. [DOI] [PubMed] [Google Scholar]
- Wallace, B.A., Lees, J.G., Orry, A.J.W., Lobley, A., and Janes, R.W. 2003. Analyses of circular dichroism spectra of membrane proteins. Protein Sci. 12 875–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venyaminov, S.Y., and Yang, J.T. 1996. Determination of protein secondary structure. In Circular dichroism and the conformational analysis of biomolecules (ed. G.D. Fasman), pp. 69–107. Plenum, New York.
- Yang, J.T., Wu, C.-S.C., and Martinez, H.M. 1986. Calculation of protein conformation from circular dichroism. Methods Enzymol. 130 208–269. [DOI] [PubMed] [Google Scholar]