

Abstract
AMP-activated protein kinase (AMPK) is a αβγ heterotrimer that is activated in response to both hormones and intracellular metabolic stress signals. AMPK is regulated by phosphorylation on the α subunit and by AMP allosteric control previously thought to be mediated by both α and γ subunits. Here we present evidence that adjacent γ subunit pairs of CBS repeat sequences (after Cystathionine Beta Synthase) form an AMP binding site related to, but distinct from the classical AMP binding site in phosphorylase, that can also bind ATP. The AMP binding site of the γ1 CBS1/CBS2 pair, modeled on the structures of the CBS sequences present in the inosine monophosphate dehydrogenase crystal structure, contains three arginine residues 70, 152, and 171 and His151. The yeast γ homolog, snf4 contains a His151Gly substitution, and when this is introduced into γ1, AMP allosteric control is substantially lost and explains why the yeast snf1p/snf4p complex is insensitive to AMP. Arg70 in γ1 corresponds to the site of mutation in human γ2 and pig γ3 genes previously identified to cause an unusual cardiac phenotype and glycogen storage disease, respectively. Mutation of any of AMP binding site Arg residues to Gln substantially abolishes AMP allosteric control in expressed AMPK holoenzyme. The Arg/Gln mutations also suppress the previously described inhibitory properties of ATP and render the enzyme constitutively active. We propose that ATP acts as an intrasteric inhibitor by bridging the α and γ subunits and that AMP functions to derepress AMPK activity.
Keywords: AMPK, AMP, CBS sequences, γ subunit, allosteric and intrasteric control
AMPK is a multisubstrate enzyme that is activated in response to both hormones and intracellular metabolic stress signals including exercise, hypoxia, and nutrient deprivation. AMPK regulates many metabolic processes including glucose transport, glycolysis, and lipid metabolism, coupling energy metabolism to physiological functions including protein synthesis and gene transcription (Kemp et al. 2003). The extent of AMPKs regulatory functions are well illustrated in lipid metabolism where it inhibits multiple steps; fatty acid synthesis by phosphorylation of acetyl-CoA carboxylase-α, cholesterol synthesis by phosphorylation of HMG-CoA reductase, triglyceride synthesis by phosphorylation of GPAT (glycerol-phosphate acyl transferase), and hormone-sensitive lipase (Hardie and Hawley 2001). In many cell types AMPK regulates β-oxidation of fatty acids by phosphorylation of acetyl CoA carboxylase-β. Furthermore, AMPK exerts powerful transcriptional control of genes involved in lipid and carbohydrate metabolism (Zhou et al. 2000; Leclerc et al. 2001; Lo et al. 2001; Zheng et al. 2001; Barthel et al. 2002; Kawaguchi et al. 2002; Stoppani et al. 2002; Ferre et al. 2003; Kemp et al. 2003; Leff 2003). AMPK can also be activated by several adipocyte-derived hormones, including adiponectin (Yamauchi et al. 2002) and leptin (Minokoshi et al. 2002), as well as by isoproterenol (Moule and Denton 1998). Several drugs, including metformin (Hawley et al. 2002) and rosiglitazone (Fryer et al. 2002), used extensively for the treatment of type II diabetes, activate AMPK, supporting the view that AMPK actions protect the body from developing diabetes. The importance of AMPK in regulating glucose metabolism has been strongly highlighted by the discovery of a series of mutations in the AMPK γ subunit genes that cause glycogen storage disease in humans (Blair et al. 2001), pigs (Milan et al. 2000), and yeast (Cannon et al. 1994).
In mammals, AMPK is a αβγ heterotrimer, with each subunit belonging to a larger isoform family comprising α1, α2, β1, β2, γ1, γ2, and γ3, exhibiting varying tissue and subcellular expression (Kemp et al. 2003). AMPK activity depends on the presence of the catalytic α subunit as well as the noncatalytic β and γ subunits (Dyck et al. 1996). AMPK is activated by phosphorylation of T172 within the α subunit activation loop (Hawley et al. 1996) by upstream protein kinases (AMPKK) that includes LKB1 (Hong et al. 2003). Once phosphorylated at T172, AMP allosteric binding can further activate AMPK. In resting muscle, AMPK is largely inactive, present in the dephospho T172 form, and following exercise is phosphorylated and activated (Park et al. 2002). Typically, low μM concentrations of AMP are required to activate AMPK. Frederich and Balschi (2002) have shown that half maximal activation of AMPK occurs at approximately 2 μM AMP using a metabolically stressed heart preparation. In addition to AMP acting as a direct allosteric activator of AMPK, it has been reported to activate the upstream AMPKK (Hawley et al. 1995), together with inhibiting T172 dephosphorylation by phosphatases (Davies et al. 1995). In this way the amplified AMP signal is proposed to act as an ultrasensitive detection system for metabolic stress (Hardie et al. 1999). The γ subunit isoforms differ in their N-terminal sequences but share the presence of four conserved repeat sequences. These correspond to a pair of repeat sequences found in cystathionine β-synthase, termed CBS sequences (Bateman 1997; see CBS domain web page at http://www.sanger.ac.uk/Users/agb/CBS/CBS.html), that are important for the allosteric control of cystathionine β-synthase. Several lines of evidence point to the γ subunit as the major site of AMP allosteric control. Photoaffinity labeling studies show AMP binding to the γ subunit (Cheung et al. 2000). This has now been extended by direct binding studies by Hardie and colleagues who have shown that each γ subunit binds 2 moles of AMP with the CBS1/2 and CBS3/4 pairs each contributing a separate binding site (Scott et al. 2003). A γ1 mutation, R70Q, in the first CBS domain, causes a loss of AMP dependence (Hamilton et al. 2001). This is similar to the D444N mutation in the first CBS domain of cystathionine β-synthase that gives rise to hereditary hyperhomocysteinemia due to a reduction in cystathionine β-synthase activity and a shift in the dose–response curve for allosteric activation by S-adenosyl methionine (Kluijtmans et al. 1996; Janosik et al. 2001; Evande et al. 2002).
Several of the mutations in γ2 that give rise to familial hypertrophic cardiomyopathy (Wolff-Parkinson-White syndrome; reviewed in Gollob et al. 2002) occur in the CBS sequences. These mutations (R302Q [CBS1], H383R [CBS2], and R351G [CBS4]) have been reported to reduce the level of AMP activation when transiently expressed in CCL13 cells (Daniel and Carling 2002).
Since the crystal structures of several enzymes containing CBS domains have been reported, including bacterial inosine monophosphate dehydrogenase (Zhang et al. 1999), with pairs of CBS sequences forming globular domains, this has allowed us to investigate whether the γ subunits contain potential binding sites for AMP related to the classical AMP binding site present in the phosphorylase crystal structure (Sprang et al. 1991). In the present study we show that the γ subunits contain prominent and highly conserved binding pockets for AMP formed by pairs of CBS sequences. The juxtaposition of γ1 CBS sequences 1 and 2 create a binding pocket comprising Arg70, His151, Arg152, and Arg171, and mutation of any of these critical residues can cause loss of AMP dependence and suppression of AMPK inhibition by ATP.
Results
Modeling γ subunit CBS sequences
The γ1 AMP binding subdomain was modeled using the previously identified CBS domain structural motif (Bateman 1997). Several examples of the CBS domains have been structurally characterized. Following review of various sequence alignments and secondary structural predictions (Stultz et al. 1993) the CBS domain structure of inosine monophosphate dehydrogenase from Streptococcus pyogenes (PDB ID: 1ZFJ; Zhang et al. 1999) was chosen for comparative modeling with AMPK γ1. Modeling was performed with the Swiss model WWW server (Peitsch 1996). Only the CBS domain repeats of the peptide sequence were modeled, and no attempt was made to model the connecting loop. The starting model was rebuilt by hand using the program TURBO (http://afmb.cnrs-mrs.fr/), and optimized by molecular dynamics calculation using the program SYBYL (Tripos Inc.). The stereochemical quality of the resulting model was checked with PROCHECK (CCP4 1994), and was found to be of acceptable quality.
Features of the phosphorylase AMP binding site
AMP is an allosteric regulator of both glycogen phosphorylase a and b (Krebs 1954). The phosphorylase a (PDB ID: 1FA9; Rath et al. 2000) and b (Sprang et al. 1991) structures are therefore suitable candidates for searching the AMP binding site for features that may be shared with AMPK (Fig. 2A ▶). In phosphorylase a, the AMP binding site residues are present in a surface cleft between two α-helices, helix 2 and helix 10. The AMP binding site is a tertiary structural element formed by both main-chain and side-chain moieties from several secondary structure elements. The binding site is highly positively charged (Fig. 2B ▶) due to the clustering of four Arg residues at one end of the binding site. Arg193 and Arg242 are held in place by salt bridge interactions with Asp306 and contribute to the surface positive charge. Arg309 and Arg310 each contribute a hydrogen bond to the AMP phosphate oxygen atoms (R309_NH2–AMP_PO 2.69 Å, R310_NH1–AMP_PO 3.33 Å). Arg309 and Arg310 are also involved in an extensive hydrogen bonding network, back-bonding to the protein core with hydrogen bonds to Tyr157, Tyr83, Asp306, and hydrogen bonds to a number of ordered water molecules. Arg309 an Arg310 also interact directly with each other. The adenosine ring of the AMP is stabilized at the opposite end of the binding cleft by a π-stack interaction with Tyr75 (AMP-Y75_Ph 3.7 Å) (Fig. 2A ▶).
Figure 2.
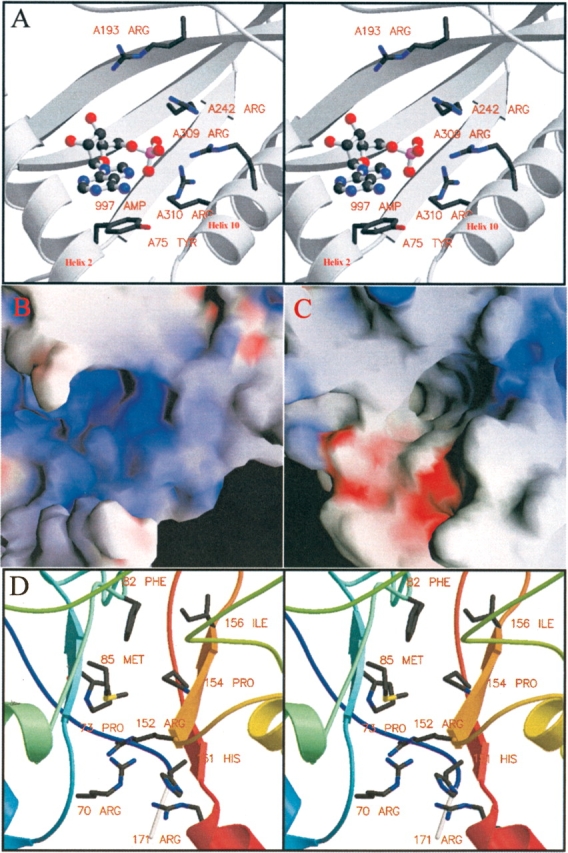
Comparison of phosphorylase and AMPK γ subunit binding pockets for AMP. (A) Stereofigure of the glycogen phosphorylase a AMP binding site; note helices 2 and 10 (Rath et al. 2000). (B) Surface representation of the glycogen phosphorylase a AMP binding site (Rath et al. 2000). Showing the large positively charged blue patch for AMP binding. (C) Surface representation of the γ subunit AMP binding domain showing the positive blue patch of Arg and His residues. (D) Stereo figure of the γ subunit AMP binding domain (note the cluster of basic residues [R70, H151, R152, R171] in the surface cleft).
Docking of AMP and ATP
An accessible surface of the γ1 subunit model (Fig. 1 ▶) was examined with the program GRASP for the presence of a suitable binding site comparable to phosphorylase (Fig. 2C ▶). A large positively charged patch was found in the middle of the cleft between the two CBS repeat sequences, CBS1 and CBS2, formed by residues Arg 70, Arg 152, Arg 171, and His 151 (Fig. 2C,D ▶). The location of the positive patch gave two options for positioning the AMP binding site. Each end of the cleft provided a reasonable structure for AMP binding. The CBS repeat motif of the γ1 subunit of AMPK is very much smaller than phosphorylase a, and therefore, the binding pocket on γ1 is created by a different secondary structure architecture as well as having differences in surface charge (Fig. 2B,C ▶). To test which putative AMP binding site in γ1 CBS1/CBS2 domain was more likely, AMP was docked into each site with the program AUTODOCK (Morris et al. 1998) and the results compared. One site was significantly favored over the other, both in docking energy and in clustering of multiple docking runs. In addition to the Arg residues in the AMP binding pocket, Pro73, Met85, His151, Pro154, and Ile156 also make contact with AMP (Figs. 3 ▶, 4A ▶).
Figure 1.
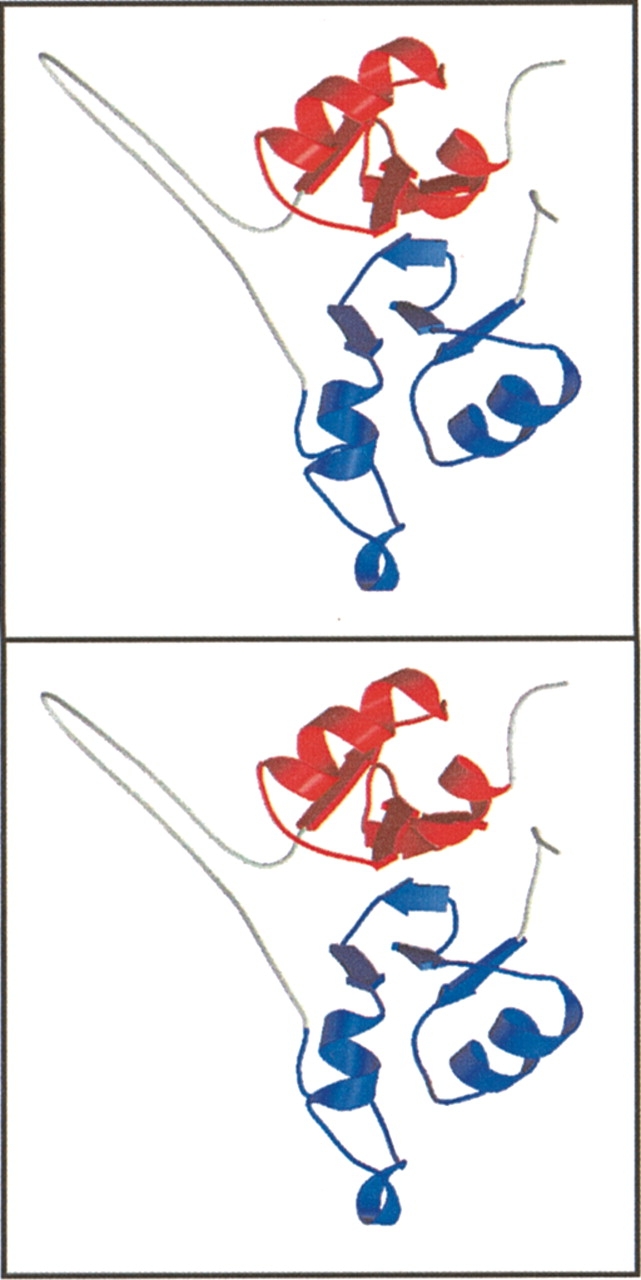
Stereoribbon representation of γ1 model. Showing CBS sequence 1 in blue and CBS sequence 2 in red, the gray loops show the unmodeled connecting loops.
Figure 3.

AMPK γ sequence alignment. Amino acid alignment of γ subunit sequences with putative AMP binding site residues contributed from CBS1/CBS2 pair marked by an asterisk. Residue numbers refer to the rat γ1 isoform.
Figure 4.

AMPK γ1 AMP binding site. (A) Stereofigure of the γ1 AMP binding site with AMP docked into the site. Residues in close proximity to the bound AMP are shown in ball and stick. (B) Surface representation of the γ subunit binding site colored by residue type (aliphatic [white], positive [blue], negative [red], and polar [green]), with docked structures of ATP (purple) and AMP (turquoise).
The AMP site was also probed with ATP and the docking results compared with those of AMP. Analysis of the docking results of both AMP and ATP showed that of 100 docking attempts with AMP, 37 docks clustered with an RMSD of less than 1 Å to the docked structure with the best docking energy. This cluster of putative AMP complex structures made chemical sense, with ionic interactions and aliphatic interactions stabilizing the substrate on the surface of the model. In particular Arg 70, Arg 152, and Arg 171 all made contributions to the binding of the phosphate group of AMP. Docking results with ATP were less well ordered, with only 6 of 100 docking attempts clustering in the same pocket as the best AMP cluster. Assessment of the clustering of the ATP structures must be viewed with the caveat that the same RMSD cutoff was applied to a molecule, which is significantly larger and with more degrees of freedom than AMP, potentially complicating comparison of the data for each ligand.
The docking energies for AMP were significantly better than those for ATP, indicating that AMP would be expected to have a lower binding constant than ATP. This suggests, as has been previously postulated, that AMP and ATP compete for binding to AMPK (Hardie et al. 1999), with AMP having a much lower off-rate from the binding site. Thus, allosteric upregulation of AMPK activity occurs while AMP is transiently bound and also in the absence of sufficient competing ATP.
The first missense mutation identified in human γ2 that causes the Wolf Parkinson White-like syndrome was R302Q (Gollob et al. 2001) in the first CBS domain, which corresponds to the position of R70 in the γ1 model structure (Figs. 2C ▶, 3 ▶) within the AMP binding site pocket. We had previously found that the R70Q mutation in the γ1 subunit was strongly activating, resulting in a high level of constitutive AMPK activity and loss of AMP dependence (Hamilton et al. 2001). The presence of R70Q in the AMP binding pocket now explains why there was a loss of AMP dependence. This also provides a reason for the reported reduction in AMP dependence of the corresponding γ2 mutants (R302Q, H383R) (Daniel and Carling 2002).
We have now tested the effect of mutating the other two Arg residues in the AMP binding pocket, R152 and R171, to Gln. Wild-type γ1 or mutant γ1 subunits were transfected into COS-7 cells with a GST-α1 construct and the native β1 subunit. The expressed AMPK holoenzyme (α1β1γ1) was purified over glutathione beads and the activity of the enzyme measured at varying AMP concentrations. In all cases the presence of the three subunits in the holoenzyme complex was confirmed by SDS-PAGE and immunoblotting with the corresponding α, β, or γ subunit antibodies. The expressed wild type enzyme from COS-7 cells was activated 1.9-fold to approximately 0.5 pmole/min by AMP, with half maximum activation KA0.5 occurring at 10.2 ± 0.6 μM (Table 1; Fig. 5 ▶). This contrasts with the high KA0.5 values for AMP activation of approximately 77 μM reported for wild-type constructs expressed in CCL13 cells (Daniel and Carling 2002). The R152Q and R171Q γ mutations increased constitutive AMPK activity to approximately 9 and 1.1 pmole/min, respectively, and there was complete loss of AMP dependence for the R70Q and R171Q mutants (Fig. 5 ▶). The R152Q mutant retained some AMP dependence with approximately 17% stimulation at 100 μM AMP. The level of activity of the AMPK carrying the R152Q mutation was approximately twice the activity of AMPK with the γ1 R70Q mutation (Fig. 5B ▶; 5 pmole/min). These results show that all three Arg residues present in the AMP modeled binding site influence AMP allosteric control. To test whether substitution of R70 with a less polar residue affected activity we made the R70V mutant. We chose Val because it is present in the yeast homolog of γ, Snf4p (Fig. 3 ▶). The presence of Val at position 70 had only a modest effect on AMP dependence increasing it from 10.2 μM to 14.8 μM and reducing the maximum stimulation from 1.9- to 1.6-fold (Table 1). This indicates that the Gln side chain at position 70 is deleterious for the allosteric control of AMPK rather than a nonspecific structural effect of mutating Arg70. The different Arg to Gln mutations in the AMP binding site resulted in an apparent increase in the maximum AMPK activity (Fig. 4 ▶). In all three cases expression and the level of Thr172 phosphorylation in the activation loop of the α catalytic subunit was higher, as assessed by antiphosphopeptide Thr172 immunoblotting (results not shown). This may indicate that the holoenzyme containing the Arg mutants in the γ subunit are better substrates for the AMPKK than the wild-type γ subunit containing enzyme, rather than having inherently higher activities.
Table 1.
Effect of γ mutations on AMP dependence
| AMPK γ subunit | KA0.5 (μM) | Fold stimulation |
| WT | 10.2 ± 0.6 | 1.9 ± 0.1 |
| F51L | 23.4 ± 3.9 | 1.5 ± 0.1 |
| F51Y | 5.4 ± 1.5 | 1.6 ± 0.1 |
| R70V | 14.8 ± 2.6 | 1.6 ± 0.1 |
| F51L/R70V | 57.7 ± 9.2 | 1.6 ± 0.01 |
| R70Q | ND | |
| R152Q | ND | |
| R171Q | ND | |
| R152Q/R171Q | ND | |
| H151G | ND | |
| H151G/R70V | ND | |
| F82L | 10.1 ± 0.4 | 1.6 ± 0.1 |
| F82Y | 9.9 ± 1.7 | 1.6 ± 0.1 |
Wild-type γ1 or mutant γ1 was cotransfected with α1 and β1 in COS-7 cells. The activity of α1β1γ1 measured in AMP concentration from 0 to 100 μM. Values are the mean ± SEM of four independent experiments. Fold stimulation, stimulation of AMPK activity by 100 μM AMP. ND, not determined. KA0.5, concentration for half maximal activation.
Figure 5.
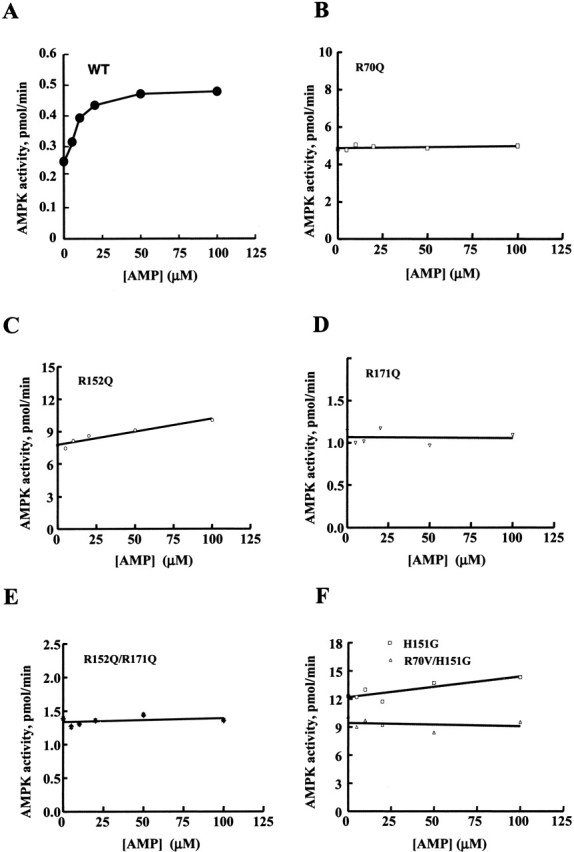
Effect of Arg mutations on AMP activation. Wild-type γ1 or mutant γ1 was cotransfected with α1 and β1 in COS-7 cells and AMPK αβγ holoenzyme was purified by Glutathione Sepharose chromatograph. AMPK activity was determined using the SAMS peptide (see Materials and Methods). The activity measured in the absence of AMP was subtracted from plus AMP values of the wild-type and Phe mutants and fitted to the one-site binding Michaelis-Menten Equation curve. The activity of the Arg mutants was AMP-independent.
Because the allosteric control by AMP was lost by mutation of Arg residues in the AMP binding site of the CBS1/CBS2 domain pair alone, we did not attempt to investigate the effect of corresponding mutations in the CBS3/CBS4 binding site. Nevertheless, it is clear that the CBS3/CBS4 binding site pair form a functionally important AMP binding site because γ2 binds 2 moles of AMP (Scott et al. 2003) and mutation R531G (equivalent to R152 in γ1 CBS2) in the CBS4 domain (Gollob et al. 2001) of the γ2 subunit, gives rise to the cardiac phenotype.
Several other mutations were made including F51L and F51Y in a region that is outside the predicted AMP binding pocket. The F51L mutation resulted in an approximate twofold increase in the KA0.5 for AMP to 24 μM. In contrast, the F51Y mutation reduced the KA0.5 for AMP to 5.4 μM (Table 1). These results indicate that mutation outside the AMP binding site can reduce or enhance the AMP binding to the γ subunit, but these changes are relatively modest and do not result in the dramatic changes in allosteric control observed when key residues are modified in the binding pocket.
In the phosphorylase a structure the AMP binding pocket contains TyrA75 (Fig. 2A ▶) in association with the adenine ring. At the base of the AMP binding pocket in the γ1 subunit there is a Phe at position 82. We mutated this residue to both Leu and Tyr and found that F82L and F82Y had KA0.5 values for AMP activation of approximately 10 μM, indistinguishable from native γ1 (Table 1). Inspection of the model of the γ AMP binding site indicates that the adenine ring may not penetrate sufficiently into the pocket (Fig. 2C,D ▶) to interact with Phe82 in marked contrast to the AMP site in phosphorylase. In the γ1 CBS1/CBS2 model the most prominent contacts between the adenine ring and the pocket side chains are Pro73, Met85, His151, Pro154, and Ile156.
As high concentrations of ATP suppress AMP stimulation of AMPK (Hardie et al. 1999), we compared the effect of increasing concentrations of ATP on both native AMPK and the R152Q mutant that retains some AMP dependence. At 100 μM AMP the wild-type AMPK activity is increased by increasing ATP to a maximum at 500 μM ATP and thereafter declines, as expected from ATP competition at the AMP allosteric site (Fig. 6A,C ▶). In contrast, the R152Q mutant shows no decrease in AMPK activity with increasing ATP concentration under these conditions (Fig. 6B,D ▶). These results indicate that mutations of the AMP binding site that weaken AMP allosteric control also suppress the inhibitory properties of ATP. This loss of ATP inhibition may contribute to enhanced activation of the mutants by the AMPK kinase.
Figure 6.

Effect of ATP concentration on AMP dependence. AMPK γ1 wild-type (A, C) and R152Q (B, D) were cotransfected with α1 and β1 in COS-7 cells. The AMPK activities were measured in the varied AMP and ATP concentrations as indicated (A, B). Changes in AMPK activity, relative to AMPK activity in the absence of AMP, are shown (C, D).
The yeast AMPK homolog snf1p kinase is present as a heterodimer of Snf1p and Snf4p when isolated from yeast, and is AMP independent (Mitchelhill et al. 1994). There is substantial similarity in the residues comprising the AMP binding site between γ CBS1/CBS2 and Snf4p (Fig. 3 ▶). For example, Arg residues equivalent to R152 and R171 are present but R70 is replaced with Val. The mutation R70V in γ1 is not sufficient to block AMP allosteric control, and therefore does not explain the Snf1p/Snf4p AMP independence. However, the most prominent difference in the AMP binding site between γ1 and Snf4p is a Gly in place of H151 (Fig. 3 ▶). Activity analysis of an AMPK complex containing the γ1-H151G mutation largely abolished AMP dependence, as shown in Figure 5F ▶. This, therefore, provides an explanation of why noone to date has been able to stimulate the Snf1p/Snf4p complex with any nucleotides (Mitchelhill et al. 1994; Wilson et al. 1996).
Discussion
Our results may provide new clues to the mechanism of allosteric control of AMPK. When ATP occupies the AMP binding site it suppresses activity and increases in cellular AMP serve to derepress enzyme activity. For many Ser/Thr and Tyr kinases activation frequently involves ordering of the activation loop to facilitate catalysis and substrate binding (Adams 2003). AMPK is largely in the inactive T172 dephosphorylated state at rest in skeletal muscle, and it is thought that the binding of AMP facilitates phosphorylation of the activation loop T172 by the AMPKK. Similarly, we expect that the presence of ATP in the γ subunit AMP binding site would suppress phosphorylation of T172 by AMPKK. The docking experiments indicate that when ATP is bound, the β and γ phosphates extend out of the AMP binding pocket, and therefore may be available to make intersubunit contacts. One possible mechanism for suppressing AMPK activity would be if the juxtaposition of the γ subunit allowed the ATP γ phosphate to bridge the α subunit activation loop region and contact with the basic residue cluster that otherwise interact with T172 once it is phosphorylated. This would prevent the correct orientation of the activation loop and suppress phosphorylation of T172 by AMPKK. Once AMP bound the γ subunit, the γ phosphate contact would be lost and T172 would be readily phosphorylated. However, if ATP reoccupied the allosteric site the γ phosphate would compete with the T172 phosphate intramolecular interactions in the catalytic subunit and suppress activity. Such competition by the γ phosphate of ATP would also expose T172 phosphate in the activation loop to dephosphorylation by phosphatases. AMP binding at the allosteric site promotes phosphorylation of T172 as well as increasing the activity of the phosphorylated enzyme. Although the γ phosphate of ATP bound to the allosteric site causes the disorder of the activation loop and phosphatase attack, equally, AMP would inhibit the dephosphorylation of T172, as has been observed experimentally (Davies et al. 1995). In this model ATP acts as an intrasteric inhibitor by bridging the α and γ subunits.
Direct binding studies by Hardie and colleagues and the modeling and mutagenesis studies reported here show that a pair of CBS sequences are required to form a functional allosteric binding domain for AMP. Based on the known structure of the pair of CBS sequences in inosine monophosphate dehydrogenase and the role of the domain in allosteric control of both cystanthionine β synthase and AMPK, it seems likely that they will be responsible for allosteric control across the diversity of proteins where they are found. Accordingly, we propose that these structures be named “Bateman modules” after their founder, Alex Bateman (Bateman 1997).
Materials and methods
Plasmid constructs and mutagenesis
AMPK mammalian expression constructs for GST-α1 and native-β1 were used as previously described (Hamilton et al. 2001). A plasmid encoding the full-length human AMPK γ1 subunit cDNA cloned in frame with an N-terminal hemagglutinin epitope tag has been previously described (Gao et al. 1996). This plasmid (pMT2-HA-PRKAγ1) encoding wild-type (WT) γ1 was used as a PCR template to generate amino acid substitutions. Mutations to the γ1 coding sequence were performed by PCR using the Quickchange XL site-directed mutagenesis kit (Stratagene) according to the manufacturer’s instructions. The substitutions generated and the 5′ to 3′ sequences of the sense strand oligonucleotides used were as follows (base changes underlined),
F51L, CCAAATTGGTTGTATTGGATACGTCCCTGCAGG; F51Y,CCAAATTGGTTGTATATGATACGTCCCTGCAGG; R70Q, GTGACTAACGGTGTACAAGCTGCCCCTTTATGG; R70V, GTGACTAACGGTGTAGTAGCTGCCCCTTTATGG; F82L, GTAAGAAGCAAAGTTTAGTGGGCATGCTGACC; F82Y, GTAAGAAGCAAAGTTATGTGGGCATGCTGACC; R152Q, GGAACAAGATCCACCAGCTGCCAGTTATTGACC; R171Q, CACCCACAAGCAGATTCTGAAGTTCC
All mutants were verified by DNA sequencing.
Plasmid DNA used in transfections was purified using a Hi-Speed plasmid purification kit (QIAGEN), and resuspended in endotoxin-free 10 mM Tris•HCl, pH 7.5, 1 mM EDTA at 1 μg/μL.
Transfections
COS-7 cells were cultured in 10-cm dishes and maintained in DME plus 10% fetal bovine serum. Transient transfections were performed with FuGENE 6 (Roche) according to the manufacturer’s instructions. Briefly, 10-cm dishes of COS-7 cells at 70% confluence were triple-transfected with a mixture of 7.5 μL of FuGENE 6 and 1 μg of each of the GST-α1, native-β1 and native γ1 (WT or mutant) constructs. Twenty-four hours posttransfection, the cells were washed with ice-cold phosphate-buffered saline, harvested in a lysis buffer (50 mM Tris•HCl, pH7.5, 1 mM EDTA, 1 mM EGTA, 1 mM DTT, 50 mM NaF, 5 mM Na Pyrophosphate, 10% Glycerol, 1% Triton X-100, 10 μg/mL Trypsin inhibitor, 2 μg/mL Aprotinin, 1 mM Benzamidine, 1 mM PMSF), and lysed by freeze thawing. The insoluble material was removed by centrifugation and the AMPK holoenzyme was purified by chromatography on Glutathione Sepharose 4B (Amersham Biosciences), followed by elution with 10 mM reduced glutathione. The α1, β1, and γ1 expression levels were assessed by SDS-PAGE of the AMPK, and immunoblotted with affinity purified polyclonal antibodies to each subunit (data not shown).
AMPK assays
Activity of AMPK protein eluted from Glutathione Sepharose 4B column was measured in a reaction containing 50 mM HEPES, pH 7.5, 10 mM MgCl2, 5% Glycerol, 0.05% Triton X-100 with 1 mM DTT, 10 μM SAMS peptide, 0.25 mM [γ-32P] ATP (500 cpm/pmole), and varying AMP concentrations as indicated. In AMP/ATP competitive experiments, the AMP and ATP concentrations were as indicated. Reactions were incubated at 30°C for 6–7 min. An aliquot was applied to P81 paper as previously described to separate the [γ-32P] ATP from the phosphorylated peptide (Glass et al. 1978).
Acknowledgments
The National Health Medical Research Council (NHMRC, Australia) National Heart Foundation (Australia), Australian Research Council (B.E.K.), and an NIH Grant (DK35712 [L.A.W.]) supported this work. D.S. is an NHMRC RD Wright Fellow, and B.E.K. is an NHMRC and Federation Fellow. We are indebted to Grahame Hardie for providing information on the AMP binding studies of the γ subunit CBS pairs.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.03340004.
References
- Adams, J.A. 2003. Activation loop phosphorylation and catalysis in protein kinases: Is there functional evidence for the autoinhibitor model? Biochemistry 42 601–607. [DOI] [PubMed] [Google Scholar]
- Barthel, A., Schmoll, D., Kruger, K.D., Roth, R.A., and Joost, H.G. 2002. Regulation of the forkhead transcription factor FKHR (FOXO1a) by glucose starvation and AICAR, an activator of AMP-activated protein kinase. Endocrinology 143 3183–3186. [DOI] [PubMed] [Google Scholar]
- Bateman, A. 1997. The structure of a domain common to archaebacteria and the homocystinuria disease protein. Trends Biochem. Sci. 22 12–13. [DOI] [PubMed] [Google Scholar]
- Blair, E., Redwood, C., Ashrafian, H., Oliveira, M., Broxholme, J., Kerr, B., Salmon, A., Ostman-Smith, I., and Watkins, H. 2001. Mutations in the γ(2) subunit of AMP-activated protein kinase cause familial hypertrophic cardiomyopathy: Evidence for the central role of energy compromise in disease pathogenesis. Hum. Mol. Genet. 10 1215–1220. [DOI] [PubMed] [Google Scholar]
- Cannon, J.F., Pringle, J.R., Fiechter, A., and Khalil, M. 1994. Characterization of glycogen-deficient glc mutants of Saccharomyces cerevisiae. Genetics 136 485–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CCP4. 1994. The CCP4 suite: Programs for protein crystallography. Acta Crystallogr. D 50 760–763. [DOI] [PubMed] [Google Scholar]
- Cheung, P.C., Salt, I.P., Davies, S.P., Hardie, D.G., and Carling, D. 2000. Characterization of AMP-activated protein kinase γ-subunit isoforms and their role in AMP binding. Biochem. J. 346 659–669. [PMC free article] [PubMed] [Google Scholar]
- Daniel, T. and Carling, D. 2002. Functional analysis of mutations in the γ 2 subunit of AMP-activated protein kinase associated with cardiac hypertrophy and Wolff-Parkinson-White syndrome. J. Biol. Chem. 277 51017–51024. [DOI] [PubMed] [Google Scholar]
- Davies, S.P., Helps, N.R., Cohen, P.T., and Hardie, D.G. 1995. 5′-AMP inhibits dephosphorylation, as well as promoting phosphorylation, of the AMP-activated protein kinase. Studies using bacterially expressed human protein phosphatase-2C α and native bovine protein phosphatase-2AC. FEBS Lett. 377 421–425. [DOI] [PubMed] [Google Scholar]
- Dyck, J.R.B., Gao, G., Widmer, J., Stapleton, D., Fernandez, C.S., Kemp, B.E., and Witters, L.A. 1996. Regulation of 5′-AMP-activated protein kinase activity by the noncatalytic β and γ subunits. J. Biol. Chem. 271 17798–17803. [DOI] [PubMed] [Google Scholar]
- Evande, R., Blom, H., Boers, G.H., and Banerjee, R. 2002. Alleviation of intrasteric inhibition by the pathogenic activation domain mutation, D444N, in human cystathionine β-synthase. Biochemistry 41 11832–11837. [DOI] [PubMed] [Google Scholar]
- Ferre, P., Azzout-Marniche, D., and Foufelle, F. 2003. AMP-activated protein kinase and hepatic genes involved in glucose metabolism. Biochem. Soc. Trans. 31 220–223. [DOI] [PubMed] [Google Scholar]
- Frederich, M. and Balschi, J.A. 2002. The relationship between AMP-activated protein kinase activity and AMP concentration in the isolated perfused rat heart. J. Biol. Chem. 277 1928–1932. [DOI] [PubMed] [Google Scholar]
- Fryer, L.G., Parbu-Patel, A., and Carling, D. 2002. The anti-diabetic drugs rosiglitazone and metformin stimulate AMP-activated protein kinase through distinct signaling pathways. J. Biol. Chem. 277 25226–25232. [DOI] [PubMed] [Google Scholar]
- Gao, G., Fernandez, C.S., Stapleton, D., Auster, A.S., Widmer, J., Dyck, J.R., Kemp, B.E., and Witters, L.A. 1996. Non-catalytic β- and γ-subunit isoforms of the 5′-AMP-activated protein kinase. J. Biol. Chem. 271 8675–8681. [DOI] [PubMed] [Google Scholar]
- Glass, D.B., Masaracchia, R.A., Feramisco, J.R., and Kemp, B.E. 1978. Isolation of phosphorylated peptides and proteins on ion exchange papers. Anal. Biochem. 87 566–575. [DOI] [PubMed] [Google Scholar]
- Gollob, M.H., Seger, J.J., Gollob, T.N., Tapscott, T., Gonzales, O., Bachinski, L., and Roberts, R. 2001. Novel PRKAG2 mutation responsible for the genetic syndrome of ventricular preexcitation and conduction system disease with childhood onset and absence of cardiac hypertrophy. Circulation 104 3030–3033. [DOI] [PubMed] [Google Scholar]
- Gollob, M.H., Green, M.S., Tang, A.S., and Roberts, R. 2002. PRKAG2 cardiac syndrome: Familial ventricular preexcitation, conduction system disease, and cardiac hypertrophy. Curr. Opin. Cardiol. 17 229–234. [DOI] [PubMed] [Google Scholar]
- Hamilton, S.R., Stapleton, D., O’Donnell Jr., J.B., Kung, J.T., Dalal, S.R., Kemp, B.E., and Witters, L.A. 2001. An activating mutation in the γ1 subunit of the AMP-activated protein kinase. FEBS Lett. 500 163–168. [DOI] [PubMed] [Google Scholar]
- Hardie, D.G. and Hawley, S.A. 2001. AMP-activated protein kinase: The energy charge hypothesis revisited. Bioessays 23 1112–1119. [DOI] [PubMed] [Google Scholar]
- Hardie, D.G., Salt, I.P., Hawley, S.A., and Davies, S.P. 1999. AMP-activated protein kinase: An ultrasensitive system for monitoring cellular energy charge. Biochem. J. 338 717–722. [PMC free article] [PubMed] [Google Scholar]
- Hawley, S.A., Selbert, M.A., Goldstein, E.G., Edelman, A.M., Carling, D., and Hardie, D.G. 1995. 5′-AMP activates the AMP-activated protein kinase cascade, and Ca2+/calmodulin activates the calmodulin-dependent protein kinase I cascade, via three independent mechanisms. J. Biol. Chem. 270 27186–27191. [DOI] [PubMed] [Google Scholar]
- Hawley, S.A., Davison, M., Woods, A., Davies, S.P., Beri, R.K., Carling, D., and Hardie, D.G. 1996. Characterization of the AMP-activated protein kinase kinase from rat liver and identification of threonine 172 as the major site at which it phosphorylates AMP-activated protein kinase. J. Biol. Chem. 271 27879–27887. [DOI] [PubMed] [Google Scholar]
- Hawley, S.A., Gadalla, A.E., Olsen, G.S., and Hardie, D.G. 2002. The antidiabetic drug metformin activates the AMP-activated protein kinase cascade via an adenine nucleotide-independent mechanism. Diabetes 51 2420–2425. [DOI] [PubMed] [Google Scholar]
- Hong, S.P., Leiper, F.C., Woods, A., Carling, D., and Carlson, M. 2003. Activation of yeast Snf1 and mammalian AMP-activated protein kinase by upstream kinases. Proc. Natl. Acad. Sci. 100 8839–8843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janosik, M., Kery, V., Gaustadnes, M., Maclean, K.N., and Kraus, J.P. 2001. Regulation of human cystathionine β-synthase by S-adenosyl-L-methionine: Evidence for two catalytically active conformations involving an autoinhibitory domain in the C-terminal region. Biochemistry 40 10625–10633. [DOI] [PubMed] [Google Scholar]
- Kawaguchi, T., Osatomi, K., Yamashita, H., Kabashima, T., and Uyeda, K. 2002. Mechanism for fatty acid “sparing” effect on glucose-induced transcription: Regulation of carbohydrate-responsive element-binding protein by AMP-activated protein kinase. J. Biol. Chem. 277 3829–3835. [DOI] [PubMed] [Google Scholar]
- Kemp, B.E., Stapleton, D., Campbell, D.J., Chen, Z.P., Murthy, S., Walter, M., Gupta, A., Adams, A.J., Katsis, F., van Denderen, B., et al. 2003. AMP-activated protein kinase, super metabolic regulator. Biochem. Soc. Trans. 31 162–168. [DOI] [PubMed] [Google Scholar]
- Kluijtmans, L.A., Boers, G.H., Stevens, E.M., Renier, W.O., Kraus, J.P., Trijbels, F.J., van den Heuvel, L.P., and Blom, H.J. 1996. Defective cystathionine β-synthase regulation by S-adenosylmethionine in a partially pyridoxine responsive homocystinuria patient. J. Clin. Invest. 98 285–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krebs, E.G. 1954. The effect of salmine on the activity of phosphorylase. Biochim. Biophys. Acta 15 508–515. [DOI] [PubMed] [Google Scholar]
- Leclerc, I., Lenzner, C., Gourdon, L., Vaulont, S., Kahn, A., and Viollet, B. 2001. Hepatocyte nuclear factor-4α involved in type 1 maturity-onset diabetes of the young is a novel target of AMP-activated protein kinase. Diabetes 50 1515–1521. [DOI] [PubMed] [Google Scholar]
- Leff, T. 2003. AMP-activated protein kinase regulates gene expression by direct phosphorylation of nuclear proteins. Biochem. Soc. Trans. 31 224–227. [DOI] [PubMed] [Google Scholar]
- Lo, W.S., Duggan, L., Tolga, N.C., Emre, Belotserkovskya, R., Lane, W.S., Shiekhattar, R., and Berger, S.L. 2001. Snf1—A histone kinase that works in concert with the histone acetyltransferase Gcn5 to regulate transcription. Science 293 1142–1146. [DOI] [PubMed] [Google Scholar]
- Milan, D., Jeon, J.T., Looft, C., Amarger, V., Robic, A., Thelander, M., Rogel-Gaillard, C., Paul, S., Iannuccelli, N., Rask, L., et al. 2000. A mutation in PRKAG3 associated with excess glycogen content in pig skeletal muscle. Science 288 1248–1251. [DOI] [PubMed] [Google Scholar]
- Minokoshi, Y., Kim, Y.B., Peroni, O.D., Fryer, L.G., Muller, C., Carling, D., and Kahn, B.B. 2002. Leptin stimulates fatty-acid oxidation by activating AMP-activated protein kinase. Nature 415 339–343. [DOI] [PubMed] [Google Scholar]
- Mitchelhill, K.I., Stapleton, D., Gao, G., House, C., Michell, B., Katsis, F., Witters, L.A., and Kemp, B.E. 1994. Mammalian AMP-activated protein kinase shares structural and functional homology with the catalytic domain of yeast Snf1 protein kinase. J. Biol. Chem. 269 2361–2364. [PubMed] [Google Scholar]
- Morris, G.M., Goodsell, D.S., Halliday, R.S., Huey, R., Hart, W.E., Belew, R.K., and Olson, A.J. 1998. Automated docking using a Lamarckian genetic algorithm and and empirical binding free energy function. J. Comput. Chem. 19 1639–1662. [Google Scholar]
- Moule, S.K. and Denton, R.M. 1998. The activation of p38 MAPK by the β-adrenergic agonist isoproterenol in rat epididymal fat cells. FEBS Lett. 439 287–290. [DOI] [PubMed] [Google Scholar]
- Nicholls, A., Sharp, K.A., and Honig, B. 1991. Protein folding and association: Insights from the interfacial and thermodynamic properties of hydrocarbons. Proteins 11 281–296. [DOI] [PubMed] [Google Scholar]
- Park, S.H., Gammon, S.R., Knippers, J.D., Paulsen, S.R., Rubink, D.S., and Winder, W.W. 2002. Phosphorylation–activity relationships of AMPK and acetyl-CoA carboxylase in muscle. J. Appl. Physiol. 92 2475–2482. [DOI] [PubMed] [Google Scholar]
- Peitsch, M.C. 1996. SWISS-MODEL: An automated comparative protein modeling server and a model repository. Fold. Des. 1 S48–S49. [Google Scholar]
- Rath, V.L., Ammirati, M., LeMotte, P.K., Fennell, K.F., Mansour, M.N., Danley, D.E., Hynes, T.R., Schulte, G.K., Wasilko, D.J., and Pandit, J. 2000. Activation of human liver glycogen phosphorylase by alteration of the secondary structure and packing of the catalytic core. Mol. Cell 6 139–148. [PubMed] [Google Scholar]
- Roussel, A., Fontecilla-Camps, J.C., and Cambillau, C. 1990. CRYStallize: A crystallographic symmetry display and handling subpackage in TOM/FRODO. J. Mol. Graphics 8 86–88, 91. [DOI] [PubMed] [Google Scholar]
- Scott, J.W., Hawley, S.A., Green, K.A., Anis, M., Stewart, G., Scullion, G.A., Norman, D.G. and Hardie, D.G. 2003. CBS domains form energy-sensing modules whose binding of adenosine ligands is disrupted by disease mutations. J. Clin. Invest. (in press). [DOI] [PMC free article] [PubMed]
- Sprang, S.R., Withers, S.G., Goldsmith, E.J., Fletterick, R.J., and Madsen, N.B. 1991. Structural basis for the activation of glycogen phosphorylase b by adenosine monophosphate. Science 254 1367–1371. [DOI] [PubMed] [Google Scholar]
- Stoppani, J., Hildebrandt, A.L., Sakamoto, K., Cameron-Smith, D., Goodyear, L.J., and Neufer, P.D. 2002. AMP-activated protein kinase activates transcription of the UCP3 and HKII genes in rat skeletal muscle. Am. J. Physiol. 283 E1239–E1248. [DOI] [PubMed] [Google Scholar]
- Stultz, C.M., White, J.V., and Smith, T.F. 1993. Structural analysis based on state-space modeling. Protein Sci. 2 305–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson, W.A., Hawley, S.A., and Hardie, D.G. 1996. Glucose repression/derepression in budding yeast: SNF1 protein kinase is activated by phosphorylation under derepressing conditions, and this correlates with a high AMP:ATP ratio. Curr. Biol. 6 1426–1434. [DOI] [PubMed] [Google Scholar]
- Yamauchi, T., Kamon, J., Minokoshi, Y., Ito, Y., Waki, H., Uchida, S., Yamashita, S., Noda, M., Kita, S., Ueki, K., et al. 2002. Adiponectin stimulates glucose utilization and fatty-acid oxidation by activating AMP-activated protein kinase. Nat. Med. 8 1288–1295. [DOI] [PubMed] [Google Scholar]
- Zhang, R., Evans, G., Rotella, F.J., Westbrook, E.M., Beno, D., Huberman, E., Joachimiak, A., and Collart, F.R. 1999. Characteristics and crystal structure of bacterial inosine-5′-monophosphate dehydrogenase. Biochemistry 38 4691–4700. [DOI] [PubMed] [Google Scholar]
- Zheng, D., MacLean, P.S., Pohnert, S.C., Knight, J.B., Olson, A.L., Winder, W.W., and Dohm, G.L. 2001. Regulation of muscle GLUT-4 transcription by AMP-activated protein kinase. J. Appl. Physiol. 91 1073–1083. [DOI] [PubMed] [Google Scholar]
- Zhou, M., Lin, B.Z., Coughlin, S., Vallega, G., and Pilch, P.F. 2000. UCP-3 expression in skeletal muscle: Effects of exercise, hypoxia, and AMP-activated protein kinase. Am. J. Physiol. 279 E622–E629. [DOI] [PubMed] [Google Scholar]