

Abstract
α and β Tubulin are well-characterized paralogs with similar structures and functions. We quantify the variability of every amino acid position in both tubulins from the aligned sequences of their numerous known orthologs. By aligning the variability profiles, we identify residues that differ significantly in variability between α and β tubulin. Most of these residues are part of well-defined secondary structures and are clustered around the nucleotide binding pocket, the site of greatest functional difference between the two paralogs. The remaining residues of large difference in variability are located in the N-terminal loop between H1 and S2. We therefore predict that certain residues in this unstructured region also contribute to a functional difference between α and β tubulin. Furthermore, we find the most restrictive variability-based alignment is nearly identical to the true structure-based alignment. Thus, by using a stringent variability-based alignment to approximate the true alignment, the method introduced here may predict sites of functional distinction between paralogous proteins even in the absence of structural information.
Keywords: sequence alignment, neutral versus functional variation, bioinformatic tools, microtubule catalytic site
Orthologs are homologous proteins with amino acid sequences that have diverged due to mutations accumulated since their separation by speciation events (Fitch 2000). Some amino acids remain unchanged, of course, even between orthologs separated by large timespans. Such conserved residues are presumably constrained by structural or functional requirements of the protein. Those that vary, on the other hand, are difficult to interpret. Variations can be neutral (i.e., irrelevant to the process of natural selection), crucial (i.e., adapting the function of a protein to the niche of its organism), or anything in between. The desire to distinguish between neutral and functional amino acid variations by strictly informatic means has motivated considerable research in recent years (Gaucher et al. 2002). An ability to identify mutations that confer subtle functional changes from sequence data would help solve the sequence structure–function relationship of known proteins and guide the rational design of protein variants.
The modern abundance of sequence data permits quantification of the variability of amino acids in many proteins. To relate this variability to function typically requires knowing the structure of the protein. For example, it is common to ask whether amino acids in the core of a protein sustain fewer variations than do those on its surface, as might be expected due to packing constraints or interactions essential for folding. Along these lines, a weak but statistically significant correlation is generally found between variability and solvent accessible surface area (Huang et al. 1996; Goldman et al. 1998; Rodionov and Blundell 1998). More intriguing, perhaps, is an apparent conservation of the three-dimensional pattern of conserved amino acids in several families of structurally homologous proteins, indicating the existence of a folding nucleus (Mirny and Shakhnovich 1999).
In this article, we demonstrate a method for extracting functional information from quantitative variability data by using paralogous proteins. The paralogs of interest here are α and β tubulin. The numerous orthologs, known structures, and ambiguous structure–function relationships of these tubulins make them an ideal and interesting test case. The results indicate that the method introduced here may be usefully applied to other protein paralogs with structures that are not yet known.
α and β tubulin form a heterodimer (αβ) that self-assembles into hollow cylindrical filaments called microtubules. Microtubules are dynamic elements of the eukaryotic skeleton that play an essential role in a variety of cellular functions. They are best known for their role in cell division, in which dramatic fluctuations in the length of individual microtubules are required to organize and separate the chromosomes (Mitchison and Kirschner 1984). These length fluctuations are fueled by the hydrolysis of one of two molecules of guanosine-triphosphate (GTP) bound to each tubulin dimer. Just how hydrolysis changes the tubulin structure so as to destabilize the microtubule is still a mystery (Nogales 2001).
In the decade before their crystal structure was known, sequence comparisons provided valuable insights into α and β tubulin structure and function (Little et al. 1981; Little and Seehaus 1988; Burns 1991). The most thorough analysis to date is based on sequences available in 1992 (Burns and Surridge 1994). These studies emphasize how well suited α and β tubulins are for variability analysis. Alignment among orthologs and between the two paralogs is unambiguous because their amino acid sequences are highly conserved and easily distinguished, and have few insertions or deletions. For quantitative analysis, it is particularly fortunate that hundreds of complete tubulin sequences are now available in public databases, with all eukaryotic phyla well represented.
Here, we quantify the variability of every amino acid in both α and β tubulin and compare a variability-based alignment of the amino acid sequences with their true structure-based alignment (Nogales et al. 1998; Löwe et al. 2001). We find that a stringent variability-based alignment effectively reproduces the true alignment, whereas a tolerant variability-based alignment can be used to identify homologous amino acids that differ significantly in variability between the two proteins. This procedure may be especially useful in directing mutagenesis studies to loci of key functional importance.
Results
Because α and β tubulin are both highly conserved and broadly sequenced, it is possible to quantify the variability of each of their residues with confidence. The statistical distribution of residue variability in both tubulins is strongly peaked at low values, with >50% of residues scoring in the bottom 10% of the variability range (Fig. 1 ▶). This is consistent with earlier reports of tubulin as one of the most highly conserved proteins (Burns and Surridge 1994).
Figure 1.

Histograms of residue variability in α tubulin (A) and β tubulin (B) are similar to one another, with a large peak at low variability.
When plotted versus sequence, the variability patterns for the two tubulins are clearly correlated (Fig. 2 ▶). This, too, is expected because variability is constrained by function, and these paralogs have extensive structural and functional homology.
Figure 2.

Residue variability, S(x), along the amino acid sequence of α tubulin (open circles) and β tubulin (filled circles). β Tubulin data are presented upside down to facilitate comparison across the sequences. Sequences listed are those of pig-brain tubulin (Krauhs et al. 1981; Postingl et al. 1981). (Residues that differ from the consensus in the data set are shaded.) Above and below the sequences, secondary structure is drawn in accordance with the most recent crystallographic model (Figure 3 ▶ in Löwe et al. 2001).
But, for all their similarity, a point-to-point comparison Sα(x) − Sβ(x), using the known structural alignment (Nogales et al. 1998; Löwe et al. 2001), reveals quantitative differences in the variability of corresponding residues. The distribution of Sα(x) − Sβ(x) has a large peak about zero, but is otherwise normal (μ = 0.01, σ = 0.18; Fig. 3 ▶). The following question arises: Are any of these differences in variability significant? Which, if any, of the quantitative differences indicates a residue that contributes to the functional difference between paralogs?
Figure 3.

Histogram of the difference in variability between corresponding residues in α- and β-tubulin (as determined from the structural alignment). Excluding the strong peak about zero, the distribution is normal (0, 0.18).
We suggest that one way to assign significance is by identifying clusters of residues with statistically large differences in variability. This we do by aligning the variability profiles. The alignment procedure minimizes the global difference in variability between the sequences by introducing/extending gaps in the profiles (see Materials and Methods). Gap placement is optimized by adding a penalty to the global score for every gap initiation event. Gap size is optimized by another penality (typically an order of magnitude smaller than the previous) proportional to the size of the gap. We monitor this variability-based alignment in terms the coefficient of correlation, Rαβ, between the profiles while varying the gap initiation penalty (Fig. 4 ▶).
Figure 4.

Variability-based sequence alignment of α and β tubulin as a function of gap initiation penalty. As the gap initiation penalty is lowered, the cross-correlation coefficient for variability between α and β tubulin increases in three distinct steps before it diverges. At each step, new gaps in the alignment appear that misalign regions of the sequence, indicating a significant difference of variability in those regions. The first alignment (I) is nearly identical to the true structural alignment. The second alignment (II) has gaps that misalign residues in the first α helix (H1) and the ninth β sheet (S9) and also realign residues in the large disordered N-terminal loop (L1). The third alignment (III) adds a pair of self-compensating gaps to the above, which misalign residues in the fourth β sheet (S4), the fourth α helix (H4), the fifth β sheet (S5), and the loops between them.
In the structure-based alignment, the correlation coefficient for variability between the tubulins is Rαβ = 0.42. This level of correlation is matched by the variability-based alignment as soon as the gap initiation penalty is low enough to allow any gaps at all (Fig. 4 ▶). This alignment (labeled I), which persists over a wide range of penalty values (1.2 < G < 3), involves two gaps in β tubulin: a small one in the disordered N-terminal loop (β, 39–40) and a slightly larger one in the loop between S9 and S10 (β, 362–365). For comparison, the structure-based alignment has two gaps in β at roughly the same positions (β, 45–46; β, 361–368) and an additional small gap α, in the disordered C terminus (α, 442–443).
The slight discrepancies between the variability and structure-based alignments are resolved when the gap initiation penalty is lowered just enough to allow one more gap into the alignment (1.0 < G < 1.2). The new gap appears in the α C terminus (α, 450–452), and the gap between S9 and S10 increases in size (β, 351–358). At the same time, however, new discrepancies arise. The smaller of the two gaps in β splits into an equivalent pair of intermediate-sized gaps piercing H1 and bracketing the N-terminal loop (β, 39–40 → β, 14–19; α, 61–64), and the larger of the two gaps in β shifts left 10 residues, into the middle of S9 (β, 362–365 → β, 351–358). These changes lead to a modest increase in the cross-correlation coefficient, Rαβ = 0.45.
When the gap initiation penalty is reduced a little more (G < 1.0), the cross-correlation coefficient makes a large and stable jump up to Rαβ = 0.52 as a new pair of self-compensating gaps appears surrounding H4 and S5 (β, 138–140; α, 175–177). This is the optimal variability-based alignment (labeled III). It persists until the gap initiation penalty becomes so low (G ≤ 0.6) that many small, closely spaced gaps arise throughout the sequences.
Discussion
Although the most restrictive variability-based alignment (I) is very similar to the true (structure-based) alignment, the optimal variability-based alignment (III) is strikingly different (Fig. 4 ▶). Three distinct regions are misaligned to accommodate multiple residues with large differences in variability between the paralogs. These misaligned regions include a total of ~100 residues located in five elements of secondary structure (H1, S4, H4, S5, and S9), two turns (T4 and T5), and the prominent disordered N-terminal loop (L1). Among these are 40 positions at which homologous residues differ in variability by >1 SD from the mean (i.e., |Sα(x) − Sβ(x)| > 0.18; Fig. 3 ▶).
The structural context of many of these 40 residues indicates that they are of particular functional importance. Half are clustered around the nucleotide binding pocket (five of these interact directly with the nucleotide), four are clustered around the taxol binding site (on β tubulin), and one participates in lateral binding between protofilaments in the microtubule (Table 1). The remainder lie in the large and enigmatic N-terminal loop (Table 2).
Table 1.
Positions of defined secondary structure and significantly different variability as identified by variability-based alignment
| x | α | Sα(x) | Sβ(x) | β |
| H1 | ||||
| 14 | V/I* | 0.42 | 0.02 | N |
| 16 | I/V* | 0.37 | 0.08 | I |
| 18 | N | 0.00 | 0.30 | A |
| 22b | E | 0.01 | 0.21 | E |
| 23b,c | L | 0.02 | 0.26 | V |
| 25b | C | 0.11 | 0.29 | S |
| 26b,c | L | 0.05 | 0.34 | D |
| S4 | ||||
| 138 | F | 0.09 | 0.34 | T |
| 139 | H | 0.25 | 0.01 | H |
| T4 | ||||
| 140a | S | 0.20 | 0.00 | S |
| 141a | F/V* | 0.31 | 0.05 | L |
| 150 | G | 0.30 | 0.02 | G |
| H4 | ||||
| 151 | S | 0.22 | 0.02 | T |
| 159b,d | V | 0.25 | 0.03 | E |
| 167 | L | 0.02 | 0.64 | L/N* |
| 168 | E | 0.41 | 0.10 | T |
| S5 | ||||
| 170 | T/S* | 0.49 | 0.01 | S |
| 171a | V/I* | 0.28 | 0.07 | V |
| 172a | Y | 0.09 | 0.46 | V |
| T5 | ||||
| 174a | A/S* | 0.23 | 0.04 | S |
| 177 | V | 0.25 | 0.06 | V |
| S9 | ||||
| 351 | F | 0.00 | 0.27 | V |
| 352a | K | 0.00 | 0.21 | K |
| 354 | G | 0.00 | 0.30 | A/S* |
| 355 | I | 0.05 | 0.28 | V |
For each position, the consensus residue and its variability are listed. When two residues appear with nearly equal frequency, both are listed
*The less variable (more conserved) homolog is emphasized in bold.
a Interacts directly with the nucleotide.
b More than 4 amino acids along the backbone from a residue that interacts directly with the nucleotide.
c Interacts directly with taxol (β tubulin only).
d Participates in lateral binding between dimers in a microtuble.
Table 2.
Positions of significantly different variability in the N-terminal loop (format as in Table 1)
| xβ | α | Sα(x) | Sβ(x) | β | xβ |
| Sequence-based alignment | |||||
| 30 | I | 0.05 | 0.30 | I | 30 |
| 32 | P | 0.20 | 0.49 | P | 32 |
| 33 | D | 0.08 | 0.43 | T | 33 |
| 35 | Q | 0.51 | 0.74 | T | 35 |
| 37 | P | 0.30 | 0.76 | H | 37 |
| 38 | S | 0.40 | 0.06 | G | 38 |
| 40 | K | 0.44 | 0.25 | S | 40 |
| 42 | I | 0.55 | 0.17 | L | 42 |
| 43 | G | 0.32 | 0.05 | Q | 43 |
| 44 | G | 0.43 | 0.13 | L | 44 |
| 48 | A | 0.34 | 0.03 | R | 48 |
| 49 | F | 0.11 | 0.35 | I | 49* |
| 53 | F | 0.00 | 0.22 | Y | 53 |
| 57 | G | 0.16 | 0.49 | S | 57 |
| 59 | G | 0.05 | 0.35 | G | 59 |
| Variability-based alignment | |||||
| 35 | Q | 0.51 | 0.08 | G | 29 |
| 40 | K | 0.44 | 0.02 | G | 34 |
| 42 | I | 0.55 | 0.12 | Y | 36 |
| 44 | G | 0.43 | 0.06 | G | 38 |
* This residue is part of a short helix (H1′) on β tubulin.
Of the 20 residues near the nucleotide binding pocket, 16 are more variable in the “N-site,” which binds GTP without catalyzing its hydrolysis, and less variable in the “E-site,” which hydrolyzes GTP as dimers assemble into protofilaments. This is consistent with the notion that catalytic function requires greater specificity than binding alone. Of the four residues that are less variable in the N-site, the tyrosine residue at position 172 in α tubulin is particularly interesting as a target for directed mutagenesis because it interacts directly with the nucleotide in the crystal structure. We speculate that it may be important in preventing hydrolysis at the N-site.
All four residues near the taxol binding site on β tubulin are more variable than their counterparts on α tubulin, which do not bind taxol. One possible interpretation is that taxol-binding residues are under a “negative selective pressure” to escape susceptibility to this natural poison. Another possibility is that cellular factors (e.g., small peptides or regulatory proteins) exploit this site to regulate microtubule stability, and the variability reflects the variety of such regulatory factors in different species. The latter explanation is particularly intriguing given recent structural evidence for at least one such factor (Kar et al. 2003).
The interpretation of variability differences is less obvious in the large N-terminal loop that connects H1 and S2. Docking the high-resolution tubulin structure into the electron density map of a microtubule puts this loop in a position to participate in the lateral bonds between dimers (Nogales et al. 1999). It is, however, the area of poorest density in the structure of β tubulin and largely absent in the structure of α tubulin (Löwe et al. 2001). Therefore, unlike the rest of the protein, alignment between these two regions is based on sequence not structural homology. Using the sequence-based alignment, 15 of the 30 positions in the loop differ significantly in variability (Table 2). In contrast, the variability-based alignment has a six-residue frameshift that reduces the number of positions with significant differences to four, all of which are less variable on β tubulin (Table 2). It is possible that the high average variability of residues in the H1-S2 loop makes their sequence-based alignment unreliable and that the variability-based alignment is a better indicator of functional homology.
In both alignments, four highly variable residues on α tubulin (Q35, K40, I42, G44) and one strongly conserved residue on β tubulin (G38) differ from their counterparts on the opposite paralog. Because similar differences in variability were so plausibly connected with functional differences in the other misaligned regions (see above), we predict that these residues in the N-terminal loop also have a role in making the biochemical functions of α and β tubulin distinct. Furthermore, because the tendency is for residues on β tubulin to be more conserved, we speculate that the functional distinction is once again related to hydrolysis and that the tenuously structured, but conserved, glycines on β tubulin are involved in the hydrolysis-driven conformational change that eventually destabilizes the microtubule.
In summary, by using α and β tubulin, we have demonstrated how amino acid variability profiles can be used to identify residues that contribute to functional differences between two paralogous proteins. Our approach is based on finding the optimal alignment of the variability profiles and comparing it with the true alignment of the paralogs to reveal on domains with numerous large differences in variability. We note that, under stringent conditions, variability-based alignment reproduces the structure-based alignment. Thus, a comparison between stringent and optimal variability-based alignments of paralogous protein sequences may be used to predict sites of functional distinction, even in the absence of structural information.
Materials and methods
Sequences
Aligned sequences of 167 α tubulins and 300 β tubulins were obtained by a Blast 2.0 search of the non-redundant database in January 2000, using pig tubulins (P02550, P02554) as queries. Sequences <90% of the length of the query tubulins (α tubulins <406 residues, β tubulins <400 residues) are considered fragments and were not used.
Quantification of variability
Variability of the residue at every position in a primary sequence was quantified by the Shannon entropy (Shannon 1948),
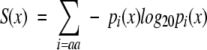 |
(1) |
where pi(x), the probability of finding amino acid i at position x in the sequence, is estimated from the relative frequency of i at x. The sum was taken over all 20 amino acids i.
We note that it is common to group the amino acids into i < 20 categories on the basis of physical character or substitution propensity (Smith and Xue 1997; Atchley et al. 1999; Mirny and Shakhnovich 1999; Plaxco et al. 2000). We experimented with several amino acid groupings. The one that minimized off diagonal elements in the substitution matrix for our tubulin sequences was: (D, E), (K, R), (P, G, A, S, T, N), (F, Y, W, H), and (I, L, M, V, C, Q). However, because even this grouping had no qualitative effect on the distribution of S(x), we chose to use the simplest i = 20 definition for our measure of variability. Also for simplicity, we ignored insertions and deletions, which, as previously noted, are rare in tubulin. Among the aligned sequences, if <20% of the sequences had an amino acid at a site, no x was assigned to that site.
Alignment
The profiles Sα(x) and Sβ(x) were aligned by using a standard minimization algorithm to identify optimal paths on a two-dimensional grid (x,y) with potential 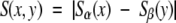 . Each path was forced to start at (x,y) = (0,0) and was assigned a score S(x,y) for each point visited plus a penalty for each vertical or horizontal move. Diagonal moves correspond to alignments between the sequences. Horizontal or vertical moves represent gaps in one of the sequences. The first horizontal or vertical move after a diagonal stretch is penalized with a relatively high initiation cost G (≥0.5). Subsequent moves in the same direction are penalized with a lower continuation cost g (typically of order G/10). For a given path the resulting score, Ω, is therefore
. Each path was forced to start at (x,y) = (0,0) and was assigned a score S(x,y) for each point visited plus a penalty for each vertical or horizontal move. Diagonal moves correspond to alignments between the sequences. Horizontal or vertical moves represent gaps in one of the sequences. The first horizontal or vertical move after a diagonal stretch is penalized with a relatively high initiation cost G (≥0.5). Subsequent moves in the same direction are penalized with a lower continuation cost g (typically of order G/10). For a given path the resulting score, Ω, is therefore
 |
(2) |
where n is the number of gaps, li is the length of the ith gap, and x′ is a position index for the aligned sequences. As both G and g are positive, the optimal path is the path with the lowest Ω (equation 2). This path is determined iteratively, by choosing whichever path to a point (x,y) from either (x − 1,y − 1) or (x, y − k) or (x − k, y), where k = 1, 2, … x minimizes Ω. The first case represents alignment, whereas the latter two represent paths with a gap that terminates at (x,y).
For given parameter set (G, g), the optimal alignment assigns an S-value (or a blank space) to every position x along a common axis for both profiles. We monitor the alignment by computing the correlation coefficient
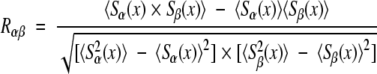 |
(3) |
where 〈 〉 denotes an average over all x. The correlation coefficient measures how predictable Sα(x) is given Sβ(x) (and vice versa). It can range in absolute value from one, if the value of Sα(x) uniquely determines the value of Sβ(x), to zero, if knowing the value of Sα(x) is of no use in predicting Sβ(x).
Acknowledgments
We thank Paul Hansma and Helen Hansma for helpful comments on an early draft of the manuscript and an anonymous referee for suggesting the alternative role of the taxol binding site. This work was supported by the NSF, partially by the CAREER program under award no. 9985493 (DKF), partially by the MRL Program under award no. DMR00-80034, and partially by the Institute for Theoretical Physics at UC Santa Barbara under award no. PHY99-07949.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.03225304.
References
- Atchley, W.R., Terhalle, W., and Dress, A. 1999. Positional dependence, cliques, and predictive motifs in the bHLH protein domain. J. Mol. Evol. 48 501–516. [DOI] [PubMed] [Google Scholar]
- Burns, R.G. 1991. α-, β-, and γ-Tubulins: Sequence comparisons and structural constraints. Cell Motil. Cytoskeleton 20 181–189. [DOI] [PubMed] [Google Scholar]
- Burns, R.G. and Surridge, C.D. 1994. Tubulin: Conservation and structure. In Microtubules (eds. J.S. Hyams and C.W. Lloyd), pp. 3–32. Wiley-Liss, New York.
- Fitch, W.M. 2000. Homology: A personal view on some of the problems. Trends Genet. 16 227–231. [DOI] [PubMed] [Google Scholar]
- Gaucher, E.A., Gu, X., Miyamoto, M.M., and Benner, S.A. 2002. Predicting functional divergence in protein evolution by site-specific rate shifts. Trends Biochem. Sci. 27 315–321. [DOI] [PubMed] [Google Scholar]
- Goldman, N., Thorne, J.L., and Jones, D.T. 1998. Assessing the impact of secondary structure and solvent accessibility on protein evolution. Genetics 149 445–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, W., Petrosino, J., Hirsch, M., Shenkin, P.S., and Palzkill, T. 1996. Amino acid sequence determinants of β-lactamase structure and activity. J. Mol. Biol. 258 688–703. [DOI] [PubMed] [Google Scholar]
- Kar, S., Fan, J., Smith, M.J., Goedert, M., and Amos, L.A. 2003. Repeat motifs of τ bind to the insides of microtubules in the absence of taxol. EMBO J. 22 70–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krauhs, E., Little, M., Kempf, T., Hofer-Warbinek, R., Ade, W., and Postingl, H. 1981. Complete amino acid sequence of β-tubulin from porcine brain. Proc. Natl. Acad. Sci. 78 4156–4160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Little, M. and Seehaus, T. 1988. Comparative analysis of tubulin sequences. Comp. Biochem. Physiol. B 90 655–670. [DOI] [PubMed] [Google Scholar]
- Little, M., Krauhs, E., and Ponstingl, H. 1981. Tubulin sequence conservation. Biosystems 14 239–246. [DOI] [PubMed] [Google Scholar]
- Löwe, J., Li, H., Downing, K.H., and Nogales, E. 2001. Refined structure of α β-tubulin at 3.5 Å resolution. J. Mol. Biol. 313 1045–1057. [DOI] [PubMed] [Google Scholar]
- Mirny, L.A. and Shakhnovich, E.I. 1999. Universally conserved positions in protein folds: Reading evolutionary signals about stability, folding kinetics and function. J. Mol. Biol. 291 177–196. [DOI] [PubMed] [Google Scholar]
- Mitchison, T. and Kirschner, M. 1984. Dynamic instability of microtubule growth. Nature 312 237–242. [DOI] [PubMed] [Google Scholar]
- Nogales, E. 2001. Structural insights into microtubule function. Ann. Rev. Biophys. Biomol. Struct. 30 397–420. [DOI] [PubMed] [Google Scholar]
- Nogales, E., Wolf, S.G., and Downing, K.H. 1998. Structure of the α β tubulin dimer by electron crystallography. Nature 391 199–203. [DOI] [PubMed] [Google Scholar]
- Nogales, E., Whittaker, M., Milligan, R.A., and Downing, K.H. 1999. High-resolution model of the microtubule. Cell 96 79–88. [DOI] [PubMed] [Google Scholar]
- Plaxco, K.W., Larson, S., Ruczinski, I., Riddle, D.S., Thayer, E.C., Buchwitz, B., Davidson, A.R., and Baker, D. 2000. Evolutionary conservation in protein folding kinetics. J. Mol. Biol. 298 303–312. [DOI] [PubMed] [Google Scholar]
- Postingl, H., Krauhs, E., Little, M., and Kempf, T. 1981. Complete amino acid sequence of α-tubulin from porcine brain. Proc. Natl. Acad. Sci. 78 2757–2761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodionov, M.A. and Blundell, T.L. 1998. Sequence and structure conservation in a protein core. Proteins 33 358–366. [PubMed] [Google Scholar]
- Shannon, C.E. 1948. The mathematical theory of communication. Bell Systems Tech. J. 27 623–656. [Google Scholar]
- Smith, D.K. and Xue, H. 1997. Sequence profiles of immunoglobulin and immunoglobulin-like domains. J. Mol. Biol. 274 530–545. [DOI] [PubMed] [Google Scholar]