

Abstract
The pathway for the in vitro equilibrium unfolding of the tubulin heterodimer by guanidinium chloride (GdmCl) has been studied using several spectroscopic techniques, specifically circular dichroism (CD), two-photon Fluorescence Correlation Spectroscopy (FCS), and time-resolved fluorescence, including lifetime and dynamic polarization. The results show that tubulin unfolding is characterized by distinct processes that occur in different GdmCl concentration ranges. From 0 to 0.5 M GdmCl, a slight alteration of the tubulin heterodimer occurs, as evidenced by a small, but reproducible increase in the rotational correlation time of the protein and a sharp decrease in the secondary structure monitored by CD. In the range 0.5–1.5 M GdmCl, significant decreases in the steady-state anisotropy and average lifetime of the intrinsic tryptophan fluorescence occur, as well as a decrease in the rotational correlation time, from 48 to 26 nsec. In the same GdmCl range, the number of protein molecules (labeled with Alexa 488), as determined by two-photon FCS measurements, increases by a factor of two, indicating dissociation of the tubulin dimer into monomers. From 1.5 to 4 M GdmCl, these monomers unfold, as evidenced by the continual decrease in the tryptophan steady-state anisotropy, average lifetime, and rotational correlation time, concomitant with secondary structural changes. These results help to elucidate the unfolding pathway of the tubulin heterodimer and demonstrate the value of FCS measurements in studies on oligomeric protein systems.
Keywords: tubulin, FCS, time-resolved fluorescence, GdmCl, unfolding
Microtubules are essential structures for cell morphology and for many cellular processes, such as development and differentiation (Roberts and Hyams 1979; Vale 1987). In vivo, microtubules are cylinders composed principally of tubulin, a heterodimeric protein of 110,000 molecular weight, consisting of α and β tubulin monomers and two sites for GTP. One of these sites is located on the amino-terminal domain of the α-tubulin, at the interface of both monomers in the heterodimer, and is denominated the nonexchangeable (N-site). The second site, denominated the exchangeable (E-site) is located at the amino-terminal region of the β-tubulin (Monasterio et al. 1995). The structure of the heterodimer, as part of a zinc-induced tubulin sheet, has been resolved at 3.7 Å using electron crystallography (Nogales et al. 1998) and refined to 3.5 Å resolution (Lowe et al. 2001). The structures of both α- and β-tubulin are basically identical, and the core of each monomer is formed by two β-sheets surrounded by α-helices. Each monomer contains four tryptophan residues widely separated in the primary sequence.
Several studies of tubulin folding under in situ conditions have shown that the nascent polypeptide of tubulin monomers requires the participation of a complex cellular machinery to form the assembled competent heterodimer. The folding mechanism proposed for tubulin includes several steps for each monomer. The newly translated monomers of tubulin first interact with a prefoldin chaperone in tandem with the molecular chaperone CCT to avoid aggregation (Yaffe et al. 1992; Hansen et al. 1999; Llorca et al. 2000). The assembly of competent α- and β-monomers requires the concerted participation at least four novel factors in addition to the prefolding and CCT chaperonines (Lopez-Fanarraga et al. 2001).
The first studies of conformational stability of calf-brain tubulin were done by Timasheff’s group (Lee et al. 1978), who compared the denaturation of tubulin by pH, SDS, urea, and GdmCl, and concluded that tubulin was fully unfolded in 5 M GdmCl. Unfolding studies on goat-brain tubulin heterodimers were carried out using urea and low pH as denaturants (Guha and Bhattacharyya 1995). In that study, the unfolding process was monitored using the intrinsic protein fluorescence and circular dichroism; the results suggested that unfolding of the tubulin dimer was a two-step process at pH 7. Furthermore, Guha and Bhattacharyya (1995) carried out their unfolding measurements at two different tubulin concentrations, to address the issue of the dimer/monomer equilibrium, and their results showed that the stability of tubulin to denaturation increased with higher protein concentration. Additional experiments from this group (Guha and Bhattacharyya 1997) supported the existence of a partially unfolded intermediate state, which was stable in 2 M urea. Andreu et al. (2002) carried out CD measurements on tubulin from bovine brain using GdmCl as a denaturant, and presented an unfolding curve similar in shape to that reported by Guha and Bhattacharyya (1997), but shifted to lower denaturant concentrations.
In the interaction of an oligomeric protein, such as the tubulin heterodimer with denaturants, it is important to consider that the denaturant may affect the intersubunit interaction, and that dissociation of the protein oligomer may occur. In the case of a protein dimer, full dissociation into monomers leads to a decrease in the particle mass by a factor of two, with a subsequent increase in the particle’s translational and rotational diffusion. Also, upon dissociation, the number of protein molecules in solution doubles. To ascertain the involvement of dimer dissociation in the tubulin unfolding process, one may approach the problem indirectly, for example, by performing studies at different protein concentrations (for example, see Tominaga et al. 1994; Guha and Bhattacharyya 1995), or utilize techniques that directly assess the protein oligomerization state.
Steady-state anisotropy is a method that reports on the mobility of fluorophores, and if the fluorophores are associated with macromolecules, in principle, can be used to monitor the oligomerization state of a dissociating system. In the case of proteins, for example, one can monitor the anisotropy of intrinsic protein fluorescence or extrinsic probes attached to the proteins to gain information on the aggregation state of the system (Hamman et al. 1996a; Sánchez et al. 1998; Jameson and Seifried 1999). One potential limitation of steady-state anisotropy, however, is the fact that the observed anisotropy is influenced by all of the processes that affect probe mobility, such as overall rotation of the protein, local domain motion, and local mobility of the probe about its point of attachment to the protein (Jameson and Seifreid 1999). In principle, these rotational modalities can be separated using time-resolved methodologies. For example, dynamic polarization, the frequency-domain equivalent to time-decay anisotropy, is a fluorescence technique that can be used to investigate rotational modalities in macromolecular systems (Gratton et al. 1984a; Jameson and Hazlett 1991; Lakowicz 1999; Valeuer 2002). In the case of a protein dimer, if the monomers maintain their approximate shape after dimer dissociation, they will have approximately half the molecular volume of the dimer, and one may expect to measure a global rotational rate approximately one-half that of the dimer, independent of any local probe mobility. In practice, however, if a system is characterized by a broad hierarchy of rotational modalities, the assignment of two rotational rates, that is, global and a local, is an extreme oversimplification, and may not accurately resolve the true global rotational rates corresponding to the protein species (Hamman et al. 1996a,b). Hence, in the case of a protein dimer in the presence of a denaturant, such as the tubulin/GdmCl system, one may still have to obtain unfolding data at different protein concentrations to be sure that a change in rotational mobility, followed by either steady-state anisotropy or dynamic polarization, is due to dimer dissociation, as opposed to significant increases in local probe mobility occurring in the intact dimer.
These potential problems can be circumvented using a more recently developed technique to follow changes in the oligomerization state of proteins, namely FCS. This method is based on intensity fluctuation of a fluorescent signal as molecules diffuse into and out of a small volume (Berland et al. 1995; Chen et al. 1999; Hess et al. 2002). Two important parameters that can be obtained from analysis of the FCS data are (1) the translational diffusion coefficient (D) and (2) the number of molecules (N). In the case of a dimer/monomer equilibrium, the more reliable parameter is N, which increases by a factor of two when the dimer is fully dissociated (Chen et al. 1999; Sánchez et al. 2001). To distinguish particles based on D, a ratio of ~1.6 between the diffusion coefficients associated with the two forms is required (Chen et al. 1999; Hess et al. 2002), but for the case of dimer to monomer dissociation, this ratio is only ~1.2.
In the present study, FCS measurements are used to separate the effect of GdmCl on local unfolding of tubulin from the overall dissociation process of dimers to monomers. The present studies also utilized the two-photon FCS method, which offers unique advantages over the one photon approach (for example, see Chen et al. 1999, and references therein). Specifically, two-photon microscopy allows one to realize a very large separation between the excitation and emission wavelengths, which facilitates rejection of scattered light, and is also characterized by a very small excitation volume that allows one to work at very low sample concentrations.
Results
Figure 1A ▶ shows the variation in the mean ellipticities at 222 nm for a 1.4 × 10−6 M solution of tubulin at 25°C as a function of GdmCl concentration. The decrease in these values, as the concentration of GdmCl increases, is evidence for a decrease in the protein’s secondary structure; a minimum secondary structural content was observed at 5.0 M GdmCl. The equilibrium unfolded curve showed at least two stages, one below 1 M GdmCl and the other between 1 and 5 M GdmCl, following the same behavior observed with bovine-brain tubulin (Andreu et al. 2002).
Figure 1.
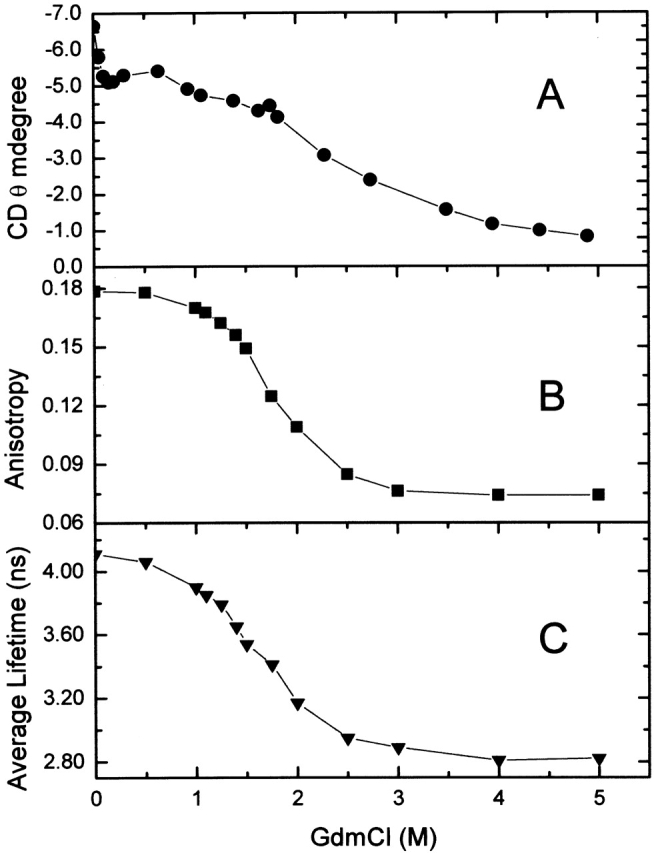
Interaction of tubulin with GdmCl. (A) Circular dichroism mean ellipticities at 222 nm for tubulin at different GdmCl concentrations. (B) Steady-state anisotropy at 295 nm excitation versus GdmCl concentration. (C) Averaged tryptophan lifetimes of tubulin versus GdmCl concentration.
Figure 1B ▶ shows the variation in the steady-state anisotropy of the intrinsic tryptophan fluorescence of tubulin as a function of GdmCl concentration. Clearly, the anisotropy decreases significantly in the range of 0 to 3.0 M GdmCl, with the most pronounced decrease occurring between 1 M and 2 M GdmCl.
Next, multifrequency phase and modulation lifetime data were obtained on tubulin solutions as a function of GdmCl concentration. Because the tubulin heterodimer contains eight tryptophan residues—four per subunit—the lifetime data was treated as an average value. Specifically, each data set was fit to three exponential components, and the average lifetime was defined as <τ> = Σ fi τi, in which fi represents the fractional contribution of the ith component to the total intensity. As shown in Figure 1C ▶, the average lifetime decreases from 4.1 to 2.8 nsec over the range of 0 to 5.0 M GdmCl.
Next, dynamic polarization data were obtained on the intrinsic tryptophan fluorescence as a function of GdmCl concentration (Fig. 2 ▶). The data were fit to two rotational correlation times—the longer correlation time was initially attributed to the global rotation of the protein, whereas the shorter correlation time was attributed to the local probe mobility. The shorter correlation time remained relatively constant, ~0.1 nsec at all GdmCl concentrations, whereas the longer correlation time showed an initial increase from 40 to 48 nsec in the 0–0.5 M GdmCl range, followed by a monotonic decrease to ~4 nsec at 2.5 M GdmCl, a value that persisted up to 5.0 M GdmCl. The correlation time values were normalized to the viscosity of the buffer without GdmCl.
Figure 2.

Rotational correlation time of tubulin versus GdmCl concentration. Data were analyzed considering two rotors; only the longer correlation time (designated as the global rotational correlation time) is shown.
Two-photon FCS data were obtained on tubulin covalently labeled with Alexa 488 (see Materials and Methods). The autocorrelation curves for fluorescein, used for instrument calibration, and for Alexa-labeled tubulin in the absence of GdmCl, are shown in Figure 3 ▶. The continuous line represents the fit, assuming a Gaussian-Lorentzian intensity profile (see Berland et al. 1995), for the explicit formulas for the point-spread function and the definition of the beam waist used, and the dots are the experimental data. In this figure, the fluctuation amplitude, G(0), has been normalized to emphasize the difference in slope presented in the autocorrelation function when there is a large difference between the diffusion coefficient of two species. The diffusion coefficients found were 300 μm2/sec and 54 μm2/sec for fluorescein and Alexa-tubulin, respectively.
Figure 3.

Normalized autocorrelation curve G(τ). Normalized autocorrelation for data taken at 100 kHz for fluorescein (•) and Alexa-labeled tubulin (○). Curved lines correspond to the best fit parameters. G(0) values have been normalized, and the instrument was calibrated with fluorescein using a diffusion coefficient of 300 μm2/sec. The Alexa-tubulin data were best fit to a diffusion coefficient of 54 μm2/sec.
Figure 4 ▶ shows the number of molecules obtained for the Alexa-tubulin samples at different concentrations of GdmCl using the experimental G(0) values (see Materials and Methods). To check the stability of the experiments at equilibrium, Alexa-tubulin in 4.0 M GdmCl was followed by FCS for at least 3 h, and the number of particles did not change. This observation is important to verify that the unfolded monomer, which may lack significant secondary structure, may have a tendency to aggregate or to stick to the glass chamber (Sánchez et al. 2001). Tubulin is a protein that easily aggregates in solution, and the protocol used in this work includes filtration by Sephacryl S300 to separate the dimers from any aggregate, just before the labeling with Alexa-488. It is important to stress that the FCS technique (as opposed to other techniques used to study dimer-monomer equilibrium) is extremely sensitive to the presence of small aggregates. In the case of Alexa 488-labeled tubulin, no aggregates were detected by FCS until 48 h after the labeling reaction; therefore, the experiments were performed before that time.
Figure 4.

Number of particles as a function of GdmCl concentration, determined by analysis of the FCS data. The tubulin concentration was 3.2 × 10−7 M.
Discussion
Previous studies on the unfolding of the tubulin heterodimer in the presence of denaturants (e.g., Lee et al. 1978; Guha and Bhattacharyya 1995, 1997; Andreu et al. 2002) have concluded that the process is multistage. The results presented here support and extend these observations. Our CD, steady-state anisotropy, and time-resolved data all reveal a significant change in tubulin’s conformation over the range from 0.5 to 1.5 M GdmCl. On the basis of previous urea denaturation studies of tubulin at two different concentrations (Guha and Bhattacharyya 1995), one could speculate that tubulin undergoes a dimer-to-monomer dissociation in this GdmCl range. In fact, the steady-state anisotropy and dynamic polarization data indicate that the rotational mobilities of tubulin’s tryptophan residues dramatically increase in this GdmCl region. In the absence of GdmCl, the global rotational correlation time resolved from the dynamic polarization data was 40 nsec. The calculated rotation correlation times for the tubulin dimer and monomer are 46 and 23 nsec, respectively, assuming spherical shapes, molecular masses of 110,000, and 55,000 daltons, respectively, a partial specific volume of 0.74 and a hydration of 0.3g/L (for example, see Jullian et al. 1989). The recent crystallographic structure of the tubulin dimer, however, indicates that its overall shape is elongated and may be better approximated as an ellipsoid of 2 : 1 axial ratio. If the tubulin dimer behaves hydrodynamically as a prolate ellipsoid of axial ratio 2 : 1, one would calculate a rotational correlation time of ~55 nsec (Weber 1952). In fact, the observed rotational correlation times in such systems will depend upon the orientation of the fluorophore’s excitation and emission dipoles with respect to the rotational axes of the ellipsoid (Brunet et al. 1994). Hence, our observed rotational correlation times seem reasonable.
The global rotational correlation time for tubulin actually increased slightly at 0.5 M GdmCl to 50 nsec, which may represent a slight expansion of the protein, or could be the result of changes in the orientation of one or more tryptophan residues with respect to the dimer’s rotational axes, leading to a preferential weighting of a slower rotational rate. The significant decrease in the global rotational correlation time of tubulin, from 40 nsec in the absence of GdmCl, to 26 nsec in the presence of 1.5 M GdmCl, can be interpreted as evidence for a dimer to monomer dissociation. However, no clear plateau in the observed rotational correlation time is evident at 1.5 M GdmCl; that is, the correlation time continues to decrease until around 2.5 M GdmCl, where a value of ~4 nsec is reached. In fact, the assignment of 1.5 M GdmCl as the approximate region where the dimer dissociation is complete, can only be inferred from the FCS results (see below).
Decreases in the steady-state anisotropy and the average fluorescence lifetime are also evident until about the 2.5–3.0 M GdmCl range. No additional significant alterations in these values are evident between 3.0 and 5.0 M GdmCl. The CD data, however, indicates some additional loss of secondary structure in the 2.5 to 5.0 M GdmCl range.
The diffusion coefficient obtained for fluorescein (300 μm2/sec) is in agreement with the literature value (Thompson 1991), and the value of 54 μm2/sec, obtained for Alexa-tubulin, would correspond, at 20°C, to the value calculated (using the Stokes-Einstein equation) for a spherical particle with a diameter of 40 Å. The actual tubulin dimer structure is nonspherical, having dimensions of 46 × 80 × 65 Å (Nogales et al. 1999), and precise calculation of the diffusion coefficient is not straightforward.
By way of contrast, the FCS results clearly indicate that a dimer-to-monomer transition occurs in the range of 0.5 to 1.5 M GdmCl. Specifically, as shown in Figure 4 ▶, the number of fluorescent particles in the observation volume increased twofold over this GdmCl range. Hence, the changes observed for the CD and intrinsic tryptophan fluorescence (Figs. 1 ▶ and 2 ▶) in the ranges of 0 to 0.5 M GdmCl and 1.5 to 4 M GdmCl can be attributed to GdmCl-induced conformational changes of tubulin dimer and monomer, respectively.
Several groups have studied the concentration-dependent dissociation of the tubulin heterodimer, and the dissociation constants reported range from around 70 to 800 nanomolar (for review, see Monasterio et al. 1995). For example, Detrich and Williams Jr. (1978), using equilibrium ultracentrifugation and small-zone gel filtration, reported a reversible, concentration-dependent dissociation of the bovine-brain heterodimer into α and β-tubulin, with a dissociation constant of 8 × 10−7 M at 5°C. Sackett et al. (1989), measuring the concentration dependence of the kinetics of tubulin digestion by subtilisin, determined a heterodimer dissociation constant (Kd) of ~1.5 × 10−7 M. Mejillano and Himes (1989), using fluorescence anisotropy and size-exclusion HPLC, found a concentration-dependent dissociation (Kd = 8.4 × 10−7 ± 0.4 × 10−7 M) of bovine-brain tubulin dimer covalently labeled with 5-[(4,6-dichlorotriazin-2-yl) amino]fluorescein, or with fluorescein isothiocyanate. However, in a recent study, Caplow and Fee (2002) used surface plasmon resonance and gel-exclusion chromatography, and they found that the dissociation rate of the tubulin dimer was extremely slow; a dissociation constant equal to 10−11 M was calculated. They also found that the dissociation was reversible and independent of GTP hydrolysis, confirming the results found by Timasheff’s group. In the present FCS study, dilution of Alexa-tubulin, in the absence of GdmCl, from the micromolar range down to 5 × 10−9 M demonstrated that the number of particles, corrected for dilution, was essentially constant until the lowest concentration reached, at which point a slight increase was noted (data not shown). These results suggest that the dissociation constant of tubulin is lower than 10−9 M, a result in agreement with the conclusions of Caplow and Fee (2002).
The lack of agreement in the reported values of tubulin’s dimer-to-monomer dissociation constant is not unique. Sánchez et al. (1998, 2001), for example, used FCS to study the dissociation of mitochondrial malate dehydrogenase (mMDH) and PLA2, respectively, and found that these proteins remained dimeric at much lower concentrations than reported by other groups using indirect methods. In the case of the mMDH dimer, the presence of aggregates and the details of the sample preparation were judged to be responsible for variation among the literature values for the dissociation constant. In the specific case of tubulin, it may also be that the variation in sample preparation and solvent conditions (temperature, ionic strength, etc.) affects the observations and results in differences between the reported dissociation constants. We wish to reiterate that the advantage of the FCS approach used in this study is that it provides a direct measurement of the number of particles in solution, and hence, provides direct information on the protein dissociation constant.
Materials and methods
The buffer used for all experiments was 50 mM HEPES (Sigma; pH 7.5) with 5 mM DTT (Boheringer Mannheim). Guanidinium chloride (ultra-pure grade) was purchased from Schwarz/Mann Biotech, and Sephacryl S-300 was purchased from Pharmacia. Alexa 488 and associated materials were purchased in labeling kits from Molecular Probes. Salts and solvents were analytical grade obtained from Merck. Distilled, deionized, and nanopure water was used throughout. Concentration of GdmCl in the stock and experimental solutions were determined with a OFFICINE Galileo No. 22701 refractometer, following the method described by Pace and Scholtz (1997).
Sample preparation
Chicken-brain tubulin purification
Brains were dissected from freshly slaughtered chickens (kindly provided by Industrial Ochagavia Ltda.), kept on ice, and used within 2 h. Tubulin was purified by the method of Weisenberg et al. (1968) and Weisenberg and Timasheff (1970), as modified by Andreu et al. (2002). The stock protein in 1 M sucrose was stored at −80°C. The experimental samples were prepared by equilibration of the stock protein by filtration through Sephacryl S-300 in the experimental buffer (specified for each experiment) at 4°C, just before use. In this step, tubulin was separated from unspecific aggregates. The protein concentration for the unlabeled material was determined using the absortivity value of 1.03 Lg−1cm−1 at 278 nm in 6M GdmCl (Na and Timasheff 1981).
Fluorescent tubulin conjugates
Before labeling, tubulin was passed through a Sephacryl S-300 column in the experimental buffer. Fluorescent conjugates of tubulin were prepared with Alexa Fluor 488 succinimidyl ester by incubation of 20 μM tubulin with the dye in a 1 : 50 molar ratio for 1 h at room temperature in 25 mM HEPES buffer (pH 7.5) containing 0.1 mM sodium bicarbonate, giving a final pH of 8. After the incubation, the free dye was removed by passing the sample through a G-25 column (NAP 5 column from Amersham Biosciences) with 25 mM HEPES as elution buffer. The Alexa/tubulin ratio was calculated using an extinction coefficient for Alexa ɛ494 = 71,000 M−1 cm−1 (Molecular Probes), and the protein concentration was determined using the Bio-Rad Protein Assay (Bio-Rad). The labeling ratio was ~4 Alexa molecules per tubulin dimer. Note that random labeling will result in a Poisson distribution of probes among the protein population. Hence, if the labeling ratio was low, for example, an average of one probe for each protein dimer, there would be a significant proportion of unlabeled monomers, which means that the increase in the number of particles after dimer dissociation would be <2. In fact, as the ratio of probe to protein increases, the measured increase in the particle number will approach 2 as the limit.
Time-resolved fluorescence measurements
Time-resolved measurements were obtained using a laser-based instrument at the Laboratory for Fluorescence Dynamics (LFD) at the University of Illinois at Urbana-Champaign, IL. In this instrument, frequency modulation of the excitation source is realized using the harmonic content approach (Gratton et al. 1984b; Alcala et al. 1985). The exciting light was from a Coherent Nd:YAG mode-locked laser, pumping a rhodamine dye laser. The dye laser was tuned to 590 nm, which was then frequency doubled to 295 nm. Emission at wavelengths >305 nm was observed through a Schott WG 320 filter. The exciting light was polarized parallel to the vertical laboratory axis, and the emission was viewed through a polarizer oriented at 55° to eliminate polarization effects on the lifetime measurements (Spencer and Weber 1970). The reference fluorophore used was p-terphenyl in ethanol, with a lifetime of 1.05 nsec. Phase and modulation values for the lifetime data were obtained as described previously (Spencer and Weber 1969; Jameson et al. 1984; Jameson and Hazlett 1991). The lifetime data were analyzed by assuming a sum of discrete exponentials, and the quality of the fits were evaluated by the χ2 values as described previously (Jameson et al. 1984; Lakowicz 1999; Valeuer 2002). The dynamic polarization data were obtained by measuring the phase delay and modulation ratios between the parallel and perpendicular emission components (Gratton et al. 1984a; Lakowicz 1999; Valeuer 2002), and these data were analyzed with a two-rotor model (Jameson and Hazlett 1991). Time-resolved data were measured for tubulin at a final concentration of 3.25 × 10−6 M at different concentrations of GdmCl, going from 0 to 4 M. Stocks solutions of the protein and GdmCl were prepared in 50 mM HEPES (pH 7.5), DTT 5 mM, and the samples at the different GdmCl concentration were incubated for 1 h on ice, and then 15 min at room temperature before measuring at 20°C.
Circular dichroism measurements
Circular dichroism spectra were taken on a JASCO J600 spectropolarimeter. Spectra were obtained for unlabeled tubulin under varying concentrations of GdmCl at a protein concentration of 1.4 × 10−6 M in 50 mM HEPES (pH 7.5), DTT 5 mM. Samples were incubated with denaturant for 1 h on ice, and 15 min at room temperature before the spectra were collected. Each spectrum was obtained at 25°C in a 0.1-cm optical path cell. Wavelengths were scanned between 250 and 215 nm at 20 nm/min with a bandwidth of 1.0 nm and a response time of 2 sec. The data presented in Figure 1A ▶ represents the average of four scans.
FCS measurements
Instrumentation
The instrumentation for the two-photon FCS experiments is similar to that described by Berland et al. (1995), with the following modifications: The experiments were carried out using a Zeiss Axiovert 135 TV microscope (Thornwood) with a 63X Plan Apochromat-oil immersion objective (NA=1.4). A mode-locked Ti: Sapphire laser (Mira 900, Coherent Inc.) pumped by an intracavity doubled Nd:YVO4:Vanadate laser (Verdi, Coherent Inc.) was used as the two-photon excitation source. For all measurements, an excitation wavelength in the range from 770 to 780 nm was used, whereas the average power at the sample ranged from 7 to 2 mW. Photon counts were detected using an Avalanche Photodiode Detector (APD; Model SPCM-AQ-151, EG&G). The output of the APD unit was connected directly to the data acquisition card. The photon counts were sampled at either 20 or 100 kHz. The recorded photon counts were later analyzed with programs written in PV-WAVE version 6.10 (Visual Numerics). Samples were mounted in a plastic homemade sample holder fabricated of Delring (Illini Plastic). A drilled hole in the center of the chamber was covered by a 1.5-mm standard microscope cover glass, and used as the window for the microscope objective. No extra treatment was done to the surface, except for intensive cleaning before each sample measurement.
Sample handling and GdmCl protocol
For each concentration of GdmCl (from 0 to 4 M), the experiment started when the Alexa-tubulin was added to the FCS chamber already containing the corresponding GdmCl concentration. The samples were mixed with a pipette, and the first FCS measurement was taken after 30 min and then again after 2 h, in some of the cases (no differences were observed between the two time points). The final protein concentration in the chamber was kept constant at 3.2 × 10−7 M. In between samples, the chamber was rinsed with buffer several times until the fluorescence reached the background counts of the buffer.
FCS data analysis
Experimental autocorrelation functions were fit assuming a Gaussian-Lorentzian intensity profile, as described in previous work, which contains the formulas for the point-spread function and the definition of the beam waist used (Berland et al. 1995; Sánchez et al. 2001). The beam waist of the excitation profile function depends on the instrument setup, and must be calibrated each time the system is aligned; for this purpose fluorescein was used. In 25 mM Tris buffer (pH 8), fluorescein has a diffusion coefficient of 300 μm2/sec (Thompson 1991). The recovered beam waist value of 0.377 μm was used to perform the analysis and G(0) values for each sample. The fluctuation amplitude, G(0), is related to the number of particles by:
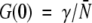 |
where γ is a geometric factor dependent upon the shape of the point spread function and N¯ is the average number of particles inside the excitation volume. The geometric factor for the Gaussian-Lorentzian model is 0.0762 (Chen et al. 1999).
Acknowledgments
We thank Maribel López for her technical help. This work was supported by FONDECYT Grant #1010848. The fluorescence experiments reported in this study were performed at the Laboratory for Fluorescence Dynamics (LFD) at the University of Illinois at Urbana-Champaign (UIUC). The LFD is supported jointly by the National Center for Research Resources of the National Institutes of Health (PHS 5 P41-RRO3155) and UIUC.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.03295604.
References
- Alcala, J.R.E., Gratton, E., and Jameson, D.M. 1985. Multifrequency phase fluorometry using the harmonic content of a mode-locked laser. Anal. Instrum. 14 225–250. [Google Scholar]
- Andreu, J.M., Oliva, M.A., and Monasterio, O. 2002. Reversible unfolding of FtsZ cell division proteins from archaea and bacteria. J. Biol. Chem. 277 43262–43270. [DOI] [PubMed] [Google Scholar]
- Berland, K.M., So, P.T.C., and Gratton, E. 1995. Two-photon fluorescence correlation spectroscopy: Method and application to the intracellular environment. Biophys. J. 68 694–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunet, J.E., Vargas, V., Gratton, E., and Jameson, D.M. 1994. Hydrodynamics of horseradish peroxidase revealed by global analysis using multiple fluorescent probes. Biophys. J. 66 446–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caplow, M. and Fee, L. 2002. Dissociation of the tubulin dimer is extremely slow, thermodynamically very unfavorable, and reversible in the absence of an energy source. Mol. Biol. Cell 13 2120–2131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, Y., Muller, J.D., Berland, K.M., and Gratton, E. 1999. Fluorescence fluctuation spectroscopy. Methods 19 234–252. [DOI] [PubMed] [Google Scholar]
- Detrich, H.W. and Willimas Jr., R.C. 1978. Reversible dissociation of the αβ dimer of tubulin. Biochemistry 17 3900–3907. [DOI] [PubMed] [Google Scholar]
- Gratton, E., Jameson, D.M., and Hall, R.D. 1984a. Multifrequency phase fluorometry. Annu. Rev. Biophys. 13 105–124. [DOI] [PubMed] [Google Scholar]
- Gratton, E., Jameson, D.M., Rosato, N., and Weber, G. 1984b. A multifrequency cross-correlation phase fluorometer using synchrotron radiation. Rev. Sci. Instrum. 55 486–494. [Google Scholar]
- Guha, S. and Bhattacharyya, B. 1995. A partially folded intermediate during tubulin unfolding: Its detection and spectroscopic characterization. Biochemistry 34 6925–6931. [DOI] [PubMed] [Google Scholar]
- ———. 1997. Refolding of urea-denatured tubulin: Recovery of nativelike structure and colchicine binding activity from partly unfolded states. Biochemistry 36 13208–13213. [DOI] [PubMed] [Google Scholar]
- Hamman, B.D., Oleinikov, A.V., Jokhadze, G.G., Traut, R.R., and Jameson, D.M. 1996a. Dimer/monomer equilibrium and domain separations of Escherichia coli ribosomal protein L7/L12. Biochemistry 35 16680–16686. [DOI] [PubMed] [Google Scholar]
- ———. 1996b. Rotational and conformational dynamics of Escherichia coli ribosomal protein L7/L12. Biochemistry 35 16672–16679. [DOI] [PubMed] [Google Scholar]
- Hansen, W.J., Cowan, N.J., and Welch, W.J. 1999. Prefoldin-nascent chain complexes in the folding of cytoskeletal proteins. J. Cell Biol. 145 265–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess, T.S., Huang, S., Heikal, A.A., and Webb, W.W. 2002. Biological and chemical applications of fluorescence correlation spectroscopy: A review. Biochemistry 41 697–705 [DOI] [PubMed] [Google Scholar]
- Jameson, D.M. and Hazlett, T.L. 1991. Time-resolved fluorescence in biology and biochemistry. In Biophysical and biochemical aspects of fluorescence spectroscopy (ed. G. Dewey), pp. 105–133. Plenum Press, New York.
- Jameson, D.M. and Seifried, S.E. 1999. Quantification of protein–protein interactions using fluorescence polarization. Methods 19 222–233. [DOI] [PubMed] [Google Scholar]
- Jameson, D.M., Gratton, E., and Hall, R.D. 1984. The measurement and analysis of heterogeneous emission by multifrequency phase and modulation spectroscopy. Appl. Spectrosc. Rev. 20 55–105. [Google Scholar]
- Jullian, C., Brunet, J.E., Thomas, V., and Jameson, D.M. 1989. Time-resolved fluorescence studies on protoporphyrin IX–apohorseradish peroxidase. Biochim. Biophys. Acta 99 206–210. [DOI] [PubMed] [Google Scholar]
- Lakowicz, J.R. 1999. Principles of fluorescence spectroscopy, 2nd ed. Kluwer Academic, New York.
- Lee, J.C., Corfman, D., Frigon, R.P., and Timasheff, S.N. 1978. Conformational study of calf brain tubulin. Arch. Biochem. Biophys. 185 4–14. [DOI] [PubMed] [Google Scholar]
- Llorca, O., Martin-Benito, J., Ritco-Vonsovici, M., Grantham, J., Hynes, G.M., Willison, K.R., Carrascosa, J.L., and Valpuesta, J.M. 2000. Eukaryotic chaperonin CCT stabilizes actin and tubulin folding intermediates in open quasi-native conformations. EMBO J. 19 5971–5979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Fanarraga, M., Avila, J., Coll, M., and Zabala, J.C. 2001. Review: Postchaperonin tubulin folding cofactors and their role in microtubule dynamics. J. Struct. Biol. 135 219–229. [DOI] [PubMed] [Google Scholar]
- Lowe, J., Li, H., Downing, K.H., and Nogales, E. 2001. Refined structure of α-β tubulin at 3.5 Å resolution. J. Mol. Biol. 313 1045–1057. [DOI] [PubMed] [Google Scholar]
- Mejillano, M.R. and Himes, R.H. 1989. Tubulin dissociation detected by fluorescence anisotropy. Biochemistry 28 6518–6524. [DOI] [PubMed] [Google Scholar]
- Monasterio, O., Andreu, J.M., and Lagos, R. 1995. Tubulin structure and function. Comments Mol. Cell. Biophys. 8 273–306. [Google Scholar]
- Na, G.C. and Timasheff, S.N. 1981. Interaction of calf brain tubulin with glycerol. J. Mol. Biol. 151 165–178. [DOI] [PubMed] [Google Scholar]
- Nogales, E., Wolf, S.G., and Downing, K.H. 1998. Structure of the αβ tubulin dimer by electron crystalography. Nature 391 199–203. [DOI] [PubMed] [Google Scholar]
- Nogales, E., Whittaker, M., Milligan, R.A., and Downing, K.H. 1999. High resolution model of the microtubule. Cell 96 79–88. [DOI] [PubMed] [Google Scholar]
- Pace, C.N. and Scholtz, J.M. 1997. Measuring the conformational stability of a protein. In Protein structure. A practical approach(ed. T.E. Creighton), 2nd ed. IRL Press, New York.
- Roberts, K. and Hyams, J.S. 1979. Microtubules. Academic Press, London.
- Sackett, D.L., Zimmerman, D.A., and Wolfe, J. 1989. Tubulin dimer dissociation and proteolytic accessibility. Biochemistry 28 2662–2667. [DOI] [PubMed] [Google Scholar]
- Sánchez, S.A., Hazlett, T.L., Brunet, J.E., and Jameson, D.M. 1998. Aggregation states of mitochondrial malate dehydrogenase. Protein Sci. 7 2184–2189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sánchez, S.A., Chen, Y., Muller, J.D., Gratton, E., and Hazlett, T.L. 2001. Solution and interface aggregation of Crotalus atrox venom phospholipase A2 by two-photon excitation fluorescence correlation spectroscopy. Biochemistry 40 6903–6911. [DOI] [PubMed] [Google Scholar]
- Spencer, R.D. and Weber, G. 1969. Measurements of subnanosecond fluorescence lifetime with a cross-correlation phase fluorometer. Ann. N.Y. Acad. Sci. 158 361–376. [Google Scholar]
- ———. 1970. Influence of Brownian rotations and energy transfer upon the measurements of fluorescence lifetimes. J. Chem. Phys. 52 1654–1663. [Google Scholar]
- Thompson, N.L. 1991. Fundamentals of fluorescence microscopy. In Topics in fluorescence spectroscopy (ed. J.R. Lakowicz), pp. 337–378. Plenum Press, New York.
- Tominaga, N., Jameson, D.M., and Uyeda, K. 1994. Reversible unfolding of fructose 6-phosphate, 2-kinase:fructose 2,6-bisphosphatase. Protein Sci. 3 1245–1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vale, R.D. 1987. Intracellular transport using microtubule-based motors. Annu. Rev. Cell Biol. 3 347–378. [DOI] [PubMed] [Google Scholar]
- Valeuer, B. 2002. Molecular fluorescence: Principles and applications. Wiley-VCH, Weinheim, Germany.
- Weber, G. 1952. Polarization of the fluorescence of macromolecules. Biochem. J. 51 145–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisenberg, R.C. and Timasheff, S.N. 1970. Aggregation of microtubule subunit protein. Effects of divalent cations, colchicine and vinblastine. Biochemistry 9 4110–4116. [DOI] [PubMed] [Google Scholar]
- Weisenberg, R.C., Borisy, G.G., and Taylor, E.W. 1968. The colchicine-binding protein of mammalian brain and its relation to microtubules. Biochemistry 7 4466–4479. [DOI] [PubMed] [Google Scholar]
- Yaffe, M.B., Farr, G.W., Miklos, D., Horwich, A.L., Sternlicht, M.L., and Sternlicht, H. 1992. TCP1 complex is a molecular chaperone in tubulin biogenesis. Nature 352245–248. [DOI] [PubMed] [Google Scholar]