

Abstract
Bacterial translation initiation factor IF2 is a multidomain protein that is an essential component of a system for ensuring that protein synthesis begins at the correct codon within a messenger RNA. Full-length IF2 from Escherichia coli and seven fragments of the protein were expressed, purified, and characterized using nuclear magnetic resonance (NMR) and circular dichroism (CD) methods. Interestingly, resonances of the 6 kD IF2N domain located at the extreme N terminus of IF2 can be clearly identified within the NMR spectra of the full-length 97-kD protein. 15N NMR relaxation rate data indicate that (1) the IF2N domain is internally well ordered and tumbles in solution in a manner that is independent of the other domains of the IF2 protein, and (2) the IF2N domain is connected to the C-terminal regions of IF2 by a flexible linker. Chemical shifts of resonances within the isolated IF2N domain do not significantly differ from those of the corresponding residues within the context of the full-length 97-kD protein, indicating that IF2N is a structurally independent unit that does not strongly interact with other regions of IF2. CD and NMR data together provide evidence that Domains I–III of IF2 have unstructured and flexible regions as well as substantial helical content; CD data indicate that the helical content of these regions decreases significantly at temperatures above 35°C. The features of structurally well-ordered N- and C-terminal domains connected by a flexible linker with significant helical content are reminiscent of another translation initiation factor, IF3.
Keywords: translation initiation, initiation factor, IF2, IF2N domain, NMR, circular dichroism, molecular dynamics
The initiation of translation in bacteria is promoted by three protein factors, IF1, IF2, and IF3. Translation initiation factors IF1 and IF2 are found in all living organisms. During the translation initiation process, IF1 binds to the A-site of the small (30S) subunit of the ribosome, IF2 binds on top of IF1, and the initiator fMet-tRNAfMet binds to the P-site. The preinitiation complex is completed by the binding of the small ribosomal subunit to the translation initiation region of the mRNA. The exact order in which each of these components binds to the small ribosomal subunit is not yet fully understood. The large (50S) ribosomal subunit joins the preinitiation complex, and GTP bound to IF2 is hydrolyzed to GDP and Pi in a ribosome-dependent reaction. The initiation factors leave the complex, which undergoes a conformational change, and is ready for the elongation phase of translation. For recent reviews of the translation initiation process, see Gualerzi et al. (2000), Boelens and Gualerzi (2002), Ramakrishnan (2002), and Sørensen et al. (2002).
Bacterial IF2 is encoded by the infB gene, which in Escherichia coli encodes three forms of the protein, designated IF2-1, IF2-2, and IF2-3, with molecular weights of 97.3 kD, 79.9 kD, and 78.8 kD, respectively (Nyengaard et al. 1991). The three forms of IF2 differ in their initiation site on the infB mRNA. Hence, the forms of IF2 have identical C termini and differ only in the absence of the first 157 and 164 amino acid residues for IF2-2 and IF2-3, respectively, as compared to IF2-1 (Mortensen et al. 1995). The cellular content of IF2-2 and IF2-3 is close to that of IF2-1 (Howe and Hershey 1982), and the presence of both the large and smaller forms is required for optimal growth of E. coli (Sacerdot et al. 1992). The presence of more than one isoform of IF2 is not peculiar to E. coli, but has been found in several other enterobacteria (Laursen et al. 2002b).
Translation initiation factor IF2 is the largest of the bacterial initiation factors, and can be divided into domains based on interspecies homology. IF2 in E. coli is composed of six domains (Mortensen et al. 1998), as shown in Figure 1 ▶. The conserved C-terminal region consists of Domains IV–VI, and a less conserved N-terminal region corresponds to Domains I–III (Steffensen et al. 1997; Sørensen et al. 2001). The most conserved parts of IF2 are involved in the binding and hydrolysis of GTP, as well as binding of fMet-tRNAfMet. Although there is no direct structural information available for Domains IV–VI of E. coli IF2, the structure of the homologous protein aIF5B from the archaea Methanobacterium thermoautotrophicum has recently been solved by X-ray methods and is shown in Figure 1 ▶ (Roll-Mecak et al. 2000); amino acid sequence homology predicts a similar structure for Domains IV–VI of bacterial IF2.
Figure 1.

Schematic representation of IF2 from E. coli and the fragments used in this study. (Top) A representation of the structure of IF2 from E. coli with the domains indicated in different colors. The cartoon models of the regions of known structure are derived from PDB entry 1ND9 for the N-terminal IF2N domain and PDB entry 1G7T for the C-terminal region. The cartoons were prepared using the program MOLMOL (Koradi et al. 1996). (Bottom) Schematic representation in which lines illustrate the content of the different fragments of IF2 characterized in this study.
The structure of Domain I of E. coli IF2 (corresponding to the first 157 residues of IF2-1) was recently investigated by nuclear magnetic resonance (NMR) methods (Laursen et al. 2003). The first 50 residues of this E. coli protein were found to form a well-ordered and compact globular domain, which has now been named IF2N in the protein families database, Pfam, and the NCBI conserved domain database. Amino acid sequence comparisons show that a similarly structured domain is present in most bacterial and plastid IF2s. In many bacteria, including E. coli, a second copy of the IF2N domain can be found just before the GTP binding G-domain of IF2. No specific function has been assigned to the IF2N domain at the present time. NMR results show that residues 51–97 do not form a regular structure and are relatively flexible, whereas residues 98–157 form a helix containing a repetitive sequence of mostly hydrophilic amino acids (Laursen et al. 2003).
A less conserved region of IF2, corresponding to Domains II and III, is located between the IF2N domain and the conserved C-terminal domains of the protein, and is not well characterized in terms of structure. Interspecies sequence comparisons show that Domains II and III vary significantly in both primary structure and length (Steffensen et al. 1997; Sørensen et al. 2001). Domain II of E. coli IF2 has been shown to interact with the ribosome (Moreno et al. 1998, 1999), and there is evidence that Domains I and II interact with the infB mRNA (Laursen et al. 2002b).
The present work describes the results of NMR and circular dichroism (CD) studies of full-length IF21 from E. coli and seven different fragments of the protein, with the aim of further characterizing the structures of the N-terminal domains.
Results
Figure 1 ▶ schematically describes the different fragments of IF2-1 that were recombinantly expressed and purified for this study. All protein fragments were stable and quite soluble in the medium ionic strength buffer used for purification. However, the fragments that did not contain Domain I were significantly less soluble in the low-ionic-strength buffers needed for the CD and NMR studies. Therefore, 100 mM NaCl or 100 mM NaF were included in the buffer for the NMR and CD studies, respectively, for the fragments that did not contain Domain I. The observation that Domain I enhances solubility has led to a proposal that this may be one function of the N-terminal region of IF2 (Sørensen et al. 2003).
Circular dichroism and secondary structure analysis
CD spectra of IF2 and its fragments all reveal spectra typical of helical proteins, with characteristic minima at 207 and 222 nm (Fig. 2 ▶). Deconvolution of the individual spectra yields the relative content of secondary structure elements in each fragment, as summarized in Table 1. The fragment containing Domains I–III has a significantly higher content of helix than any of its isolated components, specifically Domain I, II, I–II, and II–III. This suggests that additional helix structure forms when all three of the domains are present.
Figure 2.

CD spectra used for determining the relative content of secondary structure in IF2 and its fragments. The spectra were recorded in 10 mM phosphate (pH 7.5) at 20°C and the units converted to mean residue ellipticity. An additional 100 mM NaF were included in the sample buffer for the fragments corresponding to Domains II, II–III, III–VI, full-length IF2-1, and IF2-2, as required to keep the protein soluble. All spectra show the shape characteristic of proteins with substantial α-helical content, with minima at 207 and 222 nm.
Table 1.
Content of secondary structure elements in IF2 and its fragments
| Helix (%) | Strand (%) | Other (%) | |
| IF2–1 | 40 ± 3 | 12 ± 3 | 49 ± 4 |
| IF2–2 | 34 ± 2 | 16 ± 4 | 50 ± 5 |
| Domain III–VI | 42 ± 2 | 16 ± 5 | 42 ± 7 |
| Domain I | 60 ± 6 | 6 ± 2 | 35 ± 7 |
| Domain II | 50 ± 5 | 10 ± 4 | 40 ± 7 |
| Domain I–II | 54 ± 4 | 7 ± 3 | 40 ± 4 |
| Domain I–III | 70 ± 7 | 3 ± 2 | 28 ± 7 |
| Domain II–III | 39 ± 3 | 10 ± 6 | 50 ± 5 |
The relative content of secondary structure for each fragment was calculated based on the deconvolution of the CD spectra acquired at 20°C and shown in Figure 2 ▶.
To investigate the stability of the different fragments, CD spectra were acquired at temperatures ranging from 5°C–70°C. All of the fragments of IF2, with the exception of Domain III–VI and Domain II–III, begin to unfold at 35°C–40°C; a similar unfolding temperature has previously been observed for Domain I and the full-length protein (Laursen et al. 2002a, 2003). The Domain II–III fragment is less stable and starts to lose structure at even lower temperatures, whereas the Domain III–VI fragment is more stable against thermal denaturation than the other fragments. Thus, the loss in structure that occurs in the native IF2-1 and IF2-2 at 35°C–40°C can be accounted for by a loss of structure in Domains I–III in the N-terminal region of the protein.
The dependence of buffer conditions was tested by recording CD spectra of Domain I–III and full-length IF2-1 at pH 6.0 and 7.5, and MgCl2 concentrations varying between 0 and 10 mM. The concentration of NaF was also varied between 0 and 100 mM for the Domain I–III fragment. No significant difference in the CD spectra recorded at these different conditions was observed.
Nuclear magnetic resonance
NMR spectroscopy was used to further characterize the IF2 protein and its N-terminal domains. Resonances with chemical shifts that differ significantly from the values typical of random coil structure are good indicators that a protein is folded. The one-dimensional (1-D) 1H NMR spectrum of Domain I (Fig. 3 ▶, bottom) shows peaks with disperse chemical shifts; this is expected because the first 50 residues of this fragment are known to form a folded domain, based on a previous detailed structural study (Laursen et al. 2003). It is possible to identify some of the well-resolved peaks arising from Domain I within the 1-D spectra of all of the fragments containing the domain (Fig. 3 ▶). The clear observation of peaks assigned to Domain I within the spectrum of the 97-kD IF2-1 (Fig. 3 ▶, top) is rather surprising, because resonances of such high molecular weight proteins would be expected to be very broad and therefore difficult to detect; this observation provided an initial indication that Domain I tumbles independently of the rest of the protein. About 30 well-resolved resonances are observed in the two-dimensional (2-D) 15N-1H HSQC-TROSY spectrum of the full-length IF2-1 (Fig. 4 ▶); the majority of these resonances were assigned to Domain I, based on a comparison of the three-dimensional (3-D) NMR spectra of the full-length IF2-1 and of the isolated Domain I (Fig. 5 ▶).
Figure 3.
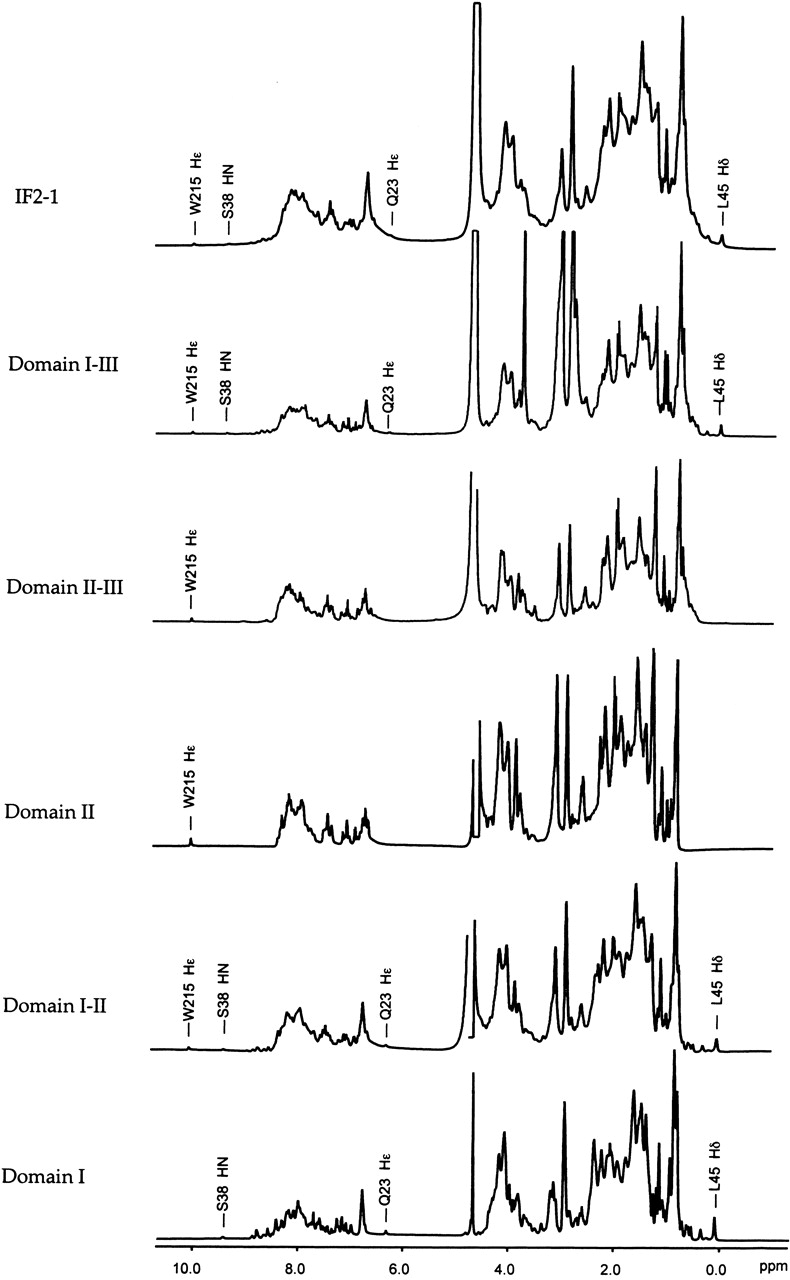
Five-hundred megahertz 1H NMR spectra of full-length IF2-1 and its fragments. Spectra were acquired at 20°C. Several peaks arising from nuclei within Domain I are easily identified in all of the fragments that contain the domain. Several of the best-resolved resonances are labeled.
Figure 4.

15N-1H HSQC-TROSY spectrum of the full-length 97-kD IF2-1 from E. coli. The spectrum was obtained at 20°C with a proton resonance frequency of 500 MHz, in 10 mM phosphate buffer (pH 6), 100 mM NaCl, and 1 mM MgCl2. The most well-resolved peaks that have been assigned are labeled. Note that almost all well-resolved peaks arise from Domain I.
Figure 5.

Sections of single slices of 3-D 15N-1H-1H HSQC-NOESY spectra. A spectrum of the full-length 97-kD IF2-1 is shown on the left, and a spectrum of the isolated Domain I containing residues 2–157 of IF2-1 is shown on the right. Both spectra were acquired at 20°C. Each slice corresponds to a 15N frequency of 124.5 ppm. Several of the best-resolved peaks are labeled. Note that the resonances of Domain I have nearly identical chemical shifts in the context of the full-length protein as they do in the isolated Domain I.
Because it is known that Domains IV–VI of IF2-1 are folded into globular structures (Fig. 1 ▶), it is expected that nuclei associated with these domains would have disperse chemical shifts. However, it is clear from Figure 4 ▶ that nearly all of the resonances with disperse chemical shifts are assignable to Domain I. The lack of observed disperse NMR signals from Domains IV–VI can be attributed to resonance broadening due to the relatively high molecular weight (and slow tumbling time) for these regions of the protein. Resonances from nuclei in flexible regions located in Domains II–VI may have short enough rotational times to be observable in the NMR spectra, but flexible regions typically have chemical shifts near the random coil values, with NMR peaks in the crowded central region of the spectrum shown in Figure 4 ▶. The disperse resonances from nuclei in the well-ordered regions of Domain I are only observable because this domain is attached to the C-terminal regions of IF2-1 by a flexible linker.
In an effort to search for evidence of structural differences between the isolated Domain I and Domain I within the context of the full-length IF2-1, the 3-D 15N-1H-1H HSQC-TOCSY and HSQC-NOESY spectra of the full-length IF2-1 and the isolated Domain I were compared (Fig. 5 ▶); we have previously assigned the chemical shifts of the isolated Domain I, available as entry 5624 in the BioMagResBank. Resonances of nearly all of the first 50 amino acids of Domain I (corresponding to the region of the protein now called the IF2N domain) were identified in the spectra and found to have essentially the same chemical shifts, whether they are in the isolated Domain I or in the context of the full-length protein (Fig. 5 ▶). No resonances were observed with significant chemical shift differences in the isolated domain versus the full-length protein. These NMR results provide evidence that the IF2N domain in the N-terminal region of Domain I does not have contacts with any of the other domains within IF2-1, and is therefore a structurally independent domain.
Resonances were assigned for only about 15 residues past amino acid 51 within the spectra of the full-length IF2-1; most of these resonances probably appear in heavily overlapping regions of the 2-D and 3-D spectra, or are unobservable due to unfavorable line widths. Assignments for D95 are indicated in Figure 5 ▶; also, the side chain Hɛ1 proton of Trp 215 was easily assigned because this is the only tryptophan residue in Domains I–III.
Homonuclear 2-D 1H-1H NOESY and TOCSY spectra of Domains I–II, II, and II–III were acquired (not shown) and revealed very few NOE cross peaks other than those already assigned to Domain I. This observation suggests that Domains II and III are not folded into globular structures, at least when they are isolated from the context of the full-length protein. NMR can potentially be used to further investigate the structural features of the combined Domains I–III, although, with a total molecular weight of over 40 kD, this would present a very challenging problem.
Dynamics of Domain I within the context of full-length IF2-1
15N relaxation rate data (T1, T2, and 15N-1H NOE) were acquired and analyzed with the purpose of further characterizing the motions within the N-terminal region of IF2-1. 15N relaxation rates were measured using 2-D 15N-1H correlated spectra of the full-length IF2-1, where resonances of 31 backbone amide nitrogens of residues in the range of 5–96 are resolved. The relaxation rate data were acquired at 20°C and 30°C with similar results. The 30°C relaxation rate data are summarized in Figure 6 ▶ and compared with data for the isolated Domain I, obtained in a previous study, also at 30°C (Laursen et al. 2003). Within full-length IF2-1, residues 5–50 (the IF2N domain) have strikingly uniform values of T1, T2, and the heteronuclear NOE, averaging 0.62 sec, 0.12 sec, and 0.56, respectively (Fig. 6 ▶). The relaxation rate data were interpreted using the Model-Free approach of Lipari and Szabo (1982), implemented in the program Modelfree 4.15 (Palmer et al. 1991; Mandel et al. 1995). The motional model that best accounts for the observed relaxation rate data is a simple isotropic diffusion model, where the protein is assumed to have an approximately spherical shape, and its tumbling is described by a single global rotational correlation time τm equal to 7.8 ns. In comparison, a rotational correlation time τm of 6.7 ns was obtained for the first 50 residues of IF2-1 when they are in the context of the isolated Domain I (Laursen et al. 2003), indicating that the rotation of the first 50 residues of IF2-1 is only slightly restricted by being attached to the full-length 97-kD protein. Order parameters (S2) for the individual amide 15N nuclei within the first 50 residues of IF2-1 were found to be uniform, averaging 0.86, a value typical of well-ordered structure, and similar to the order parameters averaging 0.81 for the same residues in the context of the isolated Domain I.
Figure 6.
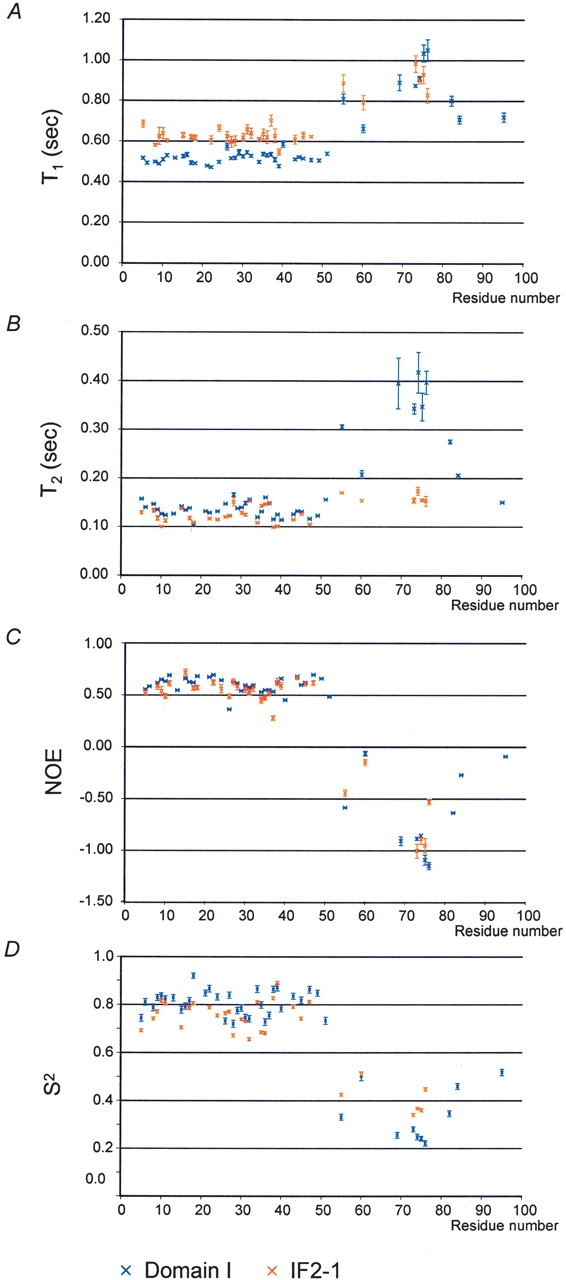
Summary of the nuclear relaxation rate data for full-length IF2-1 and the isolated Domain I. (A) T1 relaxation times. (B) T2 relaxation times. (C) Observed values for the 15N-1H heteronuclear NOE for the amide nitrogen atoms along the backbone. (D) Final optimized order parameters S2 determined for Domain I in the isolated form and in the context of the full-length IF2-1, obtained using the observed nuclear relaxation data shown in A, B and C. Data for the isolated Domain I are shown in blue, and data for Domain I within the context of IF2-1 are shown in red. Vertical bars indicate the uncertainty in the values.
Relaxation rate data was also obtained for 10 of the residues between 55 and 96 of IF2-1. These residues have much less uniform relaxation rates than the first 50 residues, and, unlike the first 50 residues, have negative NOE values (Fig. 6 ▶). These relaxation data are best modeled by significantly lower-order parameters than the first 50 residues, indicating a region that is significantly more flexible in solution. The modeling of the relaxation rate data for these residues also required an additional term (te) describing fast (picosecond) timescale movement; this term averaged 147 and 120 psec for the residues within the isolated full-length IF2-1 and the isolated Domain I, respectively. This region of the structure was also found to be flexible when it is in the context of the isolated Domain I, rather than the full-length IF2-1. As can be seen in Figure 6 ▶, several of the residues (between 70 and 77) have significantly shorter T2 relaxation times in the full-length protein than in the isolated Domain I; these differences in T2 are most consistent with differences in the time scales of rapid motions, rather than a difference of ordered versus disordered structure.
The only 15N resonance within Domain II to be specifically assigned is that of the Trp 215 side chain; this is the only tryptophan residue within Domain II. Relaxation rate data for Trp 215 indicate that it is mobile and disordered, moving independently of the folded domains.
Discussion
The complementary methods of CD and NMR were used to characterize various fragments of IF2-1, to gain new insight into the structure and dynamic properties of the three N-terminal domains of the translation factor. The NMR data all strongly support a model where the 50-residue most N-terminal domain (IF2N) is well ordered and connected to the more C-terminal regions of IF2-1 by a flexible linker. Consistent with this model, the 15N relaxation rate data indicate that residues between 55 and 96 are flexible, in the context of full-length IF2-1 as well as in the isolated 157-residue Domain I. The CD data indicate the presence of a substantial amount of helical structure in the N-terminal Domains I–III; temperature-dependence data indicates that this structure loses a significant fraction of its helical character at moderate temperatures, above 35°C to 40°C. A lack of NOE data attributable to Domains II and III suggests that the helical content of these regions may be molten globule in structure. These conclusions for Domains II–III drawn from the NMR and CD results are consistent, because a protein with the fundamental properties of a molten globule can give rise to CD signals in the far UV region (180–250 nm) typical of helix, while exhibiting poor NMR chemical shift dispersion (Kelly and Price 2000).
Biochemical data support a model with a substantial flexible and hence very accessible structure in the N-terminal region of IF2-1 (excluding the well-ordered and compact 50-residue IF2N domain). The OmpT protease cuts at least three places in Domains I–II, whereas the C-terminal Domain III–VI is stable against degradation by this same protease (Steffensen et al. 1994). The same pattern is seen for the blood coagulation factor Xa protease, which cleaves the N-terminal Domains I–III, whereas Domain IV–VI is stable against proteolytic degradation by the protease (B.S. Laursen, R.F. Andersen, H.P. Sorensen, K.K. Mortensen, and H.U. Sperling-Petersen, unpubl.). Domain I–II of IF2 is also a highly antigenic region with several epitopes, whereas the C-terminal region of the native IF2-1 protein has only a few epitopes (Mortensen et al. 1998). The presence of several epitopes and high sensitivity to proteases in the N-terminal region of IF2 indicates a structure with highly exposed residues as compared to the more compact structure of the C-terminal region of IF2, which has only a few epitopes and is more stable against proteolytic degradation.
The presence of flexible or disordered structure within the C-terminal region of Domain I and Domains II–III of IF2-1 does not indicate that these regions are functionally unimportant. Partially unfolded protein in solution is not a rare phenomenon. Despite earlier views that a 3-D structure is a prerequisite for protein function, many proteins have now been found to display functions requiring intrinsic disorder (Uversky et al. 2000; Dunker et al. 2002; Gunasekaran et al. 2003). Examples of proteins that are unstructured in solution can be found among some of the ribosomal proteins, which have globular regions as well as long extensions that penetrate into the ribosome but are unstructured in solution. Extreme cases are proteins S14 and L39e that are devoid of a globular structure, and unstructured in solution, but acquire well-defined structure within the context of the ribosome (Ramakrishnan and Moore 2001). It may not be likely that the flexible regions of IF2 penetrate into the ribosome upon binding as the ribosomal proteins do, but it is quite possible that the protein does undergo a disorder-to-order transition and become properly folded when binding to the ribosome or other possible ligands, such as the initiator fMet-tRNAfMet or certain mRNAs with which this region is known to interact (Laursen et al. 2002b).
The features of structurally well-ordered N- and C-terminal domains connected by an extended linker are reminiscent of features previously observed in other translation-associated proteins, such as ribosomal protein L19 (Biou et al. 1995; Ramakrishnan and Moore 2001) and translation initiation factor IF3 (Hua and Raleigh 1998a,b). In the case of IF3, the similarity is more striking, in that the extended region connecting the two domains is flexible in solution and has a substantial helical content that decreases with temperature (Hua and Raleigh 1998a,b), as is observed in here for the full-length IF2-1.
The G-domain (Domain IV in E. coli IF2) is in most bacteria and plastids preceded by a low complexity region (second part of Domain I to Domain III in E. coli) that links it to the IF2N domain. The length and sequence of this low complexity region varies widely between species. This may lead to the conclusion that this region is not important for the function of IF2, however, from earlier studies it is known that the region is involved in binding to the 30S ribosomal subunit, and also interacts with the infB mRNA, and hence is involved in important functions of the factor (Moreno et al. 1998; Laursen et al. 2002b). The present data indicate that this region is quite flexible. We may speculate that this flexibility is an important property of IF2 in the highly dynamic process of translation initiation. Because the IF2N domain is linked to the rest of IF2 by a flexible linker, it might be able to span a much longer distance to reach its interaction partner than previously thought.
The new information regarding the structure and dynamics of the N-terminal region of IF2 provided by this study will aid in future characterization of the functions of the conserved IF2N domain of IF2 and of the interaction between the N-terminal domains of IF2 and the ribosome.
Materials and methods
Protein cloning and expression
Domain I of IF2 (with a sequence corresponding to the first 157 residues of IF2-1) was expressed and purified as previously described (Laursen et al. 2003). The DNA coding for seven other fragments of IF2-1 was amplified by PCR using DNA from E. coli K12 as a template. DNA fragments encoding Domains I, II, I–II, and II–III of IF2-1 were inserted into the pET15b expression vector (Novagen), whereas the fragment encoding Domain I–III was inserted into the pBAD/MYC-His A expression vector (Invitrogen). Fragments encoding IF2-1, IF2-2, and IF2 Domain III–VI were inserted into the pET24d expression vector (Novagen). The IF2 Domain III–VI fragment was fused with a N-terminal histidine tag, whereas all other IF2 fragments were untagged. DNA sequencing confirmed the insertion of the correct infB fragments into each of the vectors. The proteins were expressed in BL21(DE3) cells for the pET15b constructs, BL21(DE3) pLysS for the pET24d constructs, and BL21 for the pBAD/Myc-His construct (all strains were from Novagen). Cells were grown in 2 × TY medium (16 g/L tryptone, 10 g/L yeast extract, 5 g/L NaCl at pH 7.5) for samples with the natural abundance of isotopes, and M9 minimal medium containing 0.6 g/L [15N] ammonium chloride (Cambridge Isotope Laboratories) as the only significant nitrogen source for the sample uniformly enriched with 15N. The growth medium was supplemented with 100 mg/L ampicillin when using the pET15b or the pBAD/Myc-His expression vectors, and 50 mg/L kanamycin when using the pET24d vector. The medium was further supplemented with 34 mg/L chloramphenicol when using the BL21(DE3) pLysS expression strain. In all cases, protein expression was induced when the cells reached an OD550 of 0.6–0.8; induction was with 0.1 mM IPTG for the pET expression systems and 0.2% arabinose for the pBAD expression system. The cells were allowed to grow for 5 h at 37°C after induction and finally harvested by centrifugation, washed in 0.9% NaCl, pelleted, and stored at −20°C.
Protein purification
All proteins were purified to at least 95% homogeneity as judged by SDS-PAGE after the final purification step. Samples of the IF2-1 and IF2-2 proteins were purified as described in Mortensen et al. (1991). Purification of the Domain III–VI protein fragment was carried out using an IMAC procedure as described in Sørensen et al. (2003).
Purification of Domains II, I–II, I–III, and II–III
Cell pellets were resuspended in 2.5 mL of buffer A (50 mM HEPES at pH 7.6, 10 mM MgCl2, 1 mM dithiothreitol (DTT), 0.1 mM phenylmethylsulfonylfluoride, 15 mM NaN3) per gram of cells. The cells were opened by sonication, and the solution centrifuged at 250,000g for 75 min. The supernatant was loaded onto an anion exchange column (Q-sepharose HP Amersham Biosciences) and bound protein eluted with a 0–500 mM NaCl gradient. Fractions containing the protein of interest were pooled and further chromatographed using a cation exchange column (SP-sepharose HP, Amersham Biosciences). Bound protein was eluted with a 0–500 mM NaCl gradient. This procedure gave very pure protein for Domains II, I–III, and II–III. An additional gel filtration step using a 1-m-long column (Aca54, BioSepra) was required for purification of the construct containing Domain I–II. The identities of all the purified proteins were confirmed by Western immunoblotting using monoclonal antibodies as described in Mortensen et al. (1998). The proteins were stored at −20°C in buffer A containing 100 mM NaCl and 50% glycerol. Protein samples were exhaustively dialyzed against the buffer used in the assays as described below just before use.
Circular dichroism spectroscopy
CD spectra were recorded at the UV1 photobiology beamline at the Institute for Storage Ring Facilities at Aarhus University, Denmark, using synchrotron radiation provided by the ASTRID storage ring. Spectra of all fragments shown in Figure 1 ▶ were recorded using an open 0.1-mm Hellwa suprisil quartz cell. The data were acquired using three consecutive scans where the signal was averaged for 5 sec in each scan. Each scan was performed using 1-nm intervals in the range 178–260 nm. The temperature dependence of the CD signal for each sample was determined using spectra recorded from 5°C–70°C in 5°C steps. Samples were allowed to equilibrate at each temperature for 10 min before data acquisition. Spectra were recorded in 10 mM phosphate (pH 7.5); however, the buffer contained an additional 100 mM NaF for the IF2 fragments corresponding to Domains II, II–III, III–VI, full-length IF2-1, and IF2-2, as required to keep the protein soluble. The concentration of each of the protein samples was determined using quantitative amino acid analysis, and the CD data were converted to represent mean residue ellipticity. Additional CD spectra of Domain I–III and IF2-1 were recorded at pH 6.0 and 7.5, and with varying concentrations of MgCl2 and NaF in order to detect any dependence on buffer conditions as described in the results section.
Secondary structure content in the protein fragments
The relative content of secondary structure elements in each protein fragment was determined from the CD data using the programs SELCON (Sreerama and Woody 1993), CONTINLL (Provencher and Glockner 1981), K2D (Andrade et al. 1993), CDSSTR, and VARSLC (Compton and Johnson 1986; Manavalan and Johnson 1987) at the Dichroweb Server (Lobley and Wallace 2001; Lobley et al. 2002). The server provides seven different reference sets, which were used for all the programs (with the exception of VARSLC and K2D, which do not use reference sets). The results were very consistent for all the programs and reference sets. The values in Table 1 indicate an average of all valid results from all programs. The validity of a result was determined for each program and reference set as described (Lobley and Wallace 2001; Lobley et al. 2002). The uncertainties reported for the values in Table 1 are based on the reference set with the highest deviation from the average.
NMR spectroscopy
NMR spectra were recorded at 20°C and 30°C using a 500-MHz Varian Inova spectrometer equipped with a triple-resonance probe and z-axis pulsed-field gradient. NMR samples of the IF2-1, IF2-2, and Domains II, II–III, and III–VI typically contained 10–20 mg of the protein and 10 mM phosphate (pH 6.0) in 90% H2O/10% D2O, 1 mM MgCl2, 100 mM NaCl, and 0.33 g/L NaN3. Samples of Domains I, I–II, and I–III were in the same buffer but without NaCl. Standard 1-D 1H spectra and 2-D 1H-1H NOESY and 1H-1H TOCSY spectra provided an indication of whether each protein was folded. A 2-D HSQC-TROSY spectrum (Meissner et al. 1998) and 3-D 15N-resolved HSQC-TOCSY and 15N-resolved 1H-1H HSQC-NOESY spectra were acquired for the 97-kD IF2-1 protein.
15N relaxation rates were measured for the resolved and assigned resonances of the full-length IF2-1 protein, for comparison with those of the isolated Domain I. The 15N T1 and T2 relaxation times and the 15N-1H NOE were measured using pulse sequences that feature gradient selection and sensitivity enhancement and pulses for minimizing saturation of the solvent water (Farrow et al. 1994). Six 2-D data sets with relaxation delays of 10, 260, 510, 760, 1010, and 1260 msec were acquired for the T1 relaxation measurements, and six 2-D data sets were acquired with relaxation delays of 29, 58, 87, 116, 145, and 174 msec for the T2 relaxation measurements; in each case the relaxation delay between the acquisition of each free induction decay was 3 sec. The spectra for measuring the 15N-1H NOE were acquired with either a 5-sec delay between each free induction decay or a 1-sec delay followed by a 4-sec-long series of 120° nonselective 1H pulses. The T1 and T2 data were fitted to a single exponential decay function of the form I = I0e− t/Td, in which I is the intensity of the signal at time t, I0 is the intensity at time t = 0, and Td is the decay constant T1 or T2. Rotational correlation times and order parameters were calculated using Modelfree 4.15 (Palmer et al. 1991; Mandel et al. 1995) as described (Lillemoen and Hoffman 1998). Parameters were derived using a simple isotropic diffusion model.
Acknowledgments
We thank Søren Vrønning Hoffman at the Department of Storage Ring Facilities, University of Aarhus, for help on the CD apparatus. Hans Peter Sørensen, Department of Molecular Biology, University of Aarhus, provided the plasmids for expression of IF2-1, IF2-2, and the IF2 Domains I–III and III–VI. We also thank Claus Oxvig and Lene Kristensen, Department of Molecular Biology, University of Aarhus, for performing the quantitative amino acid analysis. This work was funded by grants from the Familien Hede Nielsens Fund and the Danish Natural Science Research Council (grants no. 9901722 and 51-00-0263) to H.U.S.P. D.W.H.’s contributions to this work were supported by grant F-1353 from the Welch Foundation.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Abbreviations
CD, circular dichroism
DTT, dithiothreitol
IF, initiation factor
IF2N, IF2 N-terminal
NMR, nuclear magnetic resonance
PDB, Protein Data Bank
1-D, one-dimensional
2-D, two-dimensional
3-D, three-dimensional
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.03337604.
References
- Andrade, M.A., Chacon, P., Merelo, J.J., and Moran, F. 1993. Evaluation of secondary structure of proteins from UV circular dichroism spectra using an unsupervised learning neural network. Protein Eng. 6 383–390. [DOI] [PubMed] [Google Scholar]
- Biou, V., Shu, F., and Ramakrishnan, V. 1995. X-ray crystallography shows that translational initiation factor IF3 consists of two compact α/β domains linked by an α-helix. EMBO J. 14 4056–4064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boelens, R. and Gualerzi, C.O. 2002. Structure and function of bacterial initiation factors. Curr. Protein Pept. Sci. 3 107–119. [DOI] [PubMed] [Google Scholar]
- Compton, L.A. and Johnson Jr., W.C. 1986. Analysis of protein circular dichroism spectra for secondary structure using a simple matrix multiplication. Anal. Biochem. 155 155–167. [DOI] [PubMed] [Google Scholar]
- Dunker, A.K., Brown, C.J., Lawson, J.D., Iakoucheva, L.M., and Obradovic, Z. 2002. Intrinsic disorder and protein function. Biochemistry 41 6573–6582. [DOI] [PubMed] [Google Scholar]
- Farrow, N.A., Muhandiram, R., Singer, A.U., Pascal, S.M., Kay, C.M., Gish, G., Shoelson, S.E., Pawson, T., Forman-Kay, J.D., and Kay, L.E. 1994. Backbone dynamics of a free and phosphopeptide-complexed Src homology 2 domain studied by 15N NMR relaxation. Biochemistry 33 5984–6003. [DOI] [PubMed] [Google Scholar]
- Gualerzi, C.O., Brandi, L., Caserta, E., Teana, A.L., Spurio, R., Tomsic, J., and Pon, C.L. 2000. Translation initiation in bacteria. In The ribosome: Structure, function, antibiotics, and cellular interactions (eds. R.A. Garret et al.), pp. 477–494. ASM Press, Washington, DC.
- Gunasekaran, K., Tsai, C.J., Kumar, S., Zanuy, D., and Nussinov, R. 2003. Extended disordered proteins: Targeting function with less scaffold. Trends Biochem. Sci. 28 81–85. [DOI] [PubMed] [Google Scholar]
- Howe, J.G. and Hershey, J.W. 1982. Immunochemical analysis of molecular forms of protein synthesis initiation factors in crude cell lysates of Escherichia coli. Arch. Biochem. Biophys. 214 446–451. [DOI] [PubMed] [Google Scholar]
- Hua, Y. and Raleigh, D.P. 1998a. Conformational analysis of the interdomain linker of the central homology region of chloroplast initiation factor IF3 supports a structural model of two compact domains connected by a flexible tether. FEBS Lett. 433 153–156. [DOI] [PubMed] [Google Scholar]
- ———. 1998b. On the global architecture of initiation factor IF3: A comparative study of the linker regions from the Escherichia coli protein and the Bacillus stearothermophilus protein. J. Mol. Biol. 278 871–878. [DOI] [PubMed] [Google Scholar]
- Kelly, S.M. and Price, N.C. 2000. The use of circular dichroism in the investigation of protein structure and function. Curr. Protein Pept. Sci. 1 349–384. [DOI] [PubMed] [Google Scholar]
- Koradi, R., Billeter, M., and Wuthrich, K. 1996. MOLMOL: A program for display and analysis of macromolecular structures. J. Mol. Graph. 14 51–55. [DOI] [PubMed] [Google Scholar]
- Laursen, B.S., Siwanowicz, I., Larigauderie, G., Hedegaard, J., Ito, K., Nakamura, Y., Mortensen, K.K., and Sperling-Petersen, H.U. 2002a. Characterization of mutations in the GTP-binding domain of IF2 resulting in cold-sensitive growth of Escherichia coli. J. Mol. Biol. 326 543–551. [DOI] [PubMed] [Google Scholar]
- Laursen, B.S., Steffensen, S.A., Hedegaard, J., Moreno, J.M., Mortensen, K.K., and Sperling-Petersen, H.U. 2002b. Structural requirements of the mRNA for intracistronic translation initiation of the enterobacterial infB gene. Genes Cells 7 901–910. [DOI] [PubMed] [Google Scholar]
- Laursen, B.S., Mortensen, K.K., Sperling-Petersen, H.U., and Hoffman, D.W. 2003. A conserved structural motif at the N-terminus of bacterial translation initiation factor IF2. J. Biol. Chem. 278 16320–16328. [DOI] [PubMed] [Google Scholar]
- Lillemoen, J. and Hoffman, D.W. 1998. An investigation of the dynamics of ribosomal protein L9 using heteronuclear NMR relaxation measurements. J. Mol. Biol. 281 539–551. [DOI] [PubMed] [Google Scholar]
- Lipari, G. and Szabo, A. 1982. Model-free approach to the interpretation of nuclear magnetic resonance relaxation in macromolecules. 1. Theory and range of validity. J. Am. Chem. Soc. 104 4546–4559. [Google Scholar]
- Lobley, A. and Wallace, B.A. 2001. Dichroweb: A website for the analysis of protein secondary structure from circular dichroism spectra. Biophysical J. 80 373. [DOI] [PubMed] [Google Scholar]
- Lobley, A., Whitmore, L., and Wallace, B.A. 2002. DICHROWEB: An interactive website for the analysis of protein secondary structure from circular dichroism spectra. Bioinformatics 18 211–212. [DOI] [PubMed] [Google Scholar]
- Mandel, A.M., Akke, M., and Palmer III, A.G. 1995. Backbone dynamics of Escherichia coli ribonuclease HI: Correlations with structure and function in an active enzyme. J. Mol. Biol. 246 144–163. [DOI] [PubMed] [Google Scholar]
- Meissner, A., Schulte-Herbrueggen, T., Briand, J., and Sørensen, O.W. 1998. Double spin-state-selective coherence transfer. Application for two dimensional selection of multiplet components with long transverse relaxation times. Mol. Phys. 95 1137–1142. [Google Scholar]
- Moreno, J.M., Kildsgaard, J., Siwanowicz, I., Mortensen, K.K., and Sperling-Petersen, H.U. 1998. Binding of Escherichia coli initiation factor IF2 to 30S ribosomal subunits: A functional role for the N-terminus of the factor. Biochem. Biophys. Res. Commun. 252 465–471. [DOI] [PubMed] [Google Scholar]
- Moreno, J.M., Drskjotersen, L., Kristensen, J.E., Mortensen, K.K., and Sperling-Petersen, H.U. 1999. Characterization of the domains of E. coli initiation factor IF2 responsible for recognition of the ribosome. FEBS Lett. 455 130–134. [DOI] [PubMed] [Google Scholar]
- Mortensen, K.K., Nyengaard, N.R., Hershey, J.W., Laalami, S., and Sperling-Petersen, H.U. 1991. Superexpression and fast purification of E. coli initiation factor IF2. Biochimie 73 983–989. [DOI] [PubMed] [Google Scholar]
- Mortensen, K.K., Hajnsdorf, E., Regnier, P., and Sperling-Petersen, H.U. 1995. Improved recombinant tandem expression of translation initiation factor IF2 in RNASE E deficient E. coli cells. Biochem. Biophys. Res. Commun. 214 1254–1259. [DOI] [PubMed] [Google Scholar]
- Mortensen, K.K., Kildsgaard, J., Moreno, J.M., Steffensen, S.A., Egebjerg, J., and Sperling-Petersen, H.U. 1998. A six-domain structural model for Escherichia coli translation initiation factor IF2. Characterisation of twelve surface epitopes. Biochem. Mol. Biol. Int. 46 1027–1041. [DOI] [PubMed] [Google Scholar]
- Nyengaard, N.R., Mortensen, K.K., Lassen, S.F., Hershey, J.W., and Sperling-Petersen, H.U. 1991. Tandem translation of E. coli initiation factor IF2 beta: Purification and characterization in vitro of two active forms. Biochem. Biophys. Res. Commun. 181 1572–1579. [DOI] [PubMed] [Google Scholar]
- Palmer III, A.G., Rance, M., and Wright, P.E. 1991. Intramolecular motions of a zinc finger DNA-binding domain from Xfin characterized by proton-detected natural abundance carbon-13 heteronuclear NMR spectroscopy. J. Am. Chem. Soc. 113 4371–4380. [Google Scholar]
- Provencher, S.W. and Glockner, J. 1981. Estimation of globular protein secondary structure from circular dichroism. Biochemistry 20 33–37. [DOI] [PubMed] [Google Scholar]
- Ramakrishnan, V. 2002. Ribosome structure and the mechanism of translation. Cell 108 557–572. [DOI] [PubMed] [Google Scholar]
- Ramakrishnan, V. and Moore, P.B. 2001. Atomic structures at last: The ribosome in 2000. Curr. Opin. Struct. Biol. 11 144–154. [DOI] [PubMed] [Google Scholar]
- Roll-Mecak, A., Cao, C., Dever, T.E., and Burley, S.K. 2000. X-ray structures of the universal translation initiation factor IF2/eIF5B: Conformational changes on GDP and GTP binding. Cell 103 781–792. [DOI] [PubMed] [Google Scholar]
- Sacerdot, C., Vachon, G., Laalami, S., Morel-Deville, F., Cenatiempo, Y., and Grunberg-Manago, M. 1992. Both forms of translational initiation factor IF2 (α and β) are required for maximal growth of Escherichia coli. Evidence for two translational initiation codons for IF2 β. J. Mol. Biol. 225 67–80. [DOI] [PubMed] [Google Scholar]
- Sreerama, N. and Woody, R.W. 1993. A self-consistent method for the analysis of protein secondary structure from circular dichroism. Anal. Biochem. 209 32–44. [DOI] [PubMed] [Google Scholar]
- Steffensen, S., Poulsen, A.B., Mortensen, K.K., Korsager, B., and Sperling-Petersen, H.U. 1994. Protease activity of outer membrane protein OmpT in clinical E. coli isolates—Studies using translation initiation factor IF2 as target protein. Biochem. Mol. Biol. Int. 34 1245–1251. [PubMed] [Google Scholar]
- Steffensen, S.A., Poulsen, A.B., Mortensen, K.K., and Sperling-Petersen, H.U. 1997. E. coli translation initiation factor IF2—An extremely conserved protein. Comparative sequence analysis of the infB gene in clinical isolates of E. coli. FEBS Lett. 419 281–284. [DOI] [PubMed] [Google Scholar]
- Sørensen, H.P., Hedegaard, J., Sperling-Petersen, H.U., and Mortensen, K.K. 2001. Remarkable conservation of translation initiation factors: IF1/eIF1A and IF2/eIF5B are universally distributed phylogenetic markers. IUBMB Life 51 321–327. [DOI] [PubMed] [Google Scholar]
- Sørensen, H.P., Laursen, B.S., Mortensen, K.K., and Sperling-Petersen, H.U. 2002. Bacterial translation initiation—Mechanism and regulation. Recent Res. Dev. Biophys. Biochem. 2 243–270. [Google Scholar]
- Sørensen, H.P., Sperling-Petersen, H., and Mortensen, K.K. 2003. A favorable solubility partner for the recombinant expression of streptavidin. Protein Expr. Purif. (in press). [DOI] [PubMed]
- Uversky, V.N., Gillespie, J.R., and Fink, A.L. 2000. Why are “natively unfolded” proteins unstructured under physiologic conditions? Proteins 41 415–427. [DOI] [PubMed] [Google Scholar]