

Abstract
The type II transmembrane serine protease dipeptidyl peptidase IV (DPPIV), also known as CD26 or adenosine deaminase binding protein, is a major regulator of various physiological processes, including immune, inflammatory, nervous, and endocrine functions. It has been generally accepted that glycosylation of DPPIV and of other transmembrane dipeptidyl peptidases is a prerequisite for enzyme activity and correct protein folding. Crystallographic studies on DPPIV reveal clear N-linked glycosylation of nine Asn residues in DPPIV. However, the importance of each glycosylation site on physiologically relevant reactions such as dipeptide cleavage, dimer formation, and adenosine deaminase (ADA) binding remains obscure. Individual Asn→Ala point mutants were introduced at the nine glycosylation sites in the extracellular domain of DPPIV (residues 39–766). Crystallographic and biochemical data demonstrate that N-linked glycosylation of DPPIV does not contribute significantly to its peptidase activity. The kinetic parameters of dipeptidyl peptidase cleavage of wild-type DPPIV and the N-glycosylation site mutants were determined by using Ala-Pro-AFC and Gly-Pro-pNA as substrates and varied by <50%. DPPIV is active as a homodimer. Size-exclusion chromatographic analysis showed that the glycosylation site mutants do not affect dimerization. ADA binds to the highly glycosylated β-propeller domain of DPPIV, but the impact of glycosylation on binding had not previously been determined. Our studies indicate that glycosylation of DPPIV is not required for ADA binding. Taken together, these data indicate that in contrast to the generally accepted view, glycosylation of DPPIV is not a prerequisite for catalysis, dimerization, or ADA binding.
Keywords: dipeptidyl peptidase IV, DPPIV, CD26, glycosylation, enzyme activity, ADA, adenosine deaminase binding protein, serine protease
Dipeptidyl peptidase IV (DPPIV), also called adenosine deaminase binding protein (ADAbp) or CD26, is a 220-kD homodimeric, type II transmembrane glycoprotein, widely expressed on the surface of a variety of epithelial, endothelial, and lymphoid cell types (Abbott et al. 1994; De Meester et al. 1999; Langer and Ansorge 2002). A soluble form is also found in plasma (Iwaki-Egawa et al. 1998). The enzyme regulates various physiological processes by cleaving Xaa-Pro dipeptides from the N terminus of regulatory peptides, including many chemokines, neuropeptides, and peptide hormones (Mentlein 1999). Specific inhibition of the DPPIV dipeptidyl peptidase function increases the half-life of the incretin hormones glucagon-like peptide-1 and gastric inhibitory polypeptide, both involved in insulin secretion (for review, see Drucker 2003). Clinical evidence shows that specific DPPIV inhibitors lower blood glucose levels in type II diabetics (Ahren et al. 2002).
DPPIV is a member of the s9b family of serine peptidases. Members of the family include DPPIV, fibroblast activation protein (FAP; Sun et al. 2002), dipeptidyl peptidase 8 (DPP8; Abbott et al. 2000b), dipeptidyl peptidase 9 (DPP9; Olsen and Wagtmann 2002), dipeptidyl peptidase 10 (DPP10; Qi et al. 2003), and dipeptidyl peptidase 6 (DPP6; Wada et al. 1992). The most studied are DPPIV and FAP, sharing a sequence identity of 54% (Levy et al. 1999). Both are integral membrane proteins and require dimerization for catalytic activity. It has been reported that glycosylation of both enzymes is a prerequisite for enzyme activity (Loch et al. 1992; Fan et al. 1997; Sun et al. 2002). On the other hand, the enzyme activity of DPP8 is similar to DPPIV and FAP, but DPP8 is a nonglycosylated, cytoplasmic protein that is catalytically active as a monomer (Abbott et al. 2000b).
Based on sequence alignments, members of the s9b family of serine peptidases are thought to share a similar three-dimensional structure (Abbott et al. 2000a). Our group and others have recently solved the crystal structure of the extracellular domain of DPPIV (Engel et al. 2003; Hiramatsu et al. 2003; Rasmussen et al. 2003). The enzyme contains an eight-bladed β-propeller domain and a peptidase domain with an α/β hydrolase fold. An α-helix insertion in blade four of the β-propeller domain introduces a negatively charged pocket in the active site and determines its specificity for N-terminal peptide cleavage.
DPPIV contains nine potential N-glycosylation sites mainly present in the β-propeller domain of the molecule. Our crystallographic data show that all nine glycosylation sites are used in DPPIV (K. Aertgeerts, S. Ye, M. Tennant, B. Collins, J. Rogers, B. Sang, R. Skene, D. Webb, and G. Prasad, unpubl.). Previous studies have shown that inhibition of primary N-glycosylation of DPPIV using tunicamycin reduced the biological stability of the molecule dramatically (Loch et al. 1992). Furthermore, Fan et al. (1997) showed that glycosylation of Asn 321 is important for correct protein folding and protein trafficking. However, the contribution of each glycosylation site in DPPIV on physiologically relevant reactions such as dipeptide cleavage, dimer formation, and ADA binding still remains obscure. Therefore, we overexpressed the extracellular domain of wild-type DPPIV and introduced individual Asn→Ala point mutants at each of the nine glycosylation sites. Here we report that in contrast to the generally accepted hypothesis that glycosylation of DPPIV is required for its function, lack of glycosylation at each specific site in DPPIV did not alter its catalytic activity, dimer formation and ADA binding capacity.
Results
Structural analysis of glycosylation
Nine potential N-glycosylation sites were predicted in the extracellular domain of DPPIV by using the Prosite pattern N-{P}-[ST]-{P}; these include N85, N92, N150, N219, N229, N281, N321, N520, and N685 (human DPPIV numbering). Seven of the nine predicted glycosylation sites are conserved in human, rat, mouse, bovine, and pig DPPIV (Fig. 1A ▶). Compared with human DPPIV, rat and mouse DPPIV do not have a potential glycosylation site at residue 281; bovine and pig DPPIV lack a site at position 520. During our crystal structure determination of the extracellular domain of human DPPIV (residues 39–766), we confirmed that all nine N-glycosylation sites are used after expression and purification from Hi5 insect cells (Fig. 1B,C ▶). Rasmussen et al. (2003) and Hiramatsu et al. (2003) observed carbohydrates at seven and five of the nine potential N-linked glycosylation positions of DPPIV, respectively, when expressed by using a baculovirus system. In the crystal structure of DPPIV, obtained from pig kidney, electron density was observed for carbohydrates at 6 of the 10 potential sites (Engel et al. 2003). The crystal structure of DPPIV is composed of an eight-bladed β-propeller domain that is packed against a catalytic domain that has an α/β-hydrolase fold. Seven of the nine glycosylation sites are located in the β-propeller domain (Fig. 1B ▶). None of the sugars that were modeled in the electron density maps are in close contact to the catalytic triad or are observed within the active-site pocket of the enzyme. N85 and N219 are located at the entrance of the central pore that is formed by the β-propeller domain. (Fig. 1C ▶). None of the glycosylation sites are in close contact to a disulfide bond.
Figure 1.


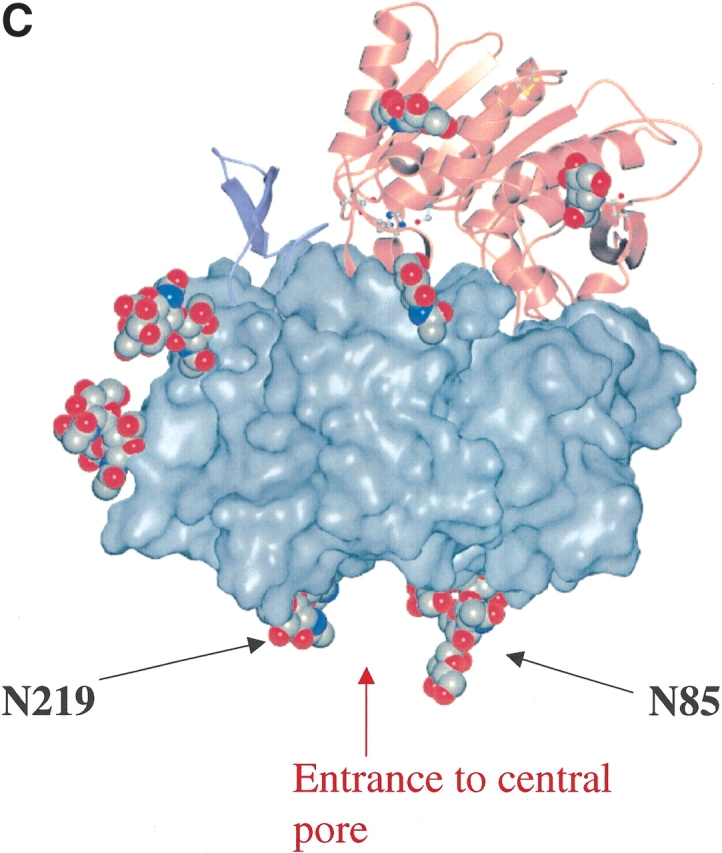
(A) Sequence alignment of human, rat, mouse, bovine, and pig DPPIV. The potential glycosylation sites predicted by using the Prosite pattern N-{P}-[ST]-{P} are indicated in bold and underlined; an asterisk indicates the catalytic triad residues: Ser 630, Asp 708, and His 740. (B) Ribbon diagram of the three-dimensional structure of DPPIV. The glycosylation sites are labeled, and the sugar molecules that were modeled into the electron density maps are shown in green as ball-and-stick representations. Seven of the nine glycosylation sites were found in the β-propeller domain of the molecule (blue), and two glycosylation sites were observed in the α/β-hydrolase domain (red) of the molecule. Disufide bonds are shown in yellow. Residues (L294, L340, V341, A342, and R343) that have been reported to play a role in ADA binding (Abbott et al. 1999) are labeled in red and shown in gold as ball-and-stick representations. Residues of the catalytic triad are shown in blue as ball-and-stick representations. (C) Three-dimensional structure of DPPIV with space-filled representation of β-propeller domain. Glycosylation sites N219 and N85 are labeled and are located at the entrance of the central pore of the β-propeller domain. The central pore (approximate diameter of 15 Å) creates an entrance to the active-site of the molecule.
In general, the major processed N-glycan produced in insect cells is the paucimannosidic structure, Man3GlcNAc2-N-Asn. The longest ordered N-glycosyl chain was modeled at position N229, containing Man2GlcNAc2-N-Asn. All nine glycosylation sites could contain Man3GlcNAc2-N-Asn. However, less complex sugars could be modeled in the calculated electron density maps, most likely because the terminal sugar moieties are not well ordered or because the longer sugar chains are heterogeneously distributed between molecules in the crystal.
To study the impact of N-glycosylation of DPPIV on physiologically important processes such as catalytic activity, dimer formation, and ADA binding, we generated nine point mutations in which asparagine at each glycosylation site was individually mutated to alanine. Alanine substitutions were chosen to avoid introducing new charge interactions or hydrogen bonds and to minimize unfavorable steric contacts.
Expression and purification of wild-type DPPIV and DPPIV glycosylation mutants
The extracellular domain of human DPPIV (residues 39–766) was overexpressed in baculovirus-infected insect cells as a soluble secreted protein. Two different insect cell lines were used to explore possible differences in glycosylation. Similarly, we expressed DPPIV N-glycosylation mutants by using recombinant baculoviruses that contained the cDNA encoding the desired mutants. From 100 mL culture medium, we obtained ~1 mg purified protein for wild-type DPPIV and a similar amount for most of the glycosylation mutants. An ~10-fold reduction in yield was found for N229A, N281A, and N321A. To check whether correct protein folding and protein trafficking into the media were affected after expression of the glycosylation mutants, we analyzed the media and cell lysates of 2 mL monolayer cultures for the presence of DPPIV protein. We could only detect the presence of wild-type DPPIV and the N-glycosylation mutants in the media and not in the cell lysates by SDS-PAGE (data not shown). We could detect enzymatic activity toward Ala-Pro-AFC in the purified cell lysates; however, it was not significantly different for the N-glycosylation mutants compared with the wild-type form of the enzyme. These data indicate that the individual removal of a glycosylation site in DPPIV does not interfere with correct protein folding and protein trafficking into the media in insect cells.
Purified samples of wild-type DPPIV and the N-glycosylation mutants gave one major band on a SDS-PAGE at ~95 kD (Fig. 2 ▶). The primary amino acid sequence has a calculated molecular mass of 86 kD, indicating the presence of posttranslational modifications that account for ~10% of the total mass of the protein.
Figure 2.
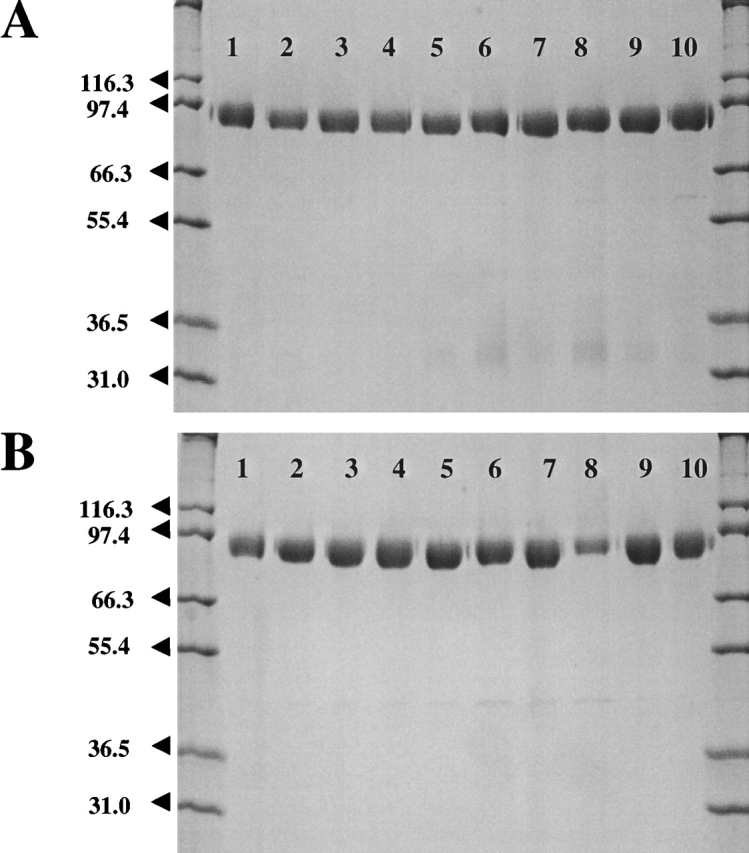
SDS-PAGE of purified wild-type DPPIV and DPPIV glycosylation mutants. (A) Mutants expressed in Sf9 insect cells. (B) Mutants expressed in Hi5 insect cells. Approximately 1000 ng purified material was loaded on a 4% to 20% gradient gel for each sample. (Lane 1) Wild-type DPPIV; (lane 2) N85A; (lane 3) N92A; (lane 4) N150A; (lane 5) N219A; (lane 6) N229A; (lane 7) N281A; (lane 8) N321A; (lane 9) N520A; (lane 10) N685A.
DPP4 glycosylation mutants are catalytically active
The extracellular domain of wild-type DPPIV (residues 39–766) expressed in Sf9 insect cells has dipeptidyl peptidase activity and has a catalytic efficiency of 1.3 × 106 M−1sec−1 and 1.2 × 105 M−1sec−1 for cleavage of the fluorogenic substrate Ala-Pro-AFC and the chromogenic substrate Gly-Pro-pNA, respectively (Table 1A). The enzymatic constants of the nine N-linked glycosylation mutants of DPPIV for enzymatic cleavage of the fluorogenic substrate Ala-Pro-AFC varied by <50%, compared with that of wild-type DPPIV. Similarly, individual kcat and Km values for all DPPIV variants toward the synthetic fluorogenic substrate varied by less than a factor of two compared with the wild-type enzyme (Table 1A). These data indicate that N-linked glycosylation of DPPIV does not significantly modulate the dipeptidyl peptidase activity of the enzyme toward small synthetic substrates. Comparable conclusions were made when the chromogenic substrate Gly-Pro-pNA was used and when the wild-type protein and the mutants were expressed in Hi5 insect cells (Table 1B). Interestingly, complete deglycosylation of DPPIV obtained by the addition of tunicamycin during protein expression resulted in active DPPIV (data not shown), consistent with the conclusion that glycosylation is not a prerequisite for enzyme activity.
Table 1A.
Kinetic constants for cleavage of the fluorogenic substrate Ala-Pro-AFC and the chromogenic substrate Gly-Pro-pNA by wild-type DPPIV and DPPIV glycosylation mutants (proteins expressed in Sf9 insect cells)
| DPPIV mutant | Km (μM) | kcat (sec−1) | kcat/Km (M−1sec−1) | Relative kcat/Km |
| Kinetic constants for cleavage of Ala-Pro-AFC | ||||
| Wild-type DPPIV | 14 | 18 | 1.3 × 106 | 1.0 |
| DPPIV-N85A | 15 | 16 | 1.1 × 106 | 0.85 |
| DPPIV-N92A | 14 | 18 | 1.3 × 106 | 1.0 |
| DPPIV-N150A | 14 | 18 | 1.3 × 106 | 1.0 |
| DPPIV-N219A | 15 | 18 | 1.2 × 106 | 0.92 |
| DPPIV-N229A | 14 | 12 | 9.1 × 105 | 0.70 |
| DPPIV-N281A | 14 | 15 | 1.1 × 106 | 0.85 |
| DPPIV-N321A | 14 | 9 | 6.5 × 105 | 0.50 |
| DPPIV-N520A | 14 | 13 | 8.9 × 105 | 0.68 |
| DPPIV-N685A | 13 | 19 | 1.4 × 106 | 1.1 |
| Kinetic constants for cleavage of Gly-Pro-pNA | ||||
| Wild-type DPPIV | 170 | 20 | 1.2 × 105 | 1.0 |
| DPPIV-N85A | 155 | 16 | 1.1 × 105 | 0.92 |
| DPPIV-N92A | 147 | 19 | 1.3 × 105 | 1.1 |
| DPPIV-N150A | 143 | 19 | 1.3 × 105 | 1.1 |
| DPPIV-N219A | 144 | 19 | 1.3 × 105 | 1.1 |
| DPPIV-N229A | 151 | 13 | 8.8 × 104 | 0.73 |
| DPPIV-N281A | 159 | 17 | 1.1 × 105 | 0.92 |
| DPPIV-N321A | 156 | 10 | 6.5 × 104 | 0.54 |
| DPPIV-N520A | 142 | 14 | 9.6 × 104 | 0.80 |
| DPPIV-N685A | 152 | 21 | 1.4 × 105 | 1.2 |
Table 1B.
Kinetic constants for cleavage of the fluorogenic substrate Ala-Pro-AFC and the chromogenic substrate Gly-Pro-pNA by wild-type DPPIV and DPPIV glycosylation mutants (proteins expressed in Hi5 insect cells)
| DPPIV mutant | Km (μM) | kcat (s−1) | kcat/Km (M−1 s−1) | Relative kcat/Km |
| Kinetic constants for cleavage of Ala-Pro-AFC | ||||
| Wild-type DPPIV | 16 | 23 | 1.4 × 106 | 1.0 |
| DPPIV-N85A | 16 | 27 | 1.7 × 106 | 1.2 |
| DPPIV-N92A | 17 | 31 | 1.9 × 106 | 1.4 |
| DPPIV-N150A | 17 | 37 | 2.2 × 106 | 1.6 |
| DPPIV-N219A | 19 | 42 | 2.2 × 106 | 1.6 |
| DPPIV-N229A | 15 | 40 | 2.7 × 106 | 1.9 |
| DPPIV-N281A | 20 | 40 | 2.0 × 106 | 1.4 |
| DPPIV-N321A | 18 | 34 | 1.8 × 106 | 1.3 |
| DPPIV-N520A | 17 | 36 | 2.1 × 106 | 1.5 |
| DPPIV-N685A | 15 | 30 | 1.9 × 106 | 1.4 |
| Kinetic constants for cleavage of Gly-Pro-pNA | ||||
| Wild-type DDPIV | 138 | 24 | 1.7 × 105 | 1.0 |
| DPPIV-N85A | 149 | 30 | 2.0 × 105 | 1.2 |
| DPPIV-N92A | 143 | 34 | 2.4 × 105 | 1.4 |
| DPPIV-N150A | 151 | 40 | 2.6 × 105 | 1.5 |
| DPPIV-N219A | 160 | 45 | 2.8 × 105 | 1.6 |
| DPPIV-N229A | 145 | 43 | 3.0 × 105 | 1.8 |
| DPPIV-N281A | 168 | 43 | 2.5 × 105 | 1.5 |
| DPPIV-N321A | 146 | 35 | 2.4 × 105 | 1.4 |
| DPPIV-N520A | 131 | 35 | 2.7 × 105 | 1.6 |
| DPPIV-N685A | 142 | 32 | 2.2 × 105 | 1.3 |
Effect of glycosylation of DPPIV on ADA binding
It has been reported that five residues located within the cysteine-rich domain of human DPPIV (L294, L340, V341, A342, and R343) are required for binding to ADA (Dong et al. 1997; Abbott et al. 1999). In the crystal structure, these residues form two hydrophobic patches that are solvent exposed and are located in the highly glycosylated β-propeller domain of the molecule. The first hydrophobic patch is formed by residue L294; the second is composed of residues L340, V341, and A342 (Fig. 1B ▶). It is worth noting that the glycosylated residues N229 and N321 are in close proximity (~15 Å) to the proposed ADA-binding site. Moreover, murine and rat DPPIV lack a glycosylation site on N281 (human DPPIV numbering) and are unable to bind ADA. It has been hypothesized from the crystal structure of porcine DPPIV that glycosylation of N279 (corresponds to N281 in the human sequence) might control ADA binding (Engel et al. 2003). No biochemical data have been reported to date on the effect of glycosylation of DPPIV on ADA binding. We measured the kinetics of wild-type DPPIV/ADA binding by using SPR measurements. Figure 3A ▶ shows a sensorgram illustrating the high-affinity binding of ADA (concentration range, 3.1 to 800 nM) to the extracellular domain of wild-type DPPIV. The association rate constant (ka) was 1.79 ± 0.014 × 105 M−1 sec−1, and the dissociation rate constant (kd) was 6.95 ± 0.55 × 10−5 sec−1. The apparent equilibrium dissociation constant (KD = kd/ka) was 0.40 ± 0.0042 nM. We also titrated the binding of the extracellular domain of wild-type DPPIV to ADA by high-performance size-exclusion chromatography (HP-SEC) using DPPIV/ADA molar ratios ranging from 2 : 1 to 1 : 2 (Fig. 3B ▶). A clear shift in retention time from 8.94 (DPPIV only) to 8.51 (DPPIV/ADA complex) was observed when equimolar concentrations of the two proteins or a molar excess of ADA over DPPIV were used. An intermediate complex was observed when a molar DPPIV/ADA ratio of 2 : 1 was used. Similarly, we used HP-SEC to analyze binding of the N-glycosylation mutants to ADA. Retention times were measured for the mutants first in the absence of ADA and then after incubation with a twofold molar excess of ADA for 2 h at 37°C. The effects of the point mutations on binding to ADA are illustrated in Table 2. Compared with wild-type DPPIV, we observed a similar shift in the retention time when the DPPIV glycosylation mutants were incubated with ADA. These data indicate that the mutations do not abolish binding to ADA, indicating that glycosylation of DPPIV is not a requirement for ADA binding.
Figure 3.


(A) BIAcore analysis: sensorgram showing binding of ADA to DPPIV. DPPIV was immobilized on a CM5 Biacore sensor chip, and ADA was injected at various concentrations (indicated on figure) over the DPPIV surface. Specific signal for binding of ADA to DPPIV was measured by subtracting response units (RU) obtained from no-protein control. Data shown are from one of four experiments. (B) Titration of binding of wild-type DPPIV to ADA by HP-SEC. Purified samples of wild-type DPPIV and ADA were incubated for 2 h at 37°C using DPPIV/ADA ratios of 2 : 1 (dashed line), 1 : 1 (dotted line), and 1 : 2 (dashed/dotted line). ADA binding was confirmed by a shift in the retention time from 8.94 (DPPIV only, full line) to 8.51 (DPPIV/ADA complex, dotted and dashed/dotted lines). An intermediate complex with a retention time of 8.67 was observed when a DPPIV/ADA ratio of 2 : 1 was used (dashed line).
Table 2.
HP-SEC analysis showing binding of wild-type DPPIV and DPPIV mutants to ADA
| DPPIV mutant | tretention no ADA | tretention + ADA | Δ(tretention no ADA − tretention + ADA) |
| Proteins expressed in Sf9 cells | |||
| Wild-type | 8.99 | 8.54 | 0.45 |
| N85A | 9.12 | 8.51 | 0.61 |
| N92A | 9.08 | 8.60 | 0.48 |
| N150A | 8.94 | 8.52 | 0.42 |
| N219A | 9.09 | 8.50 | 0.59 |
| N229A | 8.98 | 8.49 | 0.49 |
| N281A | 8.97 | 8.49 | 0.48 |
| N321A | 9.00 | 8.49 | 0.51 |
| N520A | 9.01 | 8.50 | 0.51 |
| N685A | 8.95 | 8.48 | 0.47 |
| Proteins expressed in Hi5 cells | |||
| Wild-type | 8.85 | 8.42 | 0.43 |
| N85A | 8.89 | 8.45 | 0.44 |
| N92A | 8.87 | 8.43 | 0.44 |
| N150A | 8.83 | 8.41 | 0.42 |
| N219A | 8.90 | 8.47 | 0.43 |
| N229A | 8.85 | 8.40 | 0.45 |
| N281A | 8.87 | 8.41 | 0.46 |
| N321A | 8.91 | 8.42 | 0.49 |
| N520A | 8.86 | 8.41 | 0.45 |
| N685A | 8.79 | 8.40 | 0.39 |
Effect of glycosylation on homodimer formation
DPPIV is enzymatically active as a homodimer, whereas the monomeric form of the enzyme has no activity (Bednarczyk et al. 1991; De Meester et al. 1992; Gorrell et al. 2001). HP-SEC analysis of wild-type DPPIV revealed that the protein elutes as a dimer with an apparent molecular weight (MW) of ~160 kD. As was also observed by others, the mobility of the dimer is greater than expected from the estimated MW of the monomer by SDS-PAGE (95 kD; Ajami et al. 2003). All the glycosylation mutants elute as a dimer from HP-SEC, indicating that the glycosylation sites do not individually contribute to DPPIV dimerization. The observation that glycosylation is not required for dimerization was already suggested from the crystal structure of DPPIV. The asymmetric unit is composed of two homodimers in which the monomers are related by a twofold axis. None of the glycosylated residues occupy the dimer interface (K. Aertgeerts, S. Ye, M. Tennant, B. Collins, J. Rogers, B. Sang, R. Skene, D. Webb, and G. Prasad, unpubl.).
Discussion
It has been reported that DPPIV (Fan et al. 1997) and other dipeptidyl peptidases such as DPP2 (Chiravuri et al. 2000) and FAP (Sun et al. 2002) depend on glycosylation for their enzymatic activity and/or biological stability. DPPIV has nine potential N-linked glycosylation sites. During previous studies, we crystallized wild-type human DPPIV and collected a 2.1 Å X-ray diffraction data set (K. Aertgeerts, S. Ye, M. Tennant, B. Collins, J. Rogers, B. Sang, R. Skene, D. Webb, and G. Prasad, unpubl.). We could clearly observe from the calculated electron-density maps that all nine glycosylation sites were used in DPPIV. The exact contribution of each individual glycosylation site to physiologically relevant reactions such as enzyme activity, ADA-binding, and dimer formation has not previously been addressed.
We overexpressed the extracellular domain of DPPIV in Sf9 and Hi5 insect cells and analyzed the role of N-glycosylation through individual site-directed mutagenesis of all nine glycosylation sites to alanine. The complexity of insect protein N-glycosylation pathways is intermediate between those of Saccharomyces cerevisiae and mammalian cells (for review, see Jarvis 1997; Varki et al. 1999). In contrast to mammalian cells, insect cells are unable to naturally produce complex, terminally sialylated N-glycans. When we expressed DPPIV in Hi5 and Sf9 insect cells, ~10% of the total protein molecular mass originated from oligosaccharide side chains, whereas a value of ~12% to 20% was observed when DPPIV was obtained from rat hepatocytes (Loch et al. 1992; Porwoll et al. 1998). Despite the generally known differences in glycosylation and postbiosynthetic N-glycan modifications observed between mammalian cells and insect cells, we measured kinetic parameters for dipeptide cleavage of Ala-Pro-AFC that are comparable to the ones reported for purified DPPIV obtained from mammalian cells (Dobers et al. 2002). Taken together, these data indicate that differently glycosylated DPPIV obtained from different expression systems do not affect dipeptidyl peptidase activity. Similarly, SPR analysis of ADA binding to DPPIV expressed in insect cells revealed binding constants that are comparable to the values reported for ADA binding to rabbit kidney DPPIV (Richard et al. 2002).
Nine glycosylation mutants, each removing one N-glycosylation site, expressed well and were secreted into the culture medium. Fan et al. (1997) showed that the individual replacement of residues N85, N321, and N686 with Gln decreased the biological stability and processing of rat DPPIV in CHO cells. We did observe an ~10-fold reduction in expression of N321A, but we could not detect a corresponding increase of insoluble material inside the cell. Differences in results between our data and those reported by Fan et al. could be attributable to the different expression systems used or to intrinsic differences between the rat and human versions of the enzyme.
Tunicamycin treatment of insect cells infected with the virus encoding wild-type DPPIV significantly reduced expression of the protein. This is probably due to the inhibition of protein synthesis, which occurs simultaneously with the inhibition of protein glycosylation. However, the addition of tunicamycin resulted in correctly folded and active DPPIV, as observed by DPPIV activity in the medium, whereas DPPIV activity was not found in the medium of noninfected cells. The low level of expression in the presence of tunicamycin did not allow for quantitation of the protein. Therefore, detailed reaction kinetics, dimerization, and ADA binding were not measured for the nonglycosylated enzyme.
It was apparent from the crystal structure that none of the glycosylated residues occupy or are in close contact to the active site pocket of the enzyme and therefore would not be expected to contribute directly to the substrate specificity of the enzyme. However, it is generally known that enzyme activity is highly associated with the structural integrity of the protein, which in turn can be associated with correct N-glycosylation. This indirect effect could lie in the correct dimerization of the protein because DPPIV is active as a homodimer, and no activity can be found for the monomeric form of the enzyme (Püschel et al. 1982; Bednarczyk et al. 1991; De Meester et al. 1992; Gorrell et al. 2001). Evidence to indicate that glycosylation of DPPIV could contribute to dimer formation comes from the observation that DPP8, which is 27% identical to DPPIV and shares the same substrate specificity, is a nonglycosylated protein and catalytically active as a monomer (Abbott et al. 2000b). Our data clearly indicate that compared with wild-type DPPIV, the glycosylation mutants do not change the catalytic efficiency of DPPIV for dipeptide cleavage and do not hamper dimer formation. The latter observations are not totally unexpected because we observed from the crystal structure of DPPIV that none of the glycosylation sites are in close contact to the dimer interface.
DPPIV binds ADA to the T-cell surface and thereby protects the T cell from adenosine-mediated inhibition of proliferation. ADA binding does not block the enzymatic activity of DPPIV. The binding site for ADA on DPPIV has been determined by site-directed mutagenesis and epitope mapping to reside in the glycosylation-rich β-propeller domain of the enzyme (Dong et al. 1997; Abbott et al. 1999). These studies showed that residues L294, L340, V341, A342, and R343 in DPPIV are required for ADA binding. The effect of glycosylation of DPPIV on ADA binding has not been studied previously. From the crystal structure of DPPIV, we observed that the residues required for ADA binding form two hydrophobic patches on the surface of the molecule and are in close proximity to the glycosylation sites N229 and N321 (Fig. 1B,C ▶). HP-SEC analysis demonstrates that none of the nine glycosylation mutants are deficient in ADA binding. These data clearly indicate that glycosylation of DPPIV is not involved in ADA binding and indicate that glycosylation does not interfere with DPPIV-mediated catalysis in lymphocyte biology.
In conclusion, we analyzed the involvement of each individual N-linked glycosylation site of DPPIV in enzyme catalysis, dimerization, and ADA binding. Our results indicate that in contrast to the general accepted hypothesis that glycosylation is a prerequisite for enzymatic activity of dipeptidyl peptidases, none of the nine glycosylation sites are involved in physiologically relevant reactions that we measured in vitro.
Materials and methods
Construction of wild-type DPPIV and DPPIV glycosylation mutants
The cDNA encoding human DPPIV was isolated by PCR from spleen cDNA (Clontech), and the extracellular domain (residues 39–766) was cloned into the SmaI site of a modified pFastBacHTb vector (Invitrogen). The final construct contains a baculovirus gp67 signal peptide followed by a His6 tag fused to the coding sequence corresponding to residues 39–766 of DPPIV. This construct was used to express soluble DPPIV designated as wild type. This construct was also used as a template for the PCR-based site-directed mutagenesis reactions to generate single amino acid substitutions at the nine N-linked glycosylation sites in DPPIV. The following oligonucleotide primers were used to introduce the desired mutation (mutation(s) underlined):
N85A-5′-TATTCAATGCTGAATATGGAGCAAGCTCAGTTTTCTTGGAGAA-3′, N92A-5′-GCTCAGTTTTCTTGGAGGCCAGTACATTTGATGAGTTTGG-3′, N150A-5′-TTACAGAAGAGAGGATTCCAGCAAACACACAGTGGGTCACATG-3′, N219A-5′-CTGCTCTGTGGTGGTCTCCAGCAGGCACTTTTTTAGCATATGC-3′, N229A-5′-TTTTAGCATATGCCCAATTTGCAGACACAGAAGTCCCACTTAT-3′, N281A-5′-ACTCTCTCAGCTCAGTCACCGCAGCAACTTCCATACAAATCAC-3′, N321A-5′-AGTGGCTCAGGAGGATTCAGGCATATTCGGTCATGGATATTTG-3′, N520A-5′-AACTGGACTTCATTATTTTGGCAGAAACAAAATTTTGGTATCA-3′, and N685A-5′-GACAACCTTGACCATTACAGAGCTTCAACAGTCATGAGCAGAGC-3′.
In each case the presence of the desired mutation was verified by DNA sequencing.
Expression and purification of wild-type DPPIV and DPPIV point mutants
Individual recombinant baculovirus constructs, each incorporating a distinct DPPIV N-glycosylation site mutation, were generated by transposition using the Bac-to-Bac system (GIBCO-BRL). High-titer viral stocks were generated by infection of Spodoptera frugiperda (Sf9) cells, and the expression of recombinant protein was carried out by infection of a 100 mL suspension culture of Sf9 and a 100 mL suspension culture of Trichoplusia ni (Hi5) cells (3 × 106 cells/mL; GIBCO-BRL) with a multiplicity of infection of five to 10. The culture media was harvested 72 h (for Sf9) or 48 h (for Hi5) postinfection and concentrated to 50 mL; 0.5 mL of Ni-NTA agarose beads (Qiagen) was added to the concentrated media and incubated with shaking for 2 h at 4°C. The beads were separated from the media by centrifugation and washed with 50 column volumes of wash buffer (50mM NaH2PO4, 300 mM NaCl, 20 mM imidazole, and 0.05% Tween 20 at pH 8.0). The protein was eluted with five column volumes of buffer (50 mM NaH2PO4, 300 mM NaCl, 250 mM imidazole, and 0.05% Tween 20 at pH 8.0) and then concentrated to a final volume of 600 μL by using an Apollo high-performance concentrator (Orbital Biosciences). Protein was quantitated by using a BCA assay according to the manufacturer’s protocol (Pierce).
Lectin-binding studies were performed by using the DIG Glycan differentiation kit according to the manufacturer’s protocol (Roche Molecular Diagnostics).
Analysis of protein folding and trafficking
To test the effect of glycosylation on protein secretion into the media, recombinant baculovirus was used to infect confluent monolayers of Sf9 and Hi5 cells at a multiplicity of infection of 5 to 10. Cultures were incubated for 72 h at 27°C. The culture media was harvested, and 75 μL of Ni-NTA magnetic beads was added to the samples and incubated for 2 h at 4°C with shaking on a rotary shaker. The infected cells were washed with phosphate-buffered saline and lysed with 10 mM Tris (pH 7.5), 130 mM NaCl, and 1% Triton X-100. The clarified cell lysates were gently rocked for 1 h at 4°C after the addition of 35 μL of Ni-NTA magnetic beads (Qiagen). The beads that were added to the media and to the cell lysates were removed from the solution by a magnetic separator and washed three times with 500 μL wash buffer (50 mM NaH2PO4, 300 mM NaCl, 20 mM imidazole, and 0.05% Tween 20 at pH 8.0). The bound material was eluted with 35 μL elution buffer (50 mM NaH2PO4, 300 mM NaCl, 250 mM imidazole, and 0.05% Tween 20 at pH 8.0). Samples were analyzed by SDS-PAGE. The catalytic activity was measured by adding 10 μL purified protein sample to 80 μL assay buffer (25 mM glycine, 25 mM acetic acid, 25 mM MES, 75 mM Tris, and 0.1 M NaCl at pH 8.0) in a 96-well black plate; 10 μL of 100 μM H-Ala-Pro-7-amido-4-trifluromethylcoumarin (Ala-Pro-AFC; Bachem) was added to the reaction, and release of free AFC was monitored by using a 395-nm excitation/530-nm emission filter set (SpectraMax Gemini XS, Molecular Devices).
Expression and purification of ADA
The cDNA of full-length human ADA was obtained from fetal kidney cDNA (GIBCO-BRL) and ligated into the SmaI site of a modified pFastBacHTb vector (Invitrogen). The final construct contains the cDNA encoding full-length ADA fused to an N-terminal His6-tag. Insect cell expression of ADA was done by using the Bac-to-Bac Baculovirus expression system (GIBCO-BRL) in a manner similar to that described for the expression of DPPIV. After expression, 100 mL of infected Sf9 and Hi5 cells were lysed with 25 mL lysis buffer (10 mM Tris at pH 7.5, 130 mM NaCl, and 1% TritonX-100). The samples were centrifuged, and the clarified cell lysates were applied to Ni-NTA Agarose beads (Qiagen) for binding at 4°C with shaking on a rotary shaker for 2 h; 1 mL of bead suspension was used for 25 mL of cell extract. Further purification of the sample is identical to that described for DPPIV point mutants.
Determination of catalytic activity
The determination of the catalytic constants for dipeptide cleavage was performed by using either a fluorescent or a colorimetric assay. For the fluorescent assay, 0.1 nM enzyme was mixed with 3 to 400 μM Ala-Pro-AFC (Bachem) in 20 mM Tris (pH 7.4), 20 mM KCl, 0.1 mg/mL BSA, and 1% DMSO in a 96-well half area plate and monitored kinetically at Ex400nm and Em505nm using Molecular Devices SpectraMax Gemini. For the colorimetric assay, 1 nM enzyme was mixed with 15 to 2000 μM H-Gly-Pro-p-nitro aniline (Gly-Pro-pNA; Sigma) in 20 mM Tris (pH 7.4), 20 mM KCl, 0.1 mg/mL BSA, and 1% DMSO in a 96-well half area plate and monitored kinetically at 405 nm using the Molecular Devices SpectraMax Plus. Assays were performed in duplicate for each sample. MDL data analysis toolbox was used for analysis of Michaelis-Menten kinetics.
Surface plasmon resonance experiment
Surface plasmon resonance (SPR) using the Biacore 3000 (Biacore AB) system was used to perform a detailed kinetic analysis of the interaction of DPPIV with ADA, both expressed in Hi5 insect cells. DPPIV was immobilized on a CM5 chip by using primary amine-coupling chemistry. In each experiment, ~50 to 100 response units (RU) were immobilized on flow cell 2 and flow cell 4. No-protein controls were run in flow cell 1 and 3. ADA was injected at various concentrations (ranging from 3.1 to 800 nM) in duplicate over the DPPIV surfaces for 1 min using an automated method. The running buffer was HBS-EP (0.01 M HEPES at pH 7.4, 0.15 M NaCl, 3 mM EDTA, 0.005% surfactant P20 at pH 7.4), and the detection temperature was 25°C. Binding data were then fit with BIA Evaluation software (Biacore) using a 1 : 1 model to obtain the kinetic and affinity constants.
Determination of homodimer formation and ADA binding
Homodimer formation and binding of DPPIV to ADA was analyzed by HP-SEC. The binding stoichiometry of DPPIV to ADA was initially titrated by incubating 0.3 nmole DPPIV together with 0, 0.15, 0.3, and 0.6 nmole ADA, respectively, for 2 h at 37°C. Subsequently the samples were injected onto a BioSep S3000 column (300 × 4.6 mm, Phenomenex) set up in a Summit HPLC system managed by Chromeleon software (both Dionex). The flow was 0.35 mL/min, and detection was at 280 nm. The column was calibrated by using a gel filtration standard from Biorad. Apparent molecular weights of the individual proteins and of the complex were calculated from the log relation between molecular weight and retention time. Similarly, we analyzed the binding of the DPPIV glycosylation mutants first in the absence of ADA and then in the presence of a twofold molar excess of ADA incubated for 2 h at 37°C.
Inhibition of glycosylation in the presence of tunicamycin
The optimal tunicamycin concentration was obtained by the analysis of expression and cell death in the presence of 1, 10, 25, and 50 μg/mL of the antibiotic. Optimal expression time was obtained by analysis of expression at different time points (72, 80, 95, 103, and 140 h). Finally, a suspension culture of 200 mL Sf9 and 200 mL Hi5 insect cells (3 × 106 cells/mL) was infected with recombinant virus encoding the extracellular domain of DPPIV. Tunicamycin (1 μg/mL) was added 20 min postinfection. The culture was harvested after 140 h at 27°C. The culture media was collected after centrifugation and was incubated with 700 μL Probond resin (Invitrogen) for 2 h at 4°C on a rotating platform. The cell pellet was washed with phosphate-buffered saline, and the cells were solubilized by the addition of 12 mL lysis buffer (10 mM Tris at pH 7.5, 130 mM NaCl, 1% Triton X-100). The clear cell lysate was incubated with 700 μL of Probond resin (Invitrogen) for 2 h at 4°C on a rotating platform. Further purification of the samples was identical to that described for DPPIV point mutants.
Acknowledgments
We thank Keith Wilson, Steve Kaldor, David Weitz, and Wendell Wierenga for careful reading of the manuscript and helpful suggestions.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.03352504.
References
- Abbott, C.A., Baker, E., Sutherland, G.R., and McCaughan, G.W. 1994. Genomic organization, exact localization, and tissue expression of the human CD26 (dipeptidyl peptidase IV) gene. Immunogenetics 40 331–338. [DOI] [PubMed] [Google Scholar]
- Abbott, C.A., McCaughan, G.W., Levy, M.T., Church, W.B., and Gorell, M.D. 1999. Binding to human dipeptidyl peptidase IV by adenosine deaminase and antibodies that inhibit ligand binding involves overlapping, discontinuous sites on a predicted β propeller domain. Eur. J. Biochem. 266 798–810. [DOI] [PubMed] [Google Scholar]
- Abbott, C.A., Yu, D., McCaughan, G.W., and Gorrell, M.D. 2000a. Post-proline-cleaving peptidases having DP IV like enzyme activity: Post-proline peptidases. Exp. Med. Biol. 477 103–109. [DOI] [PubMed] [Google Scholar]
- Abbott, C.A., Yu, D.M., Woollatt, E., Sutherland, G.R., McCaughan, G.W., and Gorrell, M.D. 2000b. Cloning, expression and chromosomal localization of a novel human dipeptidyl peptidase (DPP) IV homolog, DPP8. Eur. J. Biochem. 267 6140–6150. [DOI] [PubMed] [Google Scholar]
- Ahren, B., Simonsson, E., Larsson, H., Landin-Olsson, M., Torgeirsson, H., Jansson, P.A., Sandqvist, M., Bavenholm, P., Efendic, S., Eriksson, J.W., et al. 2002. Inhibition of dipeptidyl peptidase IV improves metabolic control over a 4-week study period in type 2 diabetes. Diabetes Care 25 869–875. [DOI] [PubMed] [Google Scholar]
- Ajami, K., Abbott, C.A., Obradovic, M., Gysbers, V., Kahne, T., McCaughan, G.W., and Gorrell, M.D. 2003. Structural requirements for catalysis, expression, and dimerization in the CD26/DPIV gene family. Biochemistry 42 694–701. [DOI] [PubMed] [Google Scholar]
- Bednarczyk, J.L., Carroll, S.M., Marin, C., and McIntyre, B.W. 1991. Triggering of the proteinase dipeptidyl peptidase IV (CD26) amplifies human T lymphocyte proliferation. J. Cell. Biochem. 46 206–218. [DOI] [PubMed] [Google Scholar]
- Chiravuri, M., Agarraberes, F., Mathieu, S.L., Lee, H., and Huber, B.T. 2000. Vesicular localization and characterization of a novel post-proline-cleaving aminodipeptidase, quiescent cell proline dipeptidase. J. Immunol. 165 5695–5702. [DOI] [PubMed] [Google Scholar]
- De Meester, I., Vanhoof, G., Hendriks, D., Demuth, H.U., Yaron, A., and Scharpe, S. 1992. Characterization of dipeptidyl peptidase IV (CD26) from human lymphocytes. Clin. Chim. Acta 210 23–34. [DOI] [PubMed] [Google Scholar]
- De Meester, I., Korom, S., Van Damme, J., and Scharpe, S. 1999. CD26: Let it cut or cut it down. Immunol. Today 20 367–375. [DOI] [PubMed] [Google Scholar]
- Dobers, J., Zimmermann-Kordmann, M., Leddermann, M., Schewe, T., Reutter, W., and Fan, H. 2002. Expression, purification, and characterization of human dipeptidyl peptidase IV/CD26 in Sf9 insect cells. Protein Expr. Purif. 25 527–532. [DOI] [PubMed] [Google Scholar]
- Dong, R.P., Tachibana, K., Hegen, M., Munakata, Y., Cho, D., Schlossman, S.F., and Morimoto, C. 1997. Determination of adenosine deaminase binding domain on CD26 and its immunoregulatory effect on T cell activation. J. Immunol. 159 6070–6076. [PubMed] [Google Scholar]
- Drucker, D.J. 2003. Therapeutic potential of dipeptidyl peptidase IV inhibitors for the treatment of type 2 diabetes. Expert Opin. Invest. Drugs 12 87–100. [DOI] [PubMed] [Google Scholar]
- Engel, M., Hoffmann, T., Wagner, L., Wermann, M., Heiser, U., Kiefersauer, R., Huber, R., Bode, W., Demuth, H.U., and Brandstetter, H. 2003. The crystal structure of dipeptidyl peptidase IV (CD26) reveals its functional regulation and enzymatic mechanism. Proc. Natl. Acad. Sci. 100 5063–5068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan, H., Meng, W., Kilian, C., Grams, S., and Reutter, W. 1997. Domain-specific N-glycosylation of the membrane glycoprotein dipeptidylpeptidase IV (CD26) influences its subcellular trafficking, biological stability, enzyme activity and protein folding. Eur. J. Biochem. 246 243–251. [DOI] [PubMed] [Google Scholar]
- Gorrell, M.D., Gysbers, V., and McCaughan, G.W. 2001. CD26: A multifunctional integral membrane and secreted protein of activated lymphocytes. Scan. J. Immunol. 54 249–264. [DOI] [PubMed] [Google Scholar]
- Hiramatsu, H., Kyono, K., Higashiyama, Y., Fukushima, C., Shima, H., Sugiyama, S., Inaka, K., Yamamoto, A., and Shimizu, R. 2003. The structure and function of human dipeptidyl peptidase IV, possessing a unique eight-bladed β-propeller fold. Biochem. Biophys. Res. Commun. 302 849–854. [DOI] [PubMed] [Google Scholar]
- Iwaki-Egawa, S., Watanabe, Y., Kikuya, Y., and Fujimoto, Y. 1998. Dipeptidyl peptidase IV from human serum: Purification, characterization, and N-terminal amino acid sequence. J. Biochem. 124 428–433. [DOI] [PubMed] [Google Scholar]
- Jarvis, D.L. 1997. Baculovirus expression vectors. The baculoviruses (ed. L.K. Miller), pp. 389–431. Plenium Press, New York.
- Langer, J. and Ansorge, S. 2002. Ectopeptidases: CD13/Aminopeptidase N and CD26/DipeptidylpeptidaseIV in medicine and biology. Kluwer/Plenum, New York.
- Levy, M.T., McCaughan, G.W., Abbott, C.A., Park, J.E., Cunningham, A.M., Muller, E., Rettig, W.J., and Gorrell, M.D. 1999. Fibroblast activation protein: A cell surface dipeptidyl peptidase and gelatinase expressed by stellate cells at the tissue remodelling interface in human cirrhosis. Hepatology 29 1768–1778. [DOI] [PubMed] [Google Scholar]
- Loch, N., Tauber, R., Becker, A., Hartel-Schenk, S., and Reutter, W. 1992. Biosynthesis and metabolism of dipeptidylpeptidase IV in primary cultured rat hepatocytes and Morris hepatoma 7777 cells. Eur. J. Biochem. 210 161–168. [DOI] [PubMed] [Google Scholar]
- Mentlein, R. 1999. Dipeptidyl-peptidase IV (CD26): Role in the inactivation of regulatory peptides. Regul. Pept. 85 9–24. [DOI] [PubMed] [Google Scholar]
- Olsen, C. and Wagtmann, N. 2002. Identification and characterization of human DPP9, a novel homologue of dipeptidyl peptidase IV. Gene 299 185–193. [DOI] [PubMed] [Google Scholar]
- Porwoll, S., Loch, N., Kannicht, C., Nuck, R., Grunow, D., Reutter, W., and Tauber, R. 1998. Cell surface glycoproteins undergo postbiosynthetic modification of their N-glycans by stepwise demannosylation. J. Biol. Chem. 273 1075–1085. [DOI] [PubMed] [Google Scholar]
- Püschel, G., Mentlein, R., and Heymann, E. 1982. Isolation and characterization of dipeptidyl peptidase IV from human placenta. Eur. J. Biochem. 126 359–365. [DOI] [PubMed] [Google Scholar]
- Qi, S.Y., Riviere, P.J., Trojnar, J., Junien, J.L., and Akinsanya, K.O. 2003. Cloning and characterization of dipeptidyl peptidase 10, a new member of an emerging subgroup of serine proteases. Biochem. J. 373 179–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen, H.B., Branner, S., Wiberg, F.C., and Wagtmann, N. 2003. Crystal structure of human dipeptidyl peptidase IV/CD26 in complex with a substrate analog. Nat. Struct. Biol. 10 19–25. [DOI] [PubMed] [Google Scholar]
- Richard, E., Alam, S.M., Arredondo-Vega, F.X., Patel, D.D., and Hershfield, M.S. 2002. Clustered charged amino acids of human adenosine deaminase comprise a functional epitope for binding the adenosine deaminase complexing protein CD26/dipeptidyl peptidase IV. J. Biol. Chem. 277 19720–19726. [DOI] [PubMed] [Google Scholar]
- Sun, S., Albright, C.F., Fish, B.H., George, H.J., Selling, B.H., Hollis, G.F., and Wynn, R. 2002. Expression, purification, and kinetic characterization of full-length human fibroblast activation protein. Protein Expr. Purif. 24 274–281. [DOI] [PubMed] [Google Scholar]
- Varki, A., Cummings, R., Esko, J., Freeze, H., Hart, G., and Marth, J. 1999. Essentials of glycobiology. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [PubMed]
- Wada, K., Yokotani, N., Hunter, C., Doi, K., Wenthold, R.J., and Shimasaki, S. 1992. Differential expression of two distinct forms of mRNA encoding members of a dipeptidyl aminopeptidase family. Proc. Natl. Acad. Sci. 89 197–201. [DOI] [PMC free article] [PubMed] [Google Scholar]