

Abstract
Protein interfaces are thought to be distinguishable from the rest of the protein surface by their greater degree of residue conservation. We test the validity of this approach on an expanded set of 64 protein–protein interfaces using conservation scores derived from two multiple sequence alignment types, one of close homologs/orthologs and one of diverse homologs/paralogs. Overall, we find that the interface is slightly more conserved than the rest of the protein surface when using either alignment type, with alignments of diverse homologs showing marginally better discrimination. However, using a novel surface-patch definition, we find that the interface is rarely significantly more conserved than other surface patches when using either alignment type. When an interface is among the most conserved surface patches, it tends to be part of an enzyme active site. The most conserved surface patch overlaps with 39% (± 28%) and 36% (± 28%) of the actual interface for diverse and close homologs, respectively. Contrary to results obtained from smaller data sets, this work indicates that residue conservation is rarely sufficient for complete and accurate prediction of protein interfaces. Finally, we find that obligate interfaces differ from transient interfaces in that the former have significantly fewer alignment gaps at the interface than the rest of the protein surface, as well as having buried interface residues that are more conserved than partially buried interface residues.
Keywords: interactions, binding, evolution, protein structure, sequence conservation
As structural genomics projects proceed, they are likely to yield structures of proteins that are functionally uncharacterized. Identification of active sites in enzymes and protein–protein binding sites in nonenzymatic proteins will be particularly important for elucidating function and designing inhibitors. Inhibiting protein–protein interactions with small molecules has proved particularly difficult due to their large size and lack of cavities (Toogood 2002; Gadek and Nicholas 2003). However, targeting the most critical residues may lead to improved inhibition of these interactions. Many of the residues that are critical for binding are likely to be evolutionarily conserved. Therefore, their potential impact in predicting protein–protein binding sites is an important question. Whereas there is general agreement that active/ligand binding sites are conserved across many different protein families (Grishin and Phillips 1994; Ouzounis et al. 1998; Bartlett et al. 2002), the importance of conservation is less clear for protein–protein interfaces (Grishin and Phillips 1994; Valdar and Thornton 2001b). Grishin and Phillips (1994) concluded that interface residues were only slightly more conserved than the rest of the protein sequence after examining five enzyme families. Valdar and Thornton (2001b) concluded that interface residues, particularly those completely buried in the interface, were more conserved than other surface-exposed residues after analyzing six homodimers. The distinguishing features of the latter study included the use of a similarity score rather than an identity score, the application of more robust statistical tests, and the comparison of interface residues relative to other contiguous surface patches. Although these studies are very valuable, the data sets used are small, and the results may not apply to all complexes, particularly those with heterodimeric or transient interfaces.
Nonetheless, several groups have successfully used conservation scores to predict protein–protein binding sites. Two independent groups (Elcock and McCammon 2001; Valdar and Thornton 2001a) conclude that conservation in combination with other factors can accurately discriminate genuine homodimers from crystal contacts. The majority of methods that predict protein–protein binding sites also use conservation scores (other approaches are discussed later). Those that map the conservation score to the three-dimensional structure are likely to be the most informative and include Evolutionary Trace (ET; Lichtarge et al. 1996), Consurf (Armon et al. 2001), Rate4Site (Pupko et al. 2002), and the method of Landgraf and Eisenberg (Landgraf et al. 2001). In cases in which a three-dimensional structure is unavailable, residues that are conserved for the entire family or a subfamily within the alignment are predicted to be functional (Casari et al. 1995; Livingstone and Barton 1996; Caffrey et al. 2000; Hannenhalli and Russell 2000). However, assessing the accuracy of these methods has been difficult and usually limited to a few experimentally characterized protein families. Furthermore, we are only aware of a few published experiments that confirm previously computed predictions (Stenmark et al. 1994; Bauer et al. 1999; Sowa et al. 2001).
The physical and chemical properties of protein–protein interactions have been studied on a large number of complexes by numerous groups (Chothia and Janin 1975; Argos 1988; Janin et al. 1988; Janin and Chothia 1990; Korn and Burnett 1991; Clackson and Wells 1995; Jones and Thornton 1996, 1997; Lijnzaad et al. 1996; Tsai et al. 1996, 1997a,b; Tsai and Nussinov 1997; Xu et al. 1997; Bogan and Thorn 1998; Larsen et al. 1998; Xu and Regnier 1998; Lo Conte et al. 1999; Jones et al. 2000; Sheinerman et al. 2000; Glaser et al. 2001; Chakrabarti and Janin 2002; Sheinerman and Honig 2002). In general, interfaces tend to be planar with an area that is often proportional to the total protein size (Jones and Thornton 1996). The residue composition usually differs for those complexes that are transient versus those that are obligate. This is probably due to the former relying more on salt bridges and hydrogen bonds, whereas the latter rely more on hydrophobic attractions (Jones and Thornton 1997; Lo Conte et al. 1999). There are also many examples of both geometric and electrostatic complementarity between the binding interfaces (Lawrence and Colman 1993; McCoy et al. 1997; Xu et al. 1997; Lo Conte et al. 1999; Sheinerman et al. 2000). Although the interface can be quite large, it was shown in some systems that only a small fraction of the residues contribute to the majority of the binding energy (Clackson and Wells 1995). Furthermore, these so-called hotspots of binding energy tend to have preferred residue types that often have a high degree of burial at the interface (Bogan and Thorn 1998). Interestingly, there is evidence (for 11 families) to suggest that there is a relationship between the enrichment of a residue type in a hotspot and the propensity of the corresponding residue to be conserved (Hu et al. 2000).
In this study, we examine the difference in conservation between the protein interface and the rest of the protein surface for a set of 64 protein–protein interfaces. As residue conservation depends on the choice of sequences aligned, we construct two multiple-sequence alignments (MSAs) for each protein using two different strategies. The first approach attempts to include closely related sequences, whereas the second includes a more diverse set of sequences. These MSAs are generally expected to contain orthologs and paralogs, respectively, and there are arguments for choosing either MSA type. Orthologs are expected to be almost identical in function, whereas a set of paralogs are expected to have undergone some evolutionary changes so that they can perform slightly different functions. However, nonfunctional residues are often conserved over short periods of evolutionary time, which is a source of noise that will be more prominent in orthologs. When the two approaches are examined and compared with each other, we find that the difference in conservation between the interface and the rest of surface is marginally (but not significantly) better in MSAs of diverse homologs than in MSAs of close homologs. Furthermore, we find that obligate and transient interfaces have different physico-chemical properties that influence their evolutionary rates.
Results
Nonredundant data set
The data set consists of 42 chains that form homodimers, 12 chains that form heterodimers, and 10 chains that form transient complexes as described in Table 1. As mentioned above, the MSAs of close homologs and diverse homologs are expected to contain orthologs or paralogs, respectively. A number of criteria were also used to remove distantly related or poorly aligned sequences (see Materials and Methods). Consequently, it is usually the case that only one of the chains is considered in the analysis, as the alignment for its binding partner was not satisfactory. The interface sizes ranged from 415 to 3568 Å2 for heterodimers, 550 to 4718 Å2 for homodimers, and 423 to 2361 Å2 for transient complexes. This suggests that transient interfaces are generally smaller than obligate interfaces, although it could be due to difficulties in crystallizing larger transient interfaces. Although an interface residue was defined if it had a ΔASA of 1% or more, the majority of residues (86%) lose more than 5% ASA upon complex formation. For each data set, none of the chains share significant sequence identity with the other chains (see Materials and Methods).
Table 1.
Protein interfaces used in the analysis
| Code | Protein | Species | Interface size | Residue number | Sequence number |
| Heterodimer | |||||
| 1allAB_A | phycobiliprotein allophycocyanin | Spirulina platensis | 1431 | 34 | 10,10 |
| 1hcgAB_A | coagulation factor X | Homo sapiens | 887 | 32 | 23,12 |
| 1lucAB_A | luciferase | Vibrio harveyi | 2055 | 52 | 19,8 |
| 1scuDE_E | succinyl-CoA synthetase | Escherichia coli | 1744 | 47 | 17,11 |
| 1tcoAB_B | calmodulin-dependent phosphatase | Bos taurus | 1909 | 55 | 12,12 |
| 1tcoBC_B | calmodulin-dependent phosphatase | Bos taurus | 415 | 12 | 12,12 |
| 1tcrAB_A | T cell receptor α chain | Mus musculus | 2120 | 60 | 11,7 |
| 1ubsAB_A | tryptophan synthase α subunit | Salmonella typhimurium | 1308 | 37 | 18,12 |
| 1wdcAC_C | myosin light chain | Argopecten irradians | 1856 | 46 | 10,12 |
| 2pcdBN_N | protocatechuate 3,4-dioxygenase | Pseudomonas putida | 3568 | 89 | 12,10 |
| 8atcAB_A | aspartate carbamoyltransferase | Escherichia coli | 767 | 25 | 27,13 |
| 9atcAB_B | aspartate carbamoyltransferase | Escherichia coli | 767 | 17 | 13,9 |
| Homodimer | |||||
| 1bncAB_A | acetyl-CoA carboxylase | Escherichia coli | 1224 | 35 | 40,13 |
| 1daaAB_A | D-amino acid aminotransferase | Bacillus sp YM-1 | 2302 | 57 | 25,10 |
| 1dpgAB_A | glucose-6-phosphate 1-dehydrogenase | L mesenteroides | 2285 | 59 | 13,13 |
| 1ecpBD_B | purine nucleoside phosphorylase | Escherichia coli | 1694 | 40 | 10,10 |
| 1efuBD_B | translation elongation factor Ts | Escherichia coli | 1081 | 27 | 20,15 |
| 1frpAB_A | fructose bisphosphatase | Sus scrofa | 2358 | 60 | 10,10 |
| 1fuqAB_A | fumarate hydratase | Escherichia coli | 1977 | 48 | 12,13 |
| 1gdhAB_A | D-glycerate dehydrogenase | H. methylovorum | 3127 | 72 | 46,13 |
| 1gesAB_A | pyruvate dehydrogenase | Escherichia coli | 3393 | 85 | 23,14 |
| 1glqAB_A | Glutathione S-transferase pi | Mus musculus | 1282 | 31 | 11,11 |
| 1gp1AB_A | glutathione peroxidase | Bos taurus | 775 | 18 | 20,10 |
| 1gpmBD_B | GMP synthase | Escherichia coli | 965 | 26 | 14,10 |
| 1hurAB_A | ADP-ribosylation factor 1 | Homo sapiens | 550 | 15 | 17,11 |
| 1hyhAB_A | L-lactate dehydrogenase | Weissella confusa | 818 | 23 | 22,11 |
| 1idsAC_A | superoxide dismutase | M. tuberculosis | 2182 | 52 | 18,14 |
| 1iesBE_B | ferritin | Equus caballus | 1395 | 31 | 10,10 |
| 1lehAB_A | leucine dehydrogenase | Bacillus sphaericus | 1274 | 30 | 19,10 |
| 1masAB_A | IU-nucleoside hydrolase | Crithidia fasciculata | 875 | 25 | 15,11 |
| 1mldAB_A | malate dehydrogenase | Sus scrofa | 1534 | 38 | 10,9 |
| 1nhkLR_L | nucleoside diphosphate kinase | Myxococcus xanthus | 1166 | 33 | 16,14 |
| 1oroAB_A | orotate phosphoribosyltransferase | Escherichia coli | 1217 | 35 | 23,10 |
| 1osjAB_A | 3-isopropylmalate dehydrogenase | Thermus thermophilus | 2138 | 52 | 12,12 |
| 1pkyAC_A | pyruvate kinase | Escherichia coli | 1074 | 27 | 16,14 |
| 1poly12_1 | spermidine/putrescine-binding protein | Escherichia coli | 1004 | 31 | 20,12 |
| 1qorAB_A | quinone oxidoreductase | Escherichia coli | 1194 | 32 | 22,10 |
| 1rahBD_B | aspartate carbamoyltransferase | Escherichia coli | 1143 | 27 | 12,10 |
| 1rvv12_1 | riboflavin synthase, β subunit | Bacillus subtilis | 1362 | 36 | 14,12 |
| 1scuBE_B | succinyl-CoA synthase, β subunit | Escherichia coli | 840 | 26 | 17,11 |
| 1setAB_A | seryl-tRNA synthetase | Thermus thermophilus | 2282 | 60 | 12,15 |
| 1sftAB_A | alanine racemase | G. stearothermophilus | 3151 | 83 | 10,3 |
| 1tph12_1 | triosephosphate isomerase | Gallus gallus | 1637 | 38 | 19,9 |
| 1xikAB_A | ribonucleoside-diphosphate reductase | Escherichia coli | 2976 | 69 | 11,13 |
| 2cstAB_A | aspartate aminotransferase | Gallus gallus | 3642 | 91 | 12,10 |
| 2eipAB_A | Inorganic Pyrophosphatase | Escherichia coli | 666 | 18 | 21,11 |
| 2hhmAB_A | inositol monophosphatase | Homo sapiens | 1693 | 43 | 21,9 |
| 2pcdMP_M | protocatechuate 3,4-dioxygenase | Pseudomonas putida | 1603 | 40 | 12,10 |
| 2polAB_A | DNA polymerase III, β subunit | Escherichia coli | 1271 | 30 | 25,14 |
| 3ladAB_A | lipoamide dehydrogenase | Azotobacter vinelandii | 3386 | 93 | 14,13 |
| 3mdeAB_A | acyl-CoA dehydrogenase medium chain | Sus scrofa | 1703 | 45 | 31,10 |
| 6gsvAB_A | glutathione S-transferase μ | Rattus rattus | 1309 | 32 | 17,11 |
| 8catAB_A | catalase | Bos taurus | 4718 | 108 | 10,10 |
| Transient | |||||
| 1apmIE_E | cAMP dependent kinase | Mus musculus | 1051 | 38 | 30,12 |
| 1efuAB_B | elongation factor EF-Ts | Escherichia coli | 1815 | 45 | 20,15 |
| 1g3nAB_A | cyclin-dependent kinase 4 | Homo sapiens | 885 | 23 | 10,12 |
| 1g3nAC_A | cyclin-dependent kinase 4 | Homo sapiens | 1188 | 29 | 10,12 |
| 1gotAB_B | G protein β subunit | Bos taurus | 1248 | 38 | 16,12 |
| 1k9oIE_E | trypsin | Rattus norvegicus | 914 | 33 | 16,12 |
| 1k9oIE_I | serpin | Rattus norvegicus | 914 | 17 | 14,12 |
| 1rrpAB_A | ran GTPase | Homo sapiens | 2361 | 65 | 19,11 |
| 1ughIE_E | uracil DNA glycosylase | Homo sapiens | 1096 | 31 | 35,10 |
| 1ytfAD_A | TATA binding protein | S. cerevisiae | 423 | 11 | 27,10 |
Each chain that formed an interface was assigned a code that consists of the PDB code, the chains forming the interface, and the chain that was used as part of the MSAs (e.g., lubsAB_A is PDB code IUBS, chains A and B form the interface, and chain A was aligned with related sequences). The interface size is the average size (Angstrom2) of the two interfaces, and residue number is the number of residues that are found at the interface. The numbers of sequences that are aligned to the structural template are shown for diverse and close homologs, respectively.
Comparison of interface residues with exposed noninterface residues
Figure 1 ▶ shows the difference in residue conservation between the interface and the rest of the exposed surface for both alignment types. Table 2 shows the statistics that are associated with Figure 1 ▶. The majority of proteins (40/64) are more conserved at the interface than the rest of the surface in both of the alignment types (top, right quadrant). There are six proteins for which only the MSAs of close homologs have an interface that is more conserved than the rest of the surface (top, left quadrant: 1k9oIE_I, 1lehAB_A, 1masAB_A, 1rvv12_1, 2pcdMP_M, 1gotAB_B). In four of the proteins, only the MSAs of diverse homologs have an interface that is more conserved than the rest of the surface (bottom, right quadrant: 1g3nAC_A, 8atcAB_A, 1poy12_1, 1daaAB_A). In the remaining 14 proteins, the interface is less conserved than the rest of the exposed surface for both MSA types (bottom, left quadrant). These 14 proteins can be further divided into 11 homodimers, 1bncAB_A (acetyl-CoA carboxylase), 1ecpBD_B (purine nucleoside phosphorylase), 1gp1AB_A (glutathione peroxidase), 1hyhAB_A (L-lactate dehydrogenase), 1idsAC_A (superoxide dismutase), 1nhkLR_L (nucleoside diphosphate kinase), 1qorAB_A (quinone oxidoreductase), 1rahBD_B (aspartate carbamoyltransferase), 1scuBE_B (succinyl-CoA synthase β subunit), 1xikAB_A (ribonucleoside-diphosphate reductase β subunit), 2eipAB_A (inorganic pyrophosphatase); 1 heterodimer, 1tcoBC_B (calmodulin-dependent phosphatase β subunit); and two transient complexes, 1g3nAB_A (CDK4) and 1rrpAB_A (ran GTPase). It is not entirely clear why all of these interfaces are not more conserved than the rest of the exposed surface, but it might be due to the presence of a second interface not being considered. For example, 1g3nAB_A (CDK6) forms another interface with cyclin D (1g3nAC_A; bottom, right quadrant). Combining the two interfaces of CDK6 and comparing them with the rest of the exposed surface improves the ratio of interface conservation to surface conservation. Similarly, the β subunit of the heterotrimeric G protein (top, left quadrant) forms an interface with the γ subunit as well as the α subunit (1gotAB_B). Both Ran GTPase (1rrpAB_A; bottom, left quadrant) and calcineurin A (1tcoAB_B; bottom, left quadrant) are also known to interact with several different proteins (Griffith et al. 1995; Moroianu 1999).
Figure 1.

Comparison of interface conservation with exposed noninterface conservation. The average conservation (IS; see Materials and Methods) was calculated for all interface residues and divided by the average conservation (IS) for all residues that were solvent exposed, but not part of the interface residues, using MSAs of close homologs or diverse homologs. A value of 1 or greater indicates that the protein interface is more conserved than the rest of the interface. Each data point represents one chain of a protein–protein complex, in which heterodimers are red, homodimers are blue, and transient complexes are green. Selected data points are labeled with the codes that appear in Table 1.
Table 2.
Statistics associated with Figure 1
| Diverse homologs | Close homologs | ||
| Heterodimer (12) | # Best MSAs | 8 | 4 |
| P (Best MSA) | 3.87E-02 | 9.68E-01 | |
| # interface > exposed | 11 | 10 | |
| P (Interface > Exposed) | 7.32E-04 | 8.06E-03 | |
| Homodimer (42) | # Best MSAs | 23 | 19 |
| P (Best MSA) | 3.88E-01 | 6.17E-01 | |
| # interface > exposed | 27 | 29 | |
| P (Interface > Exposed) | 4.00E-04 | 1.59E-03 | |
| Transient (10) | # Best MSAs | 4 | 6 |
| P (Best MSA) | 6.15E-01 | 3.85E-01 | |
| # interface > exposed | 6 | 7 | |
| P (Interface > Exposed) | 2.16E-01 | 3.22E-02 | |
| All (64) | # Best MSAs | 35 | 29 |
| P (Best MSA) | 1.90E-01 | 8.12E-01 | |
| # interface > exposed | 44 | 46 | |
| P (Interface > Exposed) | 3.20E-04 | 1.93E-05 |
Best MSA refers to the MSA type (close or diverse) that best distinguished between the interface and the rest of the exposed surface. The first column contains the total number of MSAs in parentheses. The P values were obtained from the Wilcoxon signed ranked test (see Materials and Methods).
An interesting example is the tetrameric succinyl-CoA synthetase (Fig. 2 ▶). The homodimeric interaction between the 41-kD subunits is not very conserved (1scuBE_B; bottom, left quadrant of Fig. 1 ▶; Fig. 2B ▶), but the same 41-kD subunit (chains B and E) forms a heterodimeric interface with a 29-kD chain that is highly conserved (1scuDE_E; top, right quadrant of Fig. 1 ▶; Fig. 2B ▶). The heterodimeric interface overlaps with the catalytic site and illustrates that two different interfaces on the same chain can evolve at very different rates.
Figure 2.
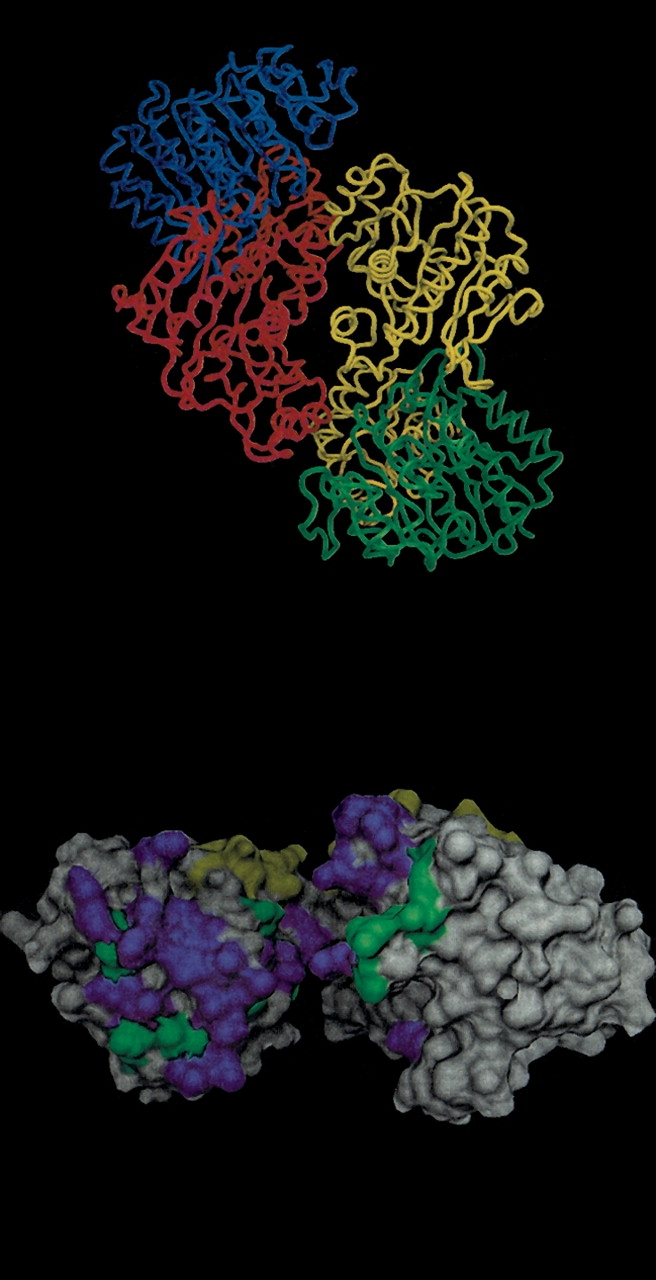
Structure of succinyl-CoA synthetase (1SCU). (A) α Carbon trace of all four chains: (Chain A) Blue, (Chain B) red, (Chain E) yellow, (Chain D) green. Chains B and E form a homodimer, whereas chain E forms a heterodimer with chain D that is identical to a heterodimer formed between chains B and chain A. (B). Molecular surface of Chain B. Residues that contact chain E are in green, and residues that contact chain A are in yellow. Highly conserved residues (IS ⩾ 0.85) are in purple and do not exist for the yellow interface (other side). Images were created with DINO (Philippsen 2002).
Table 2 shows that both MSAs of diverse homologs (44/64, P = 0.00032) and MSAs of close homologs (46/64, P = 0.0000193) are more conserved at the interface than the rest of the exposed surface. However, when compared directly, diverse homologs more often had a better ratio (interface conservation to exposed surface conservation) than close homologs (35 to 29), although this was not statistically significant (P = 0.19).
In some MSAs of diverse homologs, the interface is a lot more conserved (e.g., a ratio ⩽ 1.3) than the rest of the solvent exposed surface (Fig. 1 ▶; 1apmIE_E, 1ughIE_E, 1ubsAB_A, 1scuDE_E, 1sftAB_A, and 1pkyAC_A). These are cAMP-dependent kinase, uracil DNA glycosylase, tryptophan synthase α subunit, adenylate kinase, succinyl-CoA synthetase, alanine racemase, and pyruvate kinase, respectively. With the exception of 1ubsAB_A, their interfaces overlap with their active sites, explaining the relatively high conservation. In 1ubsAB_A, the highly conserved interface serves as a conduit in which the substrate can be passed from one active site to another.
Collectively, these results indicate that the alignment type, the presence of multiple faces, and the presence of a catalytic site at the interface can influence the conservation of the interface relative to the rest of the surface.
Comparison of interface residues with other surface patches
Despite a difference in conservation existing between the interface and the rest of the exposed surface for a statistically significant fraction of interfaces, a thorough prediction program will have to consider and rank a large number of candidate surface patches. To explore this, we generated a number of surface patches (one for almost every exposed residue), and use the Z test to examine whether the average conservation of the interface is significantly different from the conservation of all other patches on that protein (Fig. 3 ▶). With the exception of one protein (1k9oie_e), all patches had the same number of residues as the interface. In 1k9oie_e, 40% of the surface patches had fewer residues (minimum of 25 residues) than the actual interface (31 residues). The results of this test are summarized in Table 3, in which it can be seen that the majority of interfaces are not significantly more conserved than other surface patches (Z > 1.64, corresponding to the 95th percentile of the normal distribution). The MSAs of diverse homologs have slightly more significantly conserved interfaces (9/64) than MSAs of close homologs (6/64). However, the overall differences between the two alignment types are not significant.
Figure 3.

Comparison of interface conservation with the conservation for other surface patches. The average conservation (IS) of the interface is compared with the conservation of all other surface patches and expressed as a Z-value (see Materials and Methods) for MSAs of close homologs and diverse homologs. Each data point represents one chain of a protein–protein complex and is labeled according to Figure 1 ▶. Heterodimers are red, homodimers are blue, and transient complexes are green.
Table 3.
Statistics associated with Figure 3
| Diverse homologs | Close homologs | ||
| Heterodimer (12) | Z > 1.64 | 4 | 2 |
| # Best MSAs | 9 | 3 | |
| P (Best MSA) | 4.61E-02 | 9.61E-01 | |
| Homodimer (42) | Z > 1.64 | 3 | 3 |
| # Best MSAs | 20 | 22 | |
| P (Best MSA) | 7.72E-01 | 2.32E-01 | |
| Transient (10) | Z > 1.64 | 2 | 1 |
| # Best MSAs | 3 | 7 | |
| P (Best MSA) | 9.58E-01 | 5.27E-02 | |
| All (64) | Z > 1.64 | 9 | 6 |
| # Best MSAs | 32 | 32 | |
| P (Best MSA) | 6.77E-01 | 3.26E-01 |
Best MSA refers to the MSA type (close or diverse) that best distinguished between the interface and the rest of the exposed surface. The first column contains the total number of MSAs in parentheses.
There are only four significantly conserved interfaces for both alignment types, 1sftAB_A, 3mdeAB_A, 1k9oIE_E, and 1ubsAB_A. Five protein interfaces are significantly more conserved than their respective surface patches for MSAs of diverse homologs only, 1apmIE_E, 1fuqAB_A, 1tcoAB_B, 1scuDE_E, and 2pcdBN_N. Two protein interfaces are significantly more conserved than their respective surface patches for MSAs of close homologs, 2cstAB_A, and 1tcrAB_A. Assuming the correct choice of MSA type, this suggests that 11 of the 64 interfaces would have been predicted correctly. With the exception of four interfaces (1ubsAB_A, 1tcoAB_B, 2cstAB_A, and 1tcrAB_A), the remaining seven interfaces overlap with their active sites. The least conserved interfaces have already been described in Figure 1 ▶. As described in the previous section, the interface of 1ubsAB_A functions as a conduit between two active sites.
Although most interfaces are not significantly more conserved than other patches, it is possible that the most conserved patch shares some overlap with the interface. In Figure 4 ▶, we consider the most conserved surface patch in each protein and measure its overlap with the actual interface. The degree of overlap between the most conserved surface patch and the actual interface is 39% (± 28%) and 36% (± 28%) for MSAs of diverse and close homologs, respectively. The most conserved surface patch overlaps with 50% of the interface in only 17 of the 64 interfaces for both alignment types (top, right quadrant). However, in the majority of proteins (39/64), the most conserved surface patch has <50% overlap with the actual interface (bottom, left quadrant). These results suggest that protein interfaces can rarely be predicted accurately when using conservation analysis alone, regardless of the alignment type used. Again, the interface tends to be more conserved when it forms an active site.
Figure 4.

Percentage overlap between the interface and the most conserved surface patch. The most conserved surface patch was taken for each protein, and the number of residues that overlapped with the real interface was counted and expressed as a percentage. The percentages are plotted for MSAs of close homologs and diverse homologs. Each data point represents one chain of a protein–protein complex and labeled as in Figure 1 ▶. Heterodimers are red, homodimers are blue, and transient complexes are green.
Comparison of central interface residues with exposed noninterface residues
It had been shown previously for six homodimers that residues that become completely buried upon complex formation also tend to be very conserved (Valdar and Thornton 2001b). Such residues are termed “central residues,” but this does not mean they are necessarily in the center of the interface. Instead, a central residue is defined as one that has an accessible surface area of <7% when bound (B-ASA ⩽ 7%) and B-ASA should be distinguished from ASA or ΔASA (see Materials and Methods). A peripheral residue is defined as one that is only partially buried upon complex formation (B-ASA > 7%). The majority of residues (85% of peripheral residues, 94% of central residues) lose at least 5% ASA after binding. Figure 5 ▶ compares the average conservation of the central interface residues against the average conservation for the rest of the exposed surface. With the exception of 1ytfAD_A and 1tcoBC_B, the remaining 62 interfaces have a central interface. The majority (46/62) of central interfaces are more conserved than the rest of the surface for both alignment types (top, right quadrant). The difference in conservation between the central interface and the rest of the exposed surface is significant (in both alignment types) for obligate interfaces, but not transient interfaces (Table 4). Similarly, the difference in conservation between the central interface and the peripheral interface is significant for obligate interfaces but not transient interfaces (data not shown). As discussed below, this suggests that obligate binding is primarily driven by hydrophobic interactions.
Figure 5.

Comparison of central interface conservation with exposed noninterface conservation. As in Figure 1 ▶, except residues must become completely buried upon complexation (<7% relative side chain ASA) to be considered as central residues. Heterodimers are red, homodimers are blue, and transient complexes are green.
Table 4.
Statistics associated with Figure 6
| Diverse homologs | Close homologs | ||
| Heterodimer (11) | # Best MSAs | 8 | 3 |
| P (Best MSA) | 3.37E-02 | 9.73E-01 | |
| # central interface > exposed | 11 | 11 | |
| P (central Interface > Exposed) | 4.88E-04 | 4.88E-04 | |
| P (central Interface > peripheral interface) | 4.88E-04 | 4.88E-04 | |
| Homodimer (42) | # Best MSAs | 27 | 15 |
| P (Best MSA) | 1.67E-01 | 8.37E-01 | |
| # central interface > exposed | 33 | 36 | |
| P (central Interface > Exposed) | 7.30E-06 | 4.48E-07 | |
| P (central Interface > peripheral interface) | 9.98E-08 | 1.64E-06 | |
| Transient (9) | # Best MSAs | 4 | 5 |
| P (Best MSA) | 4.10E-01 | 6.33E-01 | |
| # central interface > exposed | 5 | 6 | |
| P (central Interface > Exposed) | 1.50E-01 | 1.02E-01 | |
| P (central Interface > peripheral interface) | 5.00E-01 | 1.80E-01 | |
| All (62) | # Best MSAs | 39 | 23 |
| P (Best MSA) | 3.11E-02 | 9.69E-01 | |
| # central interface > exposed | 49 | 53 | |
| P (central Interface > Exposed) | 2.067E-07 | 5.548E-09 | |
| P (central Interface > peripheral interface) | 3.333E-07 | 1.59E-08 |
Best MSA refers to the MSA type (close or diverse) that best distinguished between the interface and the rest of the exposed surface. The first column contains the total number of MSAs in parentheses. The P values were obtained from the Wilcoxon signed ranked test (see Materials and Methods).
The frequency of conserved residues at different degrees of burial at the interface
Given that central residues tend to be more conserved than peripheral residues in obligate interfaces, we decided to compare the residue preferences of conserved residues at the center and periphery. An interface residue was considered conserved when the Information score was >0.85 in MSAs of diverse homologs. Sequence logos were generated with ALPRO (Schneider and Stephens 1990).
For heterodimers, there are both similarities and subtle preferential differences between central residues (Fig. 6A ▶) and peripheral residues (Fig. 6B ▶). Leucine is the most prominent conserved residue at the central interface, but is also fairly prominent at the peripheral interface, where its B-ASA ranges from 8.2% to 33.7%. There is some evidence that residues at the protein–protein interface are less flexible than the rest of the protein surface (Cole and Warwicker 2002), and this need might be met by leucine with its limited conformational diversity (Pickett and Sternberg 1993). The aromatic residues phenylalanine and tyrosine are more prominent in the central interface than the peripheral interface. In contrast, the peripheral interface prefers conserved arginine and glycine residues. This would suggest that pi-interactions of the conserved central aromatic residues are a primary driving force for heterodimerization. The preference for conserved arginines at the peripheral interface is probably due to its ability to form hydrophobic interactions, while still requiring interactions with water or polar molecules. We speculate that the role of glycine is probably more structural, given that it is important in helix caps (Fetrow et al. 1997) and loops (Crasto and Feng 2001). The other surprise at the central interface is the preference for aspartic acid. Its is not clear to us why this is more preferred than glutamic acid, but might also be due to its high propensity to be in loops (Crasto and Feng 2001).
Figure 6.
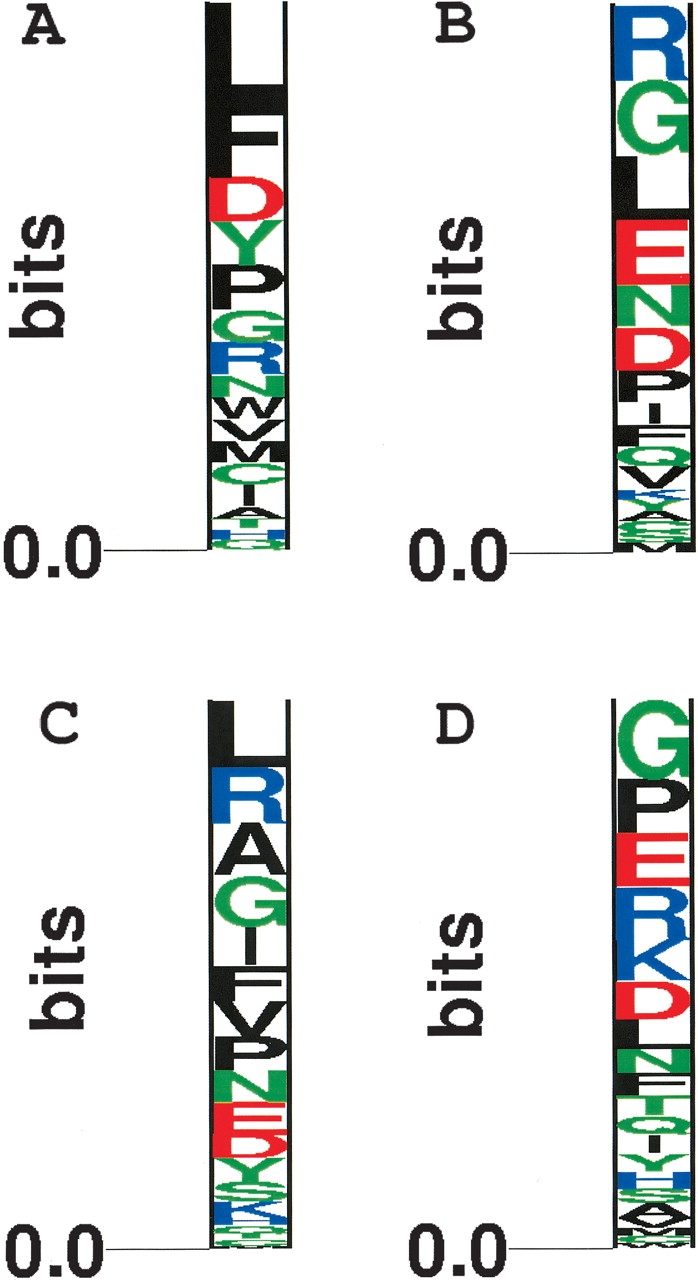
The propensity of highly conserved residues at different degrees of burial at the interface. All highly conserved residues (IS ⩾ 0.85) in MSAs of diverse homologs were classified as those with a B-ASA ⩽ 7%, and a B-ASA > 7% for heterodimers (A,B) and homodimers (C,D). Sequence logos were created with ALPRO (Schneider and Stephens 1990).
In homodimers, the central residues (Fig. 6C ▶) are predictably more hydrophobic than the interface residues (Fig. 6D ▶). However, their preference for aromatic residues is not as strong as it is in heterodimers. The highest ranked central residues are leucine and arginine, whereas the highest ranked peripheral residues are glycine and proline. With the exception of proline, the possible roles of these residues have already been mentioned. Similar to glycine, we believe that the role of proline is probably structural, given that it is a secondary structure breaker and important in loops (Crasto and Feng 2001) and helix caps (Fetrow et al. 1997).
With the exception of aspartic acid and arginine, the majority of central residues are hydrophobic. These results suggest that hydrophobic forces primarily drive packing of obligate interfaces.
The frequency of gapped alignment positions at the protein–protein interface
It is generally thought that gaps in an alignment most often correspond to loops in the protein structure. It is also well known that loops are primarily exposed and often part of an active site or protein–protein interface. Many of the residues described above are commonly found in loops (Crasto and Feng 2001). Therefore, it could be argued that a prediction method should find a way to reward a candidate surface patch that contains a loop that is believed to be part of the interface. However, many scoring schemes either ignore alignment positions with gaps or introduce a gap penalty, the argument being that a residue position is unlikely to be important if it can be deleted. In this work, our conservation score uses a gap penalty, and we were interested to know how many interface residues had one or more gaps in their alignment position compared with the number found in the rest of the exposed surface. In Figure 7 ▶, obligate interfaces (homodimers and heterodimers) tend to have fewer gaps at their interface than on the rest of their protein surface. This observation is not as striking when using alignments of close homologs (Table 5). In contrast, the number of interface gaps does not significantly differ from the number of surface gaps for transient interfaces.
Figure 7.

The number of alignment gaps at the interface versus the exposed surface. Using MSAs of diverse homologs, the X-axis is the number of exposed (noninterface) residues that have one or more alignment gaps (normalized by the total number of residues on the exposed surface). The Y-axis is the number of interface residues that has one or more alignment gaps (normalized by the total number of residues at the interface). Each data point represents one chain of a protein–protein complex. Obligate interfaces (heterodimers and homodimers) are red, and transient complexes are blue.
Table 5.
Statistics associated with Figure 7
| Diverse homologs | Close homologs | ||
| Heterodimer (12) | % interface gaps < % surface gaps | 12 | 9 |
| P (% interface gaps < % surface gaps) | 0.000244 | 0.2704 | |
| Homodimer (42) | % interface gaps < % surface gaps | 32 | 31 |
| P (% interface gaps < % surface gaps) | 4.90E-04 | 2.24E-02 | |
| Transient (10) | % interface gaps < % surface gaps | 5 | 4 |
| P (% interface gaps < % surface gaps) | 0.8125 | 0.9033 |
The first column contains the total number of MSAs in parentheses. The P values were obtained from the Wilcoxon signed ranked test (see Methods).
This result is probably not surprising if one views an obligate interface as a protein core, which are known to contain fewer gaps than the surface when multiply aligned.
Discussion
We have shown that the protein interface is usually more conserved than the rest of the exposed surface. However, a more realistic surface-patch analysis showed that the interface conservation was not sufficiently different from other surface patches to allow prediction of the interface by conservation alone. The most conserved surface patch on a protein was rarely found to share >50% residue overlap with the real interface. The results are a lot less optimistic than two previous studies that focused exclusively on protein–protein interfaces (Grishin and Phillips 1994; Valdar and Thornton 2001b), and are probably a result of our data set being significantly larger. To our knowledge, this is the first time that conservation of transient and heterodimeric interfaces has been studied. Although the number of heterodimeric and transient complexes is larger than in previous studies of homodimers, the results should still be considered preliminary. Overall, the results suggest that one will have a small chance (17/64) of correctly predicting 50% of the interface residues when the three-dimensional structure is known and either multiple alignment type is used. The success rate is likely to improve when the two interfaces form a catalytic site and will be poorer when the protein has multiple faces. The conservation of catalytic/small-ligand binding sites is well documented, and the ET method is expected to predict them accurately (Yao et al. 2003). Although there was not a significant difference between the two MSA types, we prefer the MSA of diverse homologs. They appear to be marginally better for discriminating the interface from the rest of the surface, and the number of gaps at obligate interfaces is less than the number of gaps at the rest of the surface.
Occasionally, the protein belonged to a large family in which each subgroup might be expected to differ from other subgroups at the interface. Although our information score assigns a relatively high score to these subgroup specific/tree-determinant sites, the MSAs of diverse homologs will not contain many sequences for a subgroup, whereas the MSAs of close homologs will contain many sequences for just one subgroup (see Materials and Methods). Some of the less-conserved interfaces are likely to be detected by methods that account for the phylogenetic relationships (Lichtarge et al. 1996; Armon et al. 2001; Pupko et al. 2002). Unfortunately, defining the correct subset of sequences is not trivial, particularly if the procedure is to be automated (de Sol Mesa et al. 2003). One strategy might be to define subgroups on the basis of gene duplication events, although this also has caveats. Combining parameters such as tree-determinant information with surface-patch conservation should lead to improved prediction of interfaces. Other parameters that might be combined include residue propensities (Ofran and Rost 2003), physical properties (Jones and Thornton 1997), and evolutionary models of variable residues believed to be functionally important (Hughes and Nei 1988; Pazos et al. 1997; Shirai et al. 2002). Efforts along these lines are underway.
Materials and methods
Data sets
The nontransient homodimer and heterodimer data sets were derived from a previous data set used by Glaser et al. (2001). A complex was defined as a homodimer if the two chains shared >95% sequence identity. The list of transient complexes was derived from a larger internal data set of transient complexes. Only those structures solved at a resolution of 2.5 Å or better were considered. The data set was reduced significantly after removing redundant sequences and partial structures that were not an appropriate size for patch analysis (see below). The multiple sequence alignments described below are available from http://oscar.gen.tcd.ie/~dcaffrey.
Diverse homolog selection
The objective was to have an MSA containing a diverse set of sequences that would include several paralogs whenever possible. As this is a semiautomated approach, the exact phylogeny of the sequences is unknown for each protein family. Each chain from a complex was searched against the nonredundant protein database using BLASTP with an E-Value cutoff of 0.001 (Altschul et al. 1997). Sequences from each search were clustered together when they shared >60% identity, using BLASTCLUST, which is part of the BLAST package (Altschul et al. 1997). The longest sequence from each cluster was taken and aligned to the structural template using CLUSTALW (Thompson et al. 1994). This prevented oversampling from a particular subgroup of sequences found in each protein family. To ensure that the alignments were of an adequate quality, a number of criteria were used. Sequences that had five or more gaps at positions that were otherwise populated with residues in other sequences (75% of the alignment) were removed. This process was iterated three times. To ensure that a significant portion of the protein was crystallized, we only considered alignments in which the structural template made up 85% or more of the significant sites in the alignment. A significant site was defined as a position in the alignment where >70% of sequences had a residue present. Alignments with continuous stretches of significant sites (20 or more) that were not present in the structural template were removed, as were alignments that had 10 or fewer sequences aligned to the structural template. The structural template had to contain at least 120 residues that were aligned to residues in the other sequences. The remaining structures were compared against each other for sequence redundancy using the BLASTCLUST with a cutoff of 30% identity. Finally, the alignment quality was confirmed by manual inspection with PFAAT (Johnson et al. 2003).
Close homolog selection
The objective was to have an MSA containing a set of sequences that were closely related and would typically be orthologs. Again, the semiautomated approach does not guarantee that all sequences are bona fide orthologs. Depending on the taxonomy assignment of the proteins in Table 1, the proteins were grouped as belonging to eubacteria, metazoa, or euglenezoa (Wheeler et al. 2000). For eubacteria, each of the sequences was searched against the following genomes: Bacillus anthracis (Ames), Borrelia burgdorferi, Chlamydophila pneumoniae (CWL029), Escherichia coli (K12), Haemophilus influenzae, Helicobacter pylori (J99), Listeria monocytogenes, Mycoplasma penetrans, Neisseria meningitidis (MC58), Pseudomonas aeruginosa, Salmonella typhimurium (LT2), Shigella flexneri (2a), Staphylococcus aureus (MW2), Vibrio cholerae, and Xanthomonas citri. The top hit from each genome was selected if it had an E value of e−10 or better. Sequences belonging to the metazoa group were similarly searched against species databases that were derived from the NCBI nr protein database (Homo sapiens, Mus musculus, Caenorhabditis elegans, Drosophila melanogaster, Rattus norvegicus, Anopheles gambiae, Bos taurus, Gallus gallus, Xenopus laevis, Danio rerio, Ovis aries, Sus scrofa, and Takifugu rubripes). Sequences belonging to the euglenezoa group were searched against the entire NCBI nr database, and the top hits were hand selected from appropriate species.
Residue conservation
The Shannon Entropy for a multiple alignment position can be calculated as follows:
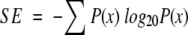 |
(1) |
in which P(x) is the relative frequency of each amino acid x in the alignment position. The base of 20 ensures that all values are bounded between zero and one (assuming that we ignore entities such as “X”, “Z”, “B”, and “−”). However, it does not account for the physicochemical similarities that are found between the different amino acids. Therefore, we calculate the Von Neumann entropy for each alignment column. The Von Neumann entropy takes a similar form to equation 1 (Lifshitz and Pitaevskii 1980; Petz 2001):
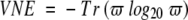 |
(2) |
in which ϖ is a density matrix with trace = 1. Apart from normalization by the trace, the density matrix is given by the product of the relative frequencies of the amino acids in each alignment position [P(x)] and an appropriate similarity matrix (e.g., BLOSUM), that is,
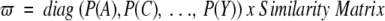 |
(3) |
The calculation of equation 2 is facilitated by first calculating the eigenvalues λi of ϖ. In this case, equation 2 is given by the simpler and more computationally efficient equation
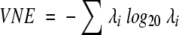 |
(4) |
In the special case in which the similarity matrix is the identity matrix, equations 2 and 4 become identical to the Shannon Entropy in equation 1. After trial and error, we found that the BLOSUM 50 target frequencies (blosum50.qij) (Henikoff and Henikoff 1992) gave results that we considered most desirable, but other matrices give appropriate results. To incorporate sequence weights, the frequency for each amino acid is computed as follows:
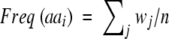 |
(5) |
in which aai is one of the 20 amino acids in the alignment position, wj is the sequence weight for sequence j to which amino acid (aaij) belongs, n is the number of sequences in the alignment, and the sequence weights sum to n. The sequence weights are computed using the method of Henikoff and Henikoff (1994), but could be derived by other means. A gap penalty is enforced using an approach similar to that used by CLUSTALX (Thompson et al. 1997). To do this, the VNE score is first transformed to its information score (IS) by subtracting it from the maximum entropy (i.e., IS = 1 −VNE). The gap penalty is the number of residues in the column, divided by the number of sequences. The information score is then multiplied by the gap penalty. An information score derived from VNE will range from 0 to 1, where a score of 1 is assigned to a 100% identical alignment column. In practice, a score will only be below 0.3 when gaps are present, as the 20 residues are not considered to be completely orthogonal. For residue propensities, we assigned an alignment position as being highly conserved when the information score was ⩾ 0.85.
Defining interface residues
Interface residues were defined as those that lost >1% relative solvent accessibility upon complex formation (ΔASA > 1%). Solvent accessibilities were calculated using the algorithm of Lee and Richards with a probe size of 1.4 Å (Lee and Richards 1971). All complexes with a total interface <1500 Å2 were manually inspected. This involved careful reading of the literature and the PDB files to ensure that all files contained genuine biological interfaces. Water molecules were not considered. Interface residues were further classified as peripheral or central on the basis of their solvent accessibility when bound (B-ASA). A peripheral residue has a B-ASA ⩾ 7%, where as a central residue, has a B-ASA <7%. To clarify, the relationship between all of these terms is as follows: ΔASA = B-ASA—Separated monomer ASA. Sequence logos for central and peripheral residues were generated for each category using ALPRO (Schneider and Stephens 1990).
Surface-patch generation
We wanted to compare the interface patch with other random surface patches to see whether the former was more conserved. A surface patch was defined by taking each solvent-exposed residue and its surrounding neighbors on the unbound protein. Thus, a protein with 100 solvent-exposed residues would have 100 surface patches. To ensure that we did not measure through the protein, the following procedure was followed. A side-chain centroid was calculated for every solvent-exposed residue on the unbound protein (a whole residue centroid for glycine) and was used to calculate distances between all exposed residues. The patch was grown from the single starting (seed) residue to include all neighboring residues that were within 7 Å of it. This process was iterated using the newly acquired residues, until the total number of residues in the patch was equal to the total number of residues in the interface. When the number of neighboring residues exceeds the number of remaining places in the patch, the residues closest to the seed residue are selected first. The patch will not always expand to an adequate size, and those with <70% of the actual interface are excluded from the analysis. The average residue conservation was calculated for each surface patch and the interface patch.
Statistical measures
The Wilcoxon-signed ranked test was used for all statistical comparisons. This test was chosen because it makes minimal assumptions about the underlying distribution, but is still able to take the magnitudes of the observed differences into account. Similar results were obtained when using the binomial and T-tests. The Z-test was used to compare the conservation of the interface relative to conservation of all other patches on the same protein.
Acknowledgments
We thank A. Cheng, Q. Cao, Y-H. Ding, P. Henstock, and S. Xi for helpful discussions. J.M. is supported by DoE/Krell Institute CSGF. We thank Research Collaboratory for Structural Bioinformatics (RCSB) and the numerous crystallographers for making their structures available in electronic format. Finally, we thank the anonymous reviewers for their suggestions.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.03323604.
References
- Altschul, S.F., Madden, T.L., Schaffer, A.A., Zhang, J., Zhang, Z., Miller, W., and Lipman, D.J. 1997. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 25 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Argos, P. 1988. An investigation of protein subunit and domain interfaces. Protein Eng. 2 101–113. [DOI] [PubMed] [Google Scholar]
- Armon, A., Graur, D., and Ben-Tal, N. 2001. ConSurf: An algorithmic tool for the identification of functional regions in proteins by surface mapping of phylogenetic information. J. Mol. Biol. 307 447–463. [DOI] [PubMed] [Google Scholar]
- Bartlett, G.J., Porter, C.T., Borkakoti, N., and Thornton, J.M. 2002. Analysis of catalytic residues in enzyme active sites. J. Mol. Biol. 324 105–121. [DOI] [PubMed] [Google Scholar]
- Bauer, B., Mirey, G., Vetter, I.R., Garcia-Ranea, J.A., Valencia, A., Wittinghofer, A., Camonis, J.H., and Cool, R.H. 1999. Effector recognition by the small GTP-binding proteins Ras and Ral. J. Biol. Chem. 274 17763–17770. [DOI] [PubMed] [Google Scholar]
- Bogan, A.A. and Thorn, K.S. 1998. Anatomy of hot spots in protein interfaces. J. Mol. Biol. 280 1–9. [DOI] [PubMed] [Google Scholar]
- Caffrey, D.R., O’Neill, L.A., and Shields, D.C. 2000. A method to predict residues conferring functional differences between related proteins: Application to MAP kinase pathways. Protein Sci. 9 655–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casari, G., Sander, C., and Valencia, A. 1995. A method to predict functional residues in proteins. Nat. Struct. Biol. 2 171–178. [DOI] [PubMed] [Google Scholar]
- Chakrabarti, P. and Janin, J. 2002. Dissecting protein–protein recognition sites. Proteins 47 334–343. [DOI] [PubMed] [Google Scholar]
- Chothia, C. and Janin, J. 1975. Principles of protein–protein recognition. Nature 256 705–708. [DOI] [PubMed] [Google Scholar]
- Clackson, T. and Wells, J.A. 1995. A hot spot of binding energy in a hormone-receptor interface. Science 267 383–386. [DOI] [PubMed] [Google Scholar]
- Cole, C. and Warwicker, J. 2002. Side-chain conformational entropy at protein–protein interfaces. Protein Sci. 11 2860–2870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crasto, C.J. and Feng, J. 2001. Sequence codes for extended conformation: A neighbor-dependent sequence analysis of loops in proteins. Proteins 42 399–413. [DOI] [PubMed] [Google Scholar]
- de Sol Mesa, D., Pazos, F., and Valencia, A. 2003. Automatic methods for predicting functionally important residues. J. Mol. Biol. 326 1289–1302. [DOI] [PubMed] [Google Scholar]
- Elcock, A.H. and McCammon, J.A. 2001. Identification of protein oligomerization states by analysis of interface conservation. Proc. Natl. Acad. Sci. 98 2990–2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fetrow, J.S., Palumbo, M.J., and Berg, G. 1997. Patterns, structures, and amino acid frequencies in structural building blocks, a protein secondary structure classification scheme. Proteins 27 249–271. [PubMed] [Google Scholar]
- Gadek, T.R. and Nicholas, J.B. 2003. Small molecule antagonists of proteins. Biochem. Pharmacol. 65 1–8. [DOI] [PubMed] [Google Scholar]
- Glaser, F., Steinberg, D.M., Vakser, I.A., and Ben-Tal, N. 2001. Residue frequencies and pairing preferences at protein–protein interfaces. Proteins 43 89–102. [PubMed] [Google Scholar]
- Griffith, J.P., Kim, J.L., Kim, E.E., Sintchak, M.D., Thomson, J.A., Fitzgibbon, M.J., Fleming, M.A., Caron, P.R., Hsiao, K., and Navia, M.A. 1995. X-ray structure of calcineurin inhibited by the immunophilin-immunosuppressant FKBP12-FK506 complex. Cell 82 507–522. [DOI] [PubMed] [Google Scholar]
- Grishin, N.V. and Phillips, M.A. 1994. The subunit interfaces of oligomeric enzymes are conserved to a similar extent to the overall protein sequences. Protein Sci. 3 2455–2458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannenhalli, S.S. and Russell, R.B. 2000. Analysis and prediction of functional sub-types from protein sequence alignments. J. Mol. Biol. 303 61–76. [DOI] [PubMed] [Google Scholar]
- Henikoff, S. and Henikoff, J.G. 1992. Amino acid substitution matrices from protein blocks. Proc. Natl. Acad. Sci. 89 10915–10919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ————. 1994. Position-based sequence weights. J. Mol. Biol. 243 574–578. [DOI] [PubMed] [Google Scholar]
- Hu, Z., Ma, B., Wolfson, H., and Nussinov, R. 2000. Conservation of polar residues as hot spots at protein interfaces. Proteins 39 331–342. [PubMed] [Google Scholar]
- Hughes, A.L. and Nei, M. 1988. Pattern of nucleotide substitution at major histocompatibility complex class I loci reveals overdominant selection. Nature 335 167–170. [DOI] [PubMed] [Google Scholar]
- Janin, J. and Chothia, C. 1990. The structure of protein–protein recognition sites. J. Biol. Chem. 265 16027–16030. [PubMed] [Google Scholar]
- Janin, J., Miller, S., and Chothia, C. 1988. Surface, subunit interfaces and interior of oligomeric proteins. J. Mol. Biol. 204 155–164. [DOI] [PubMed] [Google Scholar]
- Johnson, J.M., Mason, K., Moallemi, C., Xi, H., Somaroo, S., and Huang, E.S. 2003. Protein family annotation in a multiple alignment viewer. Bioinformatics 19 544–545. [DOI] [PubMed] [Google Scholar]
- Jones, S. and Thornton, J.M. 1996. Principles of protein–protein interactions. Proc. Natl. Acad. Sci. 93 13–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ———. 1997. Analysis of protein–protein interaction sites using surface patches. J. Mol. Biol. 272 121–132. [DOI] [PubMed] [Google Scholar]
- Jones, S., Marin, A., and Thornton, J.M. 2000. Protein domain interfaces: Characterization and comparison with oligomeric protein interfaces. Protein Eng. 13 77–82. [DOI] [PubMed] [Google Scholar]
- Korn, A.P. and Burnett, R.M. 1991. Distribution and complementarity of hydropathy in multisubunit proteins. Proteins 9 37–55. [DOI] [PubMed] [Google Scholar]
- Landgraf, R., Xenarios, I., and Eisenberg, D. 2001. Three-dimensional cluster analysis identifies interfaces and functional residue clusters in proteins. J. Mol. Biol. 307 1487–1502. [DOI] [PubMed] [Google Scholar]
- Larsen, T.A., Olson, A.J., and Goodsell, D.S. 1998. Morphology of protein–protein interfaces. Structure 6 421–427. [DOI] [PubMed] [Google Scholar]
- Lawrence, M.C. and Colman, P.M. 1993. Shape complementarity at protein/protein interfaces. J. Mol. Biol. 234 946–950. [DOI] [PubMed] [Google Scholar]
- Lee, B. and Richards, F.M. 1971. The interpretation of protein structures: Estimation of static accessibility. J. Mol. Biol. 55 379–400. [DOI] [PubMed] [Google Scholar]
- Lichtarge, O., Bourne, H.R., and Cohen, F.E. 1996. An evolutionary trace method defines binding surfaces common to protein families. J. Mol. Biol. 257 342–358. [DOI] [PubMed] [Google Scholar]
- Lifshitz, E.M. and Pitaevskii, L.P. 1980. Statistical physics, pp. 25–26. Pergamon Press, Oxford, UK.
- Lijnzaad, P., Berendsen, H.J., and Argos, P. 1996. Hydrophobic patches on the surfaces of protein structures. Proteins 25 389–397. [DOI] [PubMed] [Google Scholar]
- Livingstone, C.D. and Barton, G.J. 1996. Identification of functional residues and secondary structure from protein multiple sequence alignment. Methods Enzymol. 266 497–512. [DOI] [PubMed] [Google Scholar]
- Lo Conte, L., Chothia, C., and Janin, J. 1999. The atomic structure of protein–protein recognition sites. J. Mol. Biol. 285 2177–2198. [DOI] [PubMed] [Google Scholar]
- McCoy, A.J., Chandana Epa, V., and Colman, P.M. 1997. Electrostatic complementarity at protein/protein interfaces. J. Mol. Biol. 268 570–584. [DOI] [PubMed] [Google Scholar]
- Moroianu, J. 1999. Nuclear import and export pathways. J. Cell. Biochem. 33 76–83. [DOI] [PubMed] [Google Scholar]
- Ofran, Y. and Rost, B. 2003. Analysing six types of protein–protein interfaces. J. Mol. Biol. 325 377–387. [DOI] [PubMed] [Google Scholar]
- Ouzounis, C., Perez-Irratxeta, C., Sander, C., and Valencia, A. 1998. Are binding residues conserved? Pac. Symp. Biocomput. 3 401–412. [PubMed] [Google Scholar]
- Pazos, F., Helmer-Citterich, M., Ausiello, G., and Valencia, A. 1997. Correlated mutations contain information about protein–protein interaction. J. Mol. Biol. 271 511–523. [DOI] [PubMed] [Google Scholar]
- Petz, D. 2001. Entropy, von Neumann and the von Neumann entropy. In John von Neumann and the foundations of quantum physics. Kluwer Academic Publishers, Dordrecht.
- Philippsen, A. 2002. DINO: Visualizing structural biology. http://www.dino3d.org.
- Pickett, S.D. and Sternberg, M.J. 1993. Empirical scale of side-chain conformational entropy in protein folding. J. Mol. Biol. 231 825–839. [DOI] [PubMed] [Google Scholar]
- Pupko, T., Bell, R.E., Mayrose, I., Glaser, F., and Ben-Tal, N. 2002. Rate4Site: An algorithmic tool for the identification of functional regions in proteins by surface mapping of evolutionary determinants within their homologues. Bioinformatics Suppl. 1 S71–S77. [DOI] [PubMed] [Google Scholar]
- Schneider, T.D. and Stephens, R.M. 1990. Sequence logos: A new way to display consensus sequences. Nucleic Acids Res. 18 6097–6100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheinerman, F.B. and Honig, B. 2002. On the role of electrostatic interactions in the design of protein–protein interfaces. J. Mol. Biol. 318 161–177. [DOI] [PubMed] [Google Scholar]
- Sheinerman, F.B., Norel, R., and Honig, B. 2000. Electrostatic aspects of protein–protein interactions. Curr. Opin. Struct. Biol. 10 153–159. [DOI] [PubMed] [Google Scholar]
- Shirai, T., Matsui, Y., Shionyu-Mitsuyama, C., Yamane, T., Kamiya, H., Ishii, C., Ogawa, T., and Muramoto, K. 2002. Crystal structure of a conger eel galectin (congerin II) at 1.45Å resolution: Implication for the accelerated evolution of a new ligand-binding site following gene duplication. J. Mol. Biol. 321 879–889. [DOI] [PubMed] [Google Scholar]
- Sowa, M.E., He, W., Slep, K.C., Kercher, M.A., Lichtarge, O., and Wensel, T.G. 2001. Prediction and confirmation of a site critical for effector regulation of RGS domain activity. Nat. Struct. Biol. 8 234–237. [DOI] [PubMed] [Google Scholar]
- Stenmark, H., Valencia, A., Martinez, O., Ullrich, O., Goud, B., and Zerial, M. 1994. Distinct structural elements of rab5 define its functional specificity. EMBO J. 13 575–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson, J.D., Higgins, D.G., and Gibson, T.J. 1994. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22 4673–4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson, J.D., Gibson, T.J., Plewniak, F., Jeanmougin, F., and Higgins, D.G. 1997. The CLUSTAL_X windows interface: Flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 25 4876–4882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toogood, P.L. 2002. Inhibition of protein–protein association by small molecules: Approaches and progress. J. Med. Chem. 45 1543–1558. [DOI] [PubMed] [Google Scholar]
- Tsai, C.J. and Nussinov, R. 1997. Hydrophobic folding units at protein–protein interfaces: Implications to protein folding and to protein–protein association. Protein Sci. 6 1426–1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai, C.J., Lin, S.L., Wolfson, H.J., and Nussinov, R. 1996. Protein–protein interfaces: Architectures and interactions in protein–protein interfaces and in protein cores. Their similarities and differences. Crit. Rev. Biochem. Mol. Biol. 31 127–152. [DOI] [PubMed] [Google Scholar]
- ———. 1997a. Studies of protein–protein interfaces: A statistical analysis of the hydrophobic effect. Protein Sci. 6 53–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai, C.J., Xu, D., and Nussinov, R. 1997b. Structural motifs at protein–protein interfaces: Protein cores versus two-state and three-state model complexes. Protein Sci. 6 1793–1805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valdar, W.S. and Thornton, J.M. 2001a. Conservation helps to identify biologically relevant crystal contacts. J. Mol. Biol. 313 399–416. [DOI] [PubMed] [Google Scholar]
- ———. 2001b. Protein–protein interfaces: Analysis of amino acid conservation in homodimers. Proteins 42 108–124. [PubMed] [Google Scholar]
- Wheeler, D.L., Chappey, C., Lash, A.E., Leipe, D.D., Madden, T.L., Schuler, G.D., Tatusova, T.A., and Rapp, B.A. 2000. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 28 10–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, D., Tsai, C.J., and Nussinov, R. 1997. Hydrogen bonds and salt bridges across protein–protein interfaces. Protein Eng. 10 999–1012. [DOI] [PubMed] [Google Scholar]
- Xu, W. and Regnier, F.E. 1998. Protein–protein interactions on weak-cation-exchange sorbent surfaces during chromatographic separations. J. Chromatogr. A 828 357–364. [DOI] [PubMed] [Google Scholar]
- Yao, H., Kristensen, D.M., Mihalek, I., Sowa, M.E., Shaw, C., Kimmel, M., Kavraki, L., and Lichtarge, O. 2003. An accurate, sensitive, and scalable method to identify functional sites in protein structures. J. Mol. Biol. 326 255–261. [DOI] [PubMed] [Google Scholar]