

Abstract
The effects on folding kinetics and equilibrium stability of core mutations in the apo-mutant C112S of azurin from Pseudomonas aeruginosa were studied. A number of conserved residues within the cupredoxin family were recognized by sequential alignment as constituting a common hydrophobic core: I7, F15, L33, W48, F110, L50, V95, and V31. Of these, I7, V31, L33, and L50 were mutated for the purpose of obtaining information on the transition state and a potential folding nucleus. In addition, residue V5 in the immediate vicinity of the common core, as well as T52, separate from the core, were mutated as controls. All mutants exhibited a nonlinear dependence of activation free energy of folding on denaturant concentration, although the refolding kinetics of the V31A/C112S mutant indicated that the V31A mutation destabilizes the transition state enough to allow folding via a parallel transition state ensemble. Φ-values could be calculated for three of the six mutants, V31A/C112S, L33A/C112S, and L50A/C112S, and the fractional values of 0.63, 0.33, and 0.50 (respectively) obtained at 0.5 M GdmCl suggest that these residues are important for stabilizing the transition state. Furthermore, a linear dependence of ln kobsH2O on ΔGU−NH2O of the core mutations and the putative involvement of ground-state effects suggest the presence of native-like residual interactions in the denatured state that bias this ensemble toward a folding-competent state.
Keywords: folding, residual structure, common core, azurin, cupredoxin, β-sandwich, Greek key
The ways in which polypeptides attain their native structure is currently a research area of considerable interest. In particular, much focus is on the means by which evolutionary pressure has selected for foldable sequences that avoid non-native misfolded states, because such species dramatically increase the probability of intermolecular aggregation (Dobson 2003). Such aggregation is currently implicated in over 20 diseases, for example, Alzheimer’s disease, type II diabetes, and cataract (Stefani and Dobson 2003). However, mainly because of its complexity, protein folding is at present far from completely understood. During folding, a huge amount of intramolecular interactions such as hydrogen bonds, salt bridges, and hydrophobic interactions need to form, break, and reform to reach the native structure. Moreover, in the crowded milieu of the cell, where the density of protein is ~0.2–0.3 g/mL, these intramolecular interactions occur in competition with similar intermolecular interactions which may be detrimental to the organism (van den Berg et al. 1999), yet the process usually yields the native state in a matter of milliseconds to seconds. This inherent swiftness of the process implies a series of ordered events or intermediates, a fact that has intrigued researchers for several decades (Kim and Baldwin 1982, 1990). The order in which these intramolecular events take place cannot yet be predicted or calculated, and although molecular dynamics can now simulate the unfolding reaction with reasonable accuracy (Fersht and Daggett 2002), we still await the first molecular dynamics simulation from the completely unfolded state. Protein folding therefore continues to rely heavily on experimental work, where the tools developed thus far can give us a very detailed description of the folding pathway for a protein down to the individual role for each amino acid on the main folding pathway (Fersht 1999).
Considering the high structural similarity between homologous proteins, it is remarkable that the amino acid sequence can be so diverse among many of them. Furthermore, when analyzing the folding behavior between homologous proteins, it appears that residues participating in the transition state (TS) are no more conserved than other amino acids, although they are commonly found at identical structural positions (Clarke et al. 1999; Martinez and Serrano 1999; Larson et al. 2002). It is of particular interest, then, in any folding analysis of homologous proteins, to identify the most structurally conserved residues in a protein family and to probe their effect on the folding behavior with respect to the TS structure. Such an analysis may be achieved by a mutational approach where any change in folding behavior can be utilized as a structural probe of the TS using transition state theory in general and Hammond behavior in particular. This type of kinetic analysis is termed Φ-value analysis (Fersht 1999).
As a model protein for the folding of β-sheet proteins, we use Pseudomonas aeruginosa azurin. A recent study on the folding kinetics of apo-azurin mutant C112S identified an intermediate which manifested itself as a burst phase in the amplitude data for the refolding reaction (Sandberg et al. 2004). This intermediate was capable of binding 8-anilino-1-naphtalenesulfonate (ANS), and was therefore ascribed molten globule-like status. Nevertheless, the intermediate had only a minor influence on the refolding kinetics, and a two-state model is therefore a reasonable approximation for the folding of wild type and the C112S apo-azurin mutant. In continuing the analysis of azurin folding, we use this surrogate wild type to probe the hydrophobic core of azurin. To identify structurally conserved amino acids it is first necessary to compare the structures among several members of the family to which azurin belongs. This family of proteins is collectively referred to as the small blue copper proteins, or the cupredoxins, and all of its members are characterized by very similar eight-stranded β-structures but low pairwise structural identities (in general, 25%; Adman 1991). The most highly conserved feature in the cupredoxins is the copper-binding site, in accord with all of them being electron transfer proteins. At this site, the ion is coordinated in a trigonal pyramidal fashion with three equatorial ligands (two histidine nitrogens and one cysteine sulfur) and one axial ligand (generally a methionine sulfur), although in azurin there is also a second axial ligand (a glycine carbonyl oxygen) resulting in a trigonal bipyramidal coordination (Gray et al. 2000). Another conserved feature of most Greek key β-sandwich proteins is the tyrosine corner (Hemmingsen et al. 1994), but as it is believed to have only a stabilizing role in these domains (Hamill et al. 2000a), it is not of interest here.
Apart from these residues, a structural alignment identified a number of hydrophobic residues structurally conserved among six different cupredoxins, and these were proposed to constitute the folding nucleus in azurin (Leckner 2001). This alignment is here expanded to include three additional representative cupredoxins with very low degrees of intermolecular identity (Fig. 1 ▶). The structural analysis was based on a VAST alignment (Gibrat et al. 1996), but manual improvement was necessary. This identified eight structurally equivalent residues positioned in the central parts of the cupredoxin domain structure, which thus constitute a common core (Fig. 2 ▶). Using azurin numbering, these residues are I7, F15, L33, W48, F110, L50, V95, and V31. Because a nucleation-condensation mechanism was previously proposed to initiate the structure-formation process in other β-sandwich proteins of Greek-key topology (Clarke et al. 1999; Fowler and Clarke 2001), these conserved residues are strong candidates for such a nucleus dominated by tertiary interactions, as previously proposed (Leckner 2001). Interestingly, it was recently demonstrated that some of these residues are also part of a supersecondary substructure consisting of two pairs of interlocked strands common to all β-sandwich proteins (Kister et al. 2002). We note a similarity between the structural identities of the conserved residues in our alignment and the residues interacting in the two interlocked β-strands that are proposed to be the structural determinants of this fold: V31 and L33 of strand i (strand 3 in azurin) and W48 and L50 of strand i+1 (strand 4) are similar, but for the other interlocked pair of strands the proposed amino acids differ somewhat, where we identified V95 but not F97 of strand k (strand 6) and F110 but not Y108 of strand k+1 (strand 7). These differences are probably a reflection of our focus on a finite common core, where both F97 and Y108 deviate somewhat from the hydrophobic kernel. Nevertheless, the similarities are striking, which could imply that a similar folding mechanism is employed by all of these proteins—a type of super-pathway.
Figure 1.
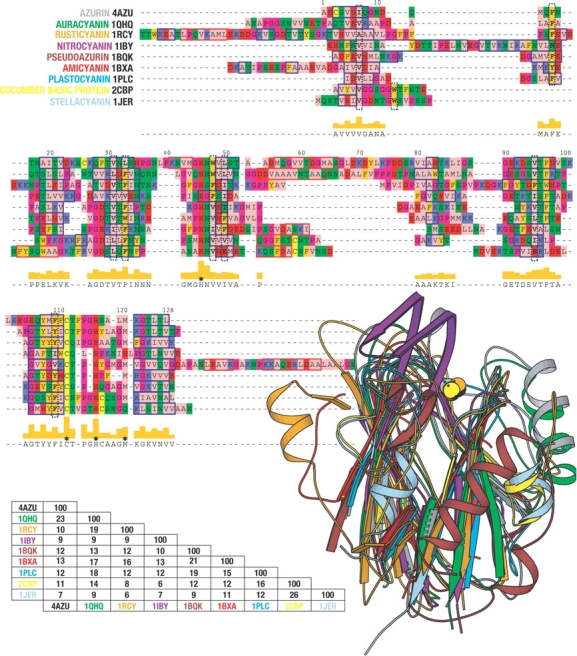
A structural alignment of nine members of the cupredoxin family. The alignment is based on a VAST (Gibrat et al. 1996) alignment, but has been manually improved. The coloring of the alignment is according to the zappo coloring scheme and was created using Pfaat (Johnson et al. 2003). The red boxes indicate β-strands and the blue boxes α-helices, as provided by dssp (Kabsch and Sander 1983). The black dashed vertical boxes indicate structurally conserved residues within the protein core. Stars indicate the position of the metal ligands. Sequence identities were calculated using Pfaat. The images were made using Molscript (Kraulis 1991).
Figure 2.
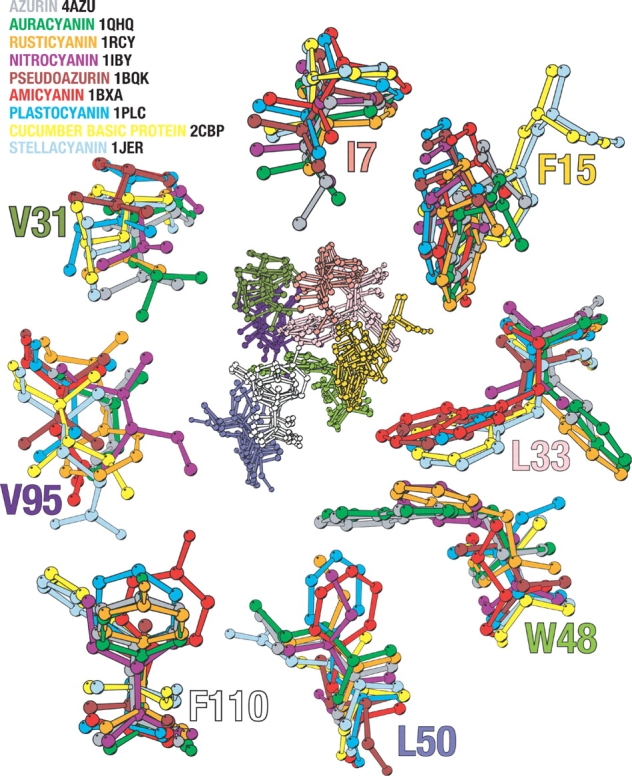
The superimposed hydrophobic core cluster of the nine different cupredoxins is seen in the center of the figure. Surrounding it are enlargements of the residues depicted in the central figure. The enlarged residues are colored according to the legend. The numbering used is according to that of P. aeruginosa azurin. Residues L33 and W48 have been enlarged together, not affecting their orientation with respect to one another. The side chain of these positions occupies the same spatial position with either position 33 or position 48 having a big bulky side chain, never both. In position 15 the case is similar; here the side chain filling the position comes from residues in another strand in stellacyanin, and basic cucumber protein compared to the other cupredoxins. The image was created using Molscript (Kraulis 1991).
We hereby present our protein engineering study that probed the influence of some of these structurally conserved residues on the folding behavior of azurin. The following mutations were thus introduced into the apo-azurin mutant C112S: I7V, V31A, L33A, and L50A, as well as the borderline case of V5A and the complete outlier T52S as controls, resulting in six double mutants. A seventh mutant was also constructed, F110A/C112S, although this species could not be expressed. All of the mutants had native-like fluorescence as well as far- and near-UV circular dichroic (CD) spectra identical to those of the wild-type protein, which all report on the maintained integrity of the native structure. Their folding behavior was then probed by their intrinsic W48 fluorescence, which is effectively reduced to zero intensity at 308 nm upon unfolding of the polypeptide chain. A linear dependence of folding rate on the destabilization of the common core suggests that these residues not only stabilize the native structure, but also the TS. Furthermore, the possible existence of ground-state effects suggests that residual interactions involving these conserved amino acids bias the denatured state toward native-like interactions. Interestingly, the most pronounced effects were observed for the residues previously proposed to constitute the structural determinants of this fold (Kister et al. 2002).
Results
Protein expression and structural characterization
The following mutations were introduced into the apo-C112S template previously described (Sandberg et al. 2002): V5A, I7V, V31A, L33A, L50A, T52S, and F110A, resulting in seven double mutants. All double mutants were expressed at wild-type levels (~100 mg/L LB medium) except the mutant F110A/C112S, which failed to express at detectable levels. F110A/C112S was therefore excluded from further analysis. The native state of all of the expressed double mutants was indistinguishable from those of the C112S apo-mutant and the wild-type protein as probed by tryptophan fluorescence and near- and far-UV CD (data not shown). Furthermore, the far-UV CD spectra in 3 M GdmCl were similar to coils for all mutants, and there was no detectable near-UV CD signal for the C112S mutant in 3 M GdmCl (data not shown). Taken together, no unusual spectroscopic features were discernible in any of the mutants studied here.
Equilibrium measurements
All equilibrium unfolding curves can be accurately described by a two-state transition model irrespective of probes used, here intrinsic tryptophan fluorescence and far-UV CD. Two independent fluorescence measurements of all double mutants were normalized prior to analysis, and the results are presented in Figure 3 ▶. Parameters obtained from these data are presented in Table 1. All double mutants are destabilized compared to C112S, for which the midpoint of denaturation, [D]50%, is 1.53 M GdmCl, in accord with previously published data (Sandberg et al. 2004). The V5A, I7V, and T52S replacements are the least destabilizing with respect to the surrogate wild-type protein C112S, which is illustrated by the lesser shift in the [D]50% (at 1.33, 1.41, and 1.40, respectively) compared to the V31A, L33A, and L50A substitutions, which destabilize the fold significantly by shifting the [D]50% to much lower concentrations (0.95, 0.87, and 0.98 M, respectively). Notably, the mU−N parameters differ somewhat between the mutants, where it is the lesser stable mutants that have the slightly higher transitional slopes.
Figure 3.

Equilibrium unfolding measured by tryptophan fluorescence for the six double mutants V5A/C112S (○), I7V/C112S (▴), V31A/C112S (▾), L33A/C112S (□), L50A/C112S (•), T52S/C112S (▪). C112S (---) is shown as reference. The tryptophan was excited at 295 nm and emission detected at 309 nm. Parameters from the fit (shown as solid lines in the figure) are presented in Table 1.
Table 1.
Thermodynamic equilibrium parameters for the GdmCl−induced unfolding of azurin mutants
| C112S | V5A/C112S | 17V/C112S | V31A/C112S | L33A/C112S | L50A/C112S | T52S/C112S | |
| [D]50% (M−1) | 1.53 ± 0 | 1.33 ± 0 | 1.40 ± 0 | 0.95 ± 0 | 0.87 ± 0 | 0.98 ± 0 | 1.41 ± 0 |
| meq (kJ mole−1 M−1) | 23.5 ± 0.3 | 25.6 ± 2 | 25.7 ± 3 | 28.7 ± 0.8 | 29.1 ± 1 | 29.7 ± 1 | 24.8 ± 1 |
| ΔGU−Neq (kJ mole−1) | 36.4 ± 0.4 | 34.1 ± 3 | 36.0 ± 4 | 27.2 ± 0.8 | 25.3 ± 0.9 | 29.2 ± 1 | 35.0 ± 1 |
Kinetic data
Refolding traces were biphasic for all species at all guanidinium chloride (GdmCl) concentrations. The slower (minor) phase has a concentration-dependent amplitude, constituting ~5% of the relative amplitude at the lowest concentration of GdmCl and ~20% close to the [D]50% for all of the mutants except the V31A/C112S and L50A/C112S mutants, where this minor phase has a maximum amplitude of close to 40% at or just below their [D]50%. The unfolding traces were also biphasic close to the [D]50%, whereas at all GdmCl concentrations typically 0.3 M higher than the [D]50% monoexponential functions could accurately describe the data. The rate constant associated with the minor phase extracted from the unfolding data is of the same magnitude as those extracted from refolding data at the same concentrations, as are their relative amplitudes. This biphasic behavior has been observed previously, and was attributed to a folding-assisted proline isomerization event impeding the folding reaction (Sandberg et al. 2004). The slow phase was therefore not analyzed further in this study, as only the fast rate constant is the one considered to faithfully report on the structure formation process.
The Chevron plots for all double mutants and the C112S reference are depicted in Figure 4 ▶, and the parameters from the fitting of equation 1 are presented in Table 2. It is clear from these data that nonlinear activation-free energies of folding are in effect in all but one mutant, V31A/C112S, where the refolding limb of the Chevron plot instead is nearly linear. In contrast, all unfolding limbs display a pronounced downward curvature. This curvature in both of the limbs cannot be ascribed to aggregation (Silow and Oliveberg 1997) nor to electrolyte artifacts arising form the fact that GdmCl is a salt (Monera et al. 1994; Makhatadze 1999) as described for the C112S mutant (Sandberg et al. 2004).
Figure 4.

Chevron plots for the six double mutants V5A/C112S (○), I7V/C112S (▴), V31A/C112S (▾), L33A/C112S (□), L50A/C112S (•), T52S/C112S (▪). Shown as reference is the fit of C112S (–––) (raw data not shown). The solid lines are the least-squares fitting of equation 1 to the data.
Table 2.
Kinetic parameters for folding and unfolding of the azurin mutants in GdmCl
| C112S | V5A/C112S | I7V/C112S | V31A/C112S | L33A/C112S | L50A/C112S | T52S/C112S | |
| kfH2O (s−1) | 62.9 ± 8 | 36.7 ± 4 | 53.8 ± 5 | 8.20 ± 1 | 19.1 ± 4 | 23.5 ± 4 | 59.2 ± 6 |
| kuH2O (s−1) | (5.68 ± 4) • 10−5 | (2.96 ± 2) • 10−5 | (6.56 ± 4) • 10−5 | (7.04 ± 4) • 10−5 | (8.29 ± 2) • 10−3 | (4.22 ± 2) • 10−4 | (6.90 ± 2) • 10−4 |
| mf (M−1) | −1.94 ± 0.3 − (0.904 ± 0.2) [D] | −2.39 ± 0.3 − (0.858 ± 0.2) [D] | −2.67 ± 0.2 − (0.744 ± 0.2) [D] | −4.36 ± 0.5 − (0.209 ± 0.5) [D] | −2.67 ± 1 − (2.21 ± 1) [D] | −3.62 ± 0.6 − (2.22 ± 0.5) [D] | −1.60 ± 0.3 − (1.32 ± 0.2) [D] |
| mu (M−1) | 6.97 ± 0.6 − (0.871 ± 0.1) [D] | 8.97 ± 0.7 − (1.43 ± 0.2) [D] | 6.71 ± 0.6 − (0.839 ± 0.1) [D] | 9.20 ± 0.7 − (1.72 ± 0.2) [D] | 6.29 ± 0.6 − (0.997 ± 0.2) [D] | 6.06 ± 0.7 − (0.922 ± 0.2) [D] | 5.78 ± 0.5 − (0.706 ± 0.1) [D] |
| Δ GU−Nkin (kJ mole−1) | 33.9 ± 2 | 34.3 ± 2 | 33.2 ± 2 | 28.4 ± 1 | 18.8 ± 1 | 26.2 ± 1 | 27.7 ± 1 |
| Φf (H2O) | — | — | — | 0.53 | 0.26 | 0.32 | — |
| Φf (0.5 M GdmCl) | — | — | — | 0.63 | 0.33 | 0.50 | — |
| βTS | 0.53 | 0.47 | 0.52 | 0.44 | 0.59 | 0.65 | 0.58 |
Extrapolating the fitted kinetic traces to time zero reveals a concentration-dependent burst phase accumulating at GdmCl concentrations below ~0.7 M for the C112S derivative and the most stable mutants: V5A/C112S, I7V/C112S, and T52S/C112S (Fig. 5 ▶). This burst appearance of a fluorescent species is not as apparent in the extrapolated refolding data for the least stable double mutants V31A/C112S, L33A/C112S, and L50A/C112S. It is difficult to ascertain whether the observed lift in the baseline of L33A/C112S corresponds to a nonlinear pretransitional baseline or a proper burst phase in this species.
Figure 5.

Start (○) and end (•) values for the refolding traces. The start values for fluorescence in kinetics experiments were obtained by extrapolating the fit of the kinetic traces back to time 0. Fluorescence intensities are normalized to correct for differences in concentration.
Discussion
The effect of core mutations on the integrity of the structure
In this study, we introduced and analyzed the effect of deletion mutations in the common core of the cupredoxin domain. From a structural alignment, the following residues in P. aeruginosa azurin were identified and mutated accordingly: I7V, V31A, L33A, L50A, and F110A as well as V5A and T52S as controls. These mutations were introduced into the previously characterized apo-C112S mutant (Sandberg et al. 2004). This wild-type surrogate behaves like the wild-type protein, but complications arising from metal contamination and cysteine oxidation reactions that may compromise the homogeneity of the protein solutions are completely abolished by this conservative replacement. When these double mutants are expressed and purified, all but one are obtained in wild-type amounts, which indicate that they adopt stable tertiary structures resistant to proteases. The exception appears to be the double mutant F110A/C112S, probably due to a combination of the large cavity-creating effect of removing a phenyl group and its probable involvement in stabilization of both the transition state and the native structure (see discussion below). Indeed, previous stability measurements on the F110S mutant introduced into Cu2+-coordinating wild-type azurin also revealed this mutation to be extremely destabilizing (Mei et al. 1999).
The fluorescent probe W48 was also identified as a common core residue, but was left intact for technical reasons. This residue is an excellent probe for monitoring the integrity of the native state, because the native-state fluorescence at 308 nm is the least red-shifted of all known protein tryptophans, as both water and charged groups are denied close access to the indole (Eftink 1991; Vivian and Callis 2001). The native-like fluorescence spectra of all mutants is thus a strong indication that the hydrophobic environment around the tryptophan is similar to that of the wild-type protein. That the native state structure is maintained in these mutants is corroborated by the identical far- and near-UV CD spectra (data not shown).
The impact on stability
The equilibrium unfolding data of all mutants can be accurately described by a two-state model. The mU−N-values obtained from individual least-squares fitting of the data for the different mutants differ to some extent and will be discussed further below. Interestingly, the mutants with the lower [D]50%’s have slightly higher mU−N-values, which might be a reflection of different amounts of surface area exposed upon unfolding, because the m-value is directly proportional to this parameter (Myers et al. 1995).
All mutants show a decrease in [D]50% relative to the reference protein C112S, but a slight difference in mU−N-value for the mutants compensates, resulting in very similar ΔGU−NH2O for three of the mutants: V5A/C112S, I7V/C112S, and T52S/C112S (Table 1). The minor decrease in Δ ΔGU−NH2O for these mutants makes the subsequent Φ-value analysis very sensitive, and the calculated values are thus not very reliable (Sanchez and Kiefhaber 2003b). All of the common core residues examined here, except I7, were found to contribute significantly to the stability of the native fold, whereas the other mutations had only a minor effect. Interestingly, the V31 and L33 of strand 3 and L50 of strand 4 are part of the interlocked pair of strands previously proposed to constitute part of the structural determinants of all β-sandwich proteins (Kister et al. 2002).
Folding by the major phase
Two phases are needed to accurately describe the refolding reactions of all apo-azurin mutants in this study, but only the faster (major) phase is considered in detail here, as it is believed to report faithfully on attainment of tertiary structure by the major folding trajectory, whereas the slower (minor) phase is believed to reflect an isomerization reaction (Sandberg et al. 2004). Fortunately, the two phases differ enough in amplitude and rate magnitude that they can easily be separated from each other. Furthermore, the previously detected intermediate in the refolding reaction of C112S (Sandberg et al. 2004) may be neglected on the grounds of it having only a minor effect on the refolding kinetics. We therefore believe that the two-state approximation can be extended to encompass the core mutants in the present study, because none of the double mutants show the same pronounced concentration dependence of the burst phase as does the C112S wild-type surrogate protein, leading us to believe that the intermediate does not accumulate in significant amounts. This is also in accord with the absence of a “kink” in the refolding limbs of all species studied here. Hence, there is no evidence that any of the core mutations stabilizes the intermediate, and all data are treated as apparent two-state kinetic transitions with observed curvature in the Chevron plots being attributed to movement of the TS (Oliveberg 1998).
The minor decrease in Δ ΔGU−NH2O upon mutation of the noncore residues V5 and T52, and also of the I7, precludes Φ-value analysis. For the more destabilized common core mutants, the Φ-values extracted at 0.5 M GdmCl for the folding kinetics are fractional, not exceeding 0.63, and are thus indicative of an expanded native-like TS, in agreement with what was found previously for β-sandwich proteins (Hamill et al. 2000b; Cota et al. 2001; Fowler and Clarke 2001). However, the effect of the mutations in the common core on folding behavior is most easily seen by considering the correlation between stability and folding rate. For the Ig-like domain, a linear correlation between folding rate and stability across remotely homologous proteins indicates that the folding pathway is determined from the common elements of structure that also define the fold (Clarke et al. 1999). For such a mechanism, mutations that are not part of the TS should thus not alter the folding rate, whereas mutations that are implicated in the TS should. When plotting ln kfH2O against ΔGU−NH2O, there appears to be a discernible effect between residues affecting the energy of the TS and those that do not (Fig. 6 ▶). A notable exception from this linear dependency is V31A/C112S. Also, V31A/C112S is an exception as judged by the refolding kinetics. In the Chevron plot, V31A/C112S shows upward curvature compared to the expected two-state folding behavior (Fig. 7 ▶), indicating that this mutant folds via two parallel pathways. The second TS only becomes kinetically accessible in cases where the structure of the main TS is severely destabilized, as we believe is the case for this mutant.
Figure 6.

ln kfH2O varies linearly with ΔGU−NH2O, as seen here for the V5A/C112S (○), I7V/C112S (▴), L33A/C112S (□), L50A/C112S (•), T52S/C112S (▪), and C112S (+) mutants. The V31A/C112S (▾) mutant deviates from this correlation.
Figure 7.

Chevron plots, ln kobs plotted against [GdmCl], for the V31A/C112S (▾) mutant. The dashed line is the reconstructed refolding limb obtained from unfolding kinetic data and the folding free energy from equilibrium data, thus showing the expected refolding kinetics for a two-state reaction.
Upon closer examination of the folding data, it appears that a ground-state effect may contribute negatively to the interpretation of the Φ-values. A ground-state effect can be detected by a comparison of the m-values obtained from kinetics with those from equilibrium (Sanchez and Kiefhaber 2003a). The equilibrium m-value (meq) can be viewed as a measure of the total reaction coordinate length in going from U to N, and the mf and mu parameters extracted from the kinetic data should thus ultimately add up to the meq, as they report on the reaction coordinate in going from U to the TS and the N to the TS, respectively. If the structure of U is altered so that more accessible surface area is exposed upon unfolding, this should show up as an increase in the meq and also in the mf, whereas mu should remain unchanged. When comparing the m-values across the mutants (Fig. 8 ▶), it cannot be ruled out that these ommon core mutations also affect the activation free energies of folding by disrupting residual structure in the denatured state. More mutations are needed to verify the generality of this observation. As nucleation appears to be coupled to hydrophobic condensation in the folding of β-sandwich proteins (Clarke et al. 1999; Hamill et al. 2000b; Cota et al. 2001; Fowler and Clarke 2001), the influence of residual structure on the process may be a general feature in the folding of these proteins because of the strong need to mitigate the high entropic cost of bringing together the nucleus. It is possible that this hints at a general mechanism adopted by these proteins by way of the highly conserved supersecondary β-sandwich motif.
Figure 8.

Dependence of meq (•), mu (▴), and mf (□) for the seven mutants on ΔGU−Nkin. Linear fits of the data show dependence of meq and mf on ΔGU−Nkin, whereas mu is in principle constant.
Materials and methods
Chemicals
Guanidinium chloride (GdmCl) was of highest grade from Apollo Scientific. The concentration of GdmCl was determined by index of refraction (Nozaki 1972), and the pH of the GdmCl-containing solutions was adjusted as proposed (Acevedo et al. 2002).
Mutagenesis and expression
The C112S apo-azurin derivative used as template for the core mutations has been described (Sandberg et al. 2002). All other mutations were introduced with the Quik-Change kit from Stratagene, and the integrity of all mutant species was verified by DNA sequencing. Proteins were expressed in Escherichia coli and purified as described (Karlsson et al. 1989).
Equilibrium fluorescence
All equilibrium fluorescence measurements were done on 4–6 μM protein samples in 10 mM phosphate buffer (pH 7.0) at 20°C. Emission spectra of native protein samples were collected between 300 nm and 460 nm on a SPEX Fluorolog τ2 spectrofluorometer using excitation at 295 nm. For the GdmCl-titration curves, two independent series were measured by monitoring the emission at 309 nm using excitation at 295 nm on either a SPEX Fluorolog τ3 spectrofluorometer or a Bio-Logic MOS-450 spectrometer. Samples for equilibrium folding curves were prepared by titrating 4 μM of protein sample in 0 M GdmCl with the appropriate amount of a 4 μM protein solution in 7.0 M GdmCl, or they were prepared individually and equilibrated overnight—no qualitative difference in parameters was observed, indicating that the equilibrium had gone to completion.
Equilibrium CD
All equilibrium CD measurements were recorded on a Jasco J-810 spectropolarimeter. Far-UV spectra were recorded from 260 to 185 nm on 3 μM protein samples in 10 mM phosphate buffer (pH 7.0) using a 3-mm path length cuvette, whereas near-UV samples were collected from 320 to 260 nm on 0.1 mM protein samples using a 1-cm path-length cuvette. For the GdmCl titrations, 3 μM samples in 10 mM phosphate buffer (pH 7.0) were prepared individually and equilibrated overnight. For these samples, the CD signal was detected at 220 nm at 20°C.
Equilibrium data analysis
All fitting of analytical functions to the data in this article were done with a least-squares fitting procedure as described (Santoro and Bolen 1988; Clarke and Fersht 1993) using the Igor Pro program (Wavemetrics). ΔGU−NH2O can then be calculated from the product of the [D]50% and the mU−N (Clarke and Fersht 1993).
Stopped-flow kinetics
All solutions were made in 10 mM phosphate buffer (pH 7.0), and the temperature was 20°C. The kinetics for folding/unfolding were measured on a Bio-Logic μSFM-20/MPS-52 stopped-flow apparatus with a theoretical dead time of 10.7 msec. Folding/unfolding was initiated by rapid mixing of one part protein (unfolding, 40–60 μM protein in buffer; folding, 40–60 μM in 2–3 M GdmCl) and nine parts buffer with various concentrations of GdmCl. The tryptophan was excited at 295 nm, and fluorescence was detected using a Bio-Logic MOS-450 spectrometer equipped with a 310±2 nm interference filter (Melles Griot). The kinetics were detected for typically between 16 sec and 40 sec, although some points required longer detection times to accurately resolve the slower phase.
Typically six kinetic traces were averaged and the parameters extracted by fitting an exponential decay function to the data (typically two for the refolding and one for the unfolding data). The observed rate constant, kobs, was analyzed by least-squares minimization of the following equation to the Chevron plots (ln kobs against [D]):
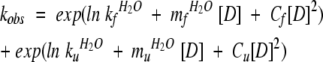 |
(1) |
where mf/uH2O is the linear dependence of folding/unfolding and Cf/u is the parameter describing the curvature observed in the folding/unfolding limbs in the Chevron plot.
Calculation of the β-Tanford values (βTS), which reflects buried surface area of the transition state (TS) relative to the unfolded state, was done according to:
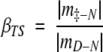 |
(2) |
where |m‡−N| is δΔG‡−N/δ[D], and |mD−N| is δΔGD−N/δ[D].
Φ-values were calculated by taking the folding rate constants and folding free energies at both 0 M GdmCl and 0.5 M GdmCl. At 0.5 M GdmCl, the contribution from the intermediate as judged by the appearance of burst phases is negligible and, furthermore, the [D] is sufficiently away from the transition region of the L33A/C112S mutant (which 0.7 M GdmCl was deemed not to be). All Φ-values were then calculated as (Fersht 1999):
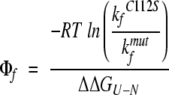 |
(3) |
where kfC112S is the folding rate constant of the C112S mutant, and kfmut is the folding rate constant for the double mutant. The Δ ΔGU−N in this equation is the difference in equilibrium ΔGU−N between the C112S mutant and the double mutant. Hence, the C112S mutant is used as a surrogate wild-type reference.
Acknowledgments
This work has been funded by the Swedish Research Council and the Carl Trygger Foundation.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Abbreviations
ANS, 8-anilino-1-naphtalenesulfonate
CD, circular dichroism;
GdmCl, guanidinium chloride
TS, transition state.
Article published online ahead of print. Article and publication date are at http://www.proteinscience.org/cgi/doi/10.1110/ps.04849004.
References
- Acevedo, O., Guzman-Casado, M., Garcia-Mira, M.M., Ibarra-Molero, B., and Sanchez-Ruiz, J.M. 2002. pH corrections in chemical denaturant solutions. Anal. Biochem. 306 158–161. [DOI] [PubMed] [Google Scholar]
- Adman, E.T. 1991. Copper protein structures. Adv. Protein Chem. 42 145–197. [DOI] [PubMed] [Google Scholar]
- Clarke, J. and Fersht, A.R. 1993. Engineered disulfide bonds as probes of the folding pathway of barnase: Increasing the stability of proteins against the rate of denaturation. Biochemistry 32 4322–4329. [DOI] [PubMed] [Google Scholar]
- Clarke, J., Cota, E., Fowler, S.B., and Hamill, S.J. 1999. Folding studies of immunoglobulin-like β-sandwich proteins suggest that they share a common folding pathway. Structure Fold. Des. 7 1145–1153. [DOI] [PubMed] [Google Scholar]
- Cota, E., Steward, A., Fowler, S.B., and Clarke, J. 2001. The folding nucleus of a fibronectin type III domain is composed of core residues of the immunoglobulin-like fold. J. Mol. Biol. 305 1185–1194. [DOI] [PubMed] [Google Scholar]
- Dobson, C.M. 2003. Protein folding and misfolding. Nature 426 884–890. [DOI] [PubMed] [Google Scholar]
- Eftink, M.R. 1991. Fluorescence techniques for studying protein structure. Methods Biochem. Anal. 35 127–205. [DOI] [PubMed] [Google Scholar]
- Fersht, A. 1999. Structure and mechanism in protein science. A guide to enzyme catalysis and protein folding. W.H. Freeman and Company, New York.
- Fersht, A.R. and Daggett, V. 2002. Protein folding and unfolding at atomic resolution. Cell 108 573–582. [DOI] [PubMed] [Google Scholar]
- Fowler, S.B. and Clarke, J. 2001. Mapping the folding pathway of an immunoglobulin domain: Structural detail from Phi value analysis and movement of the transition state. Structure Camb. 9 355–366. [DOI] [PubMed] [Google Scholar]
- Gibrat, J.F., Madej, T., and Bryant, S.H. 1996. Surprising similarities in structure comparison. Curr. Opin. Struct. Biol. 6 377–385. [DOI] [PubMed] [Google Scholar]
- Gray, H.B., Malmstrom, B.G., and Williams, R.J. 2000. Copper coordination in blue proteins. J. Biol. Inorg. Chem. 5 551–559. [DOI] [PubMed] [Google Scholar]
- Hamill, S.J., Cota, E., Chothia, C., and Clarke, J. 2000a. Conservation of folding and stability within a protein family: The tyrosine corner as an evolutionary cul-de-sac. J. Mol. Biol. 295 641–649. [DOI] [PubMed] [Google Scholar]
- Hamill, S.J., Steward, A., and Clarke, J. 2000b. The folding of an immunoglobulin-like Greek key protein is defined by a common-core nucleus and regions constrained by topology. J. Mol. Biol. 297 165–178. [DOI] [PubMed] [Google Scholar]
- Hemmingsen, J.M., Gernert, K.M., Richardson, J.S., and Richardson, D.C. 1994. The tyrosine corner: A feature of most Greek key β-barrel proteins. Protein Sci. 3 1927–1937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson, J.M., Mason, K., Moallemi, C., Xi, H., Somaroo, S., and Huang, E.S. 2003. Protein family annotation in a multiple alignment viewer. Bioinformatics 19 544–545. [DOI] [PubMed] [Google Scholar]
- Kabsch, W. and Sander, C. 1983. Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 22 2577–2637. [DOI] [PubMed] [Google Scholar]
- Karlsson, B.G., Pascher, T., Nordling, M., Arvidsson, R.H., and Lundberg, L.G. 1989. Expression of the blue copper protein azurin from Pseudomonas aeruginosa in Escherichia coli. FEBS Lett. 246 211–217. [DOI] [PubMed] [Google Scholar]
- Kim, P.S. and Baldwin, R.L. 1982. Specific intermediates in the folding reactions of small proteins and the mechanism of protein folding. Annu. Rev. Biochem. 51 459–489. [DOI] [PubMed] [Google Scholar]
- ———. 1990. Intermediates in the folding reactions of small proteins. Annu. Rev. Biochem. 59 631–660. [DOI] [PubMed] [Google Scholar]
- Kister, A.E., Finkelstein, A.V., and Gelfand, I.M. 2002. Common features in structures and sequences of sandwich-like proteins. Proc. Natl. Acad. Sci. 99 14137–14141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraulis, P.J. 1991. Molscript—A program to produce both detailed and schematic plots of protein structures. J. Appl. Crystallogr. 24 946–950. [Google Scholar]
- Larson, S.M., Ruczinski, I., Davidson, A.R., Baker, D., and Plaxco, K.W. 2002. Residues participating in the protein folding nucleus do not exhibit preferential evolutionary conservation. J. Mol. Biol. 316 225–233. [DOI] [PubMed] [Google Scholar]
- Leckner, J. 2001. “Folding and structure of azurin—The influence of a metal.” Ph.D. thesis, Department of Molecular Biotechnology, Chalmers University of Technology, Göteborg, Sweden.
- Makhatadze, G.I. 1999. Thermodynamics of protein interactions with urea and guanidinium hydrochloride. J. Phys. Chem. B 103 4781–4785. [Google Scholar]
- Martinez, J.C. and Serrano, L. 1999. The folding transition state between SH3 domains is conformationally restricted and evolutionarily conserved. Nat. Struct. Biol. 6 1010–1016. [DOI] [PubMed] [Google Scholar]
- Mei, G., Di Venere, A., Campeggi, F.M., Gilardi, G., Rosato, N., De Matteis, F., and Finazzi-Agro, A. 1999. The effect of pressure and guanidine hydrochloride on azurins mutated in the hydrophobic core. Eur. J. Biochem. 265 619–626. [DOI] [PubMed] [Google Scholar]
- Monera, O.D., Kay, C.M., and Hodges, R.S. 1994. Protein denaturation with guanidine hydrochloride or urea provides a different estimate of stability depending on the contributions of electrostatic interactions. Protein Sci. 3 1984–1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers, J.K., Pace, C.N., and Scholtz, J.M. 1995. Denaturant m values and heat capacity changes: Relation to changes in accessible surface areas of protein unfolding. Protein Sci. 4 2138–2148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nozaki, Y. 1972. The preparation of guanidine hydrochloride. Methods Enzymol. 26 43–50. [DOI] [PubMed] [Google Scholar]
- Oliveberg, M. 1998. Alternative explanations for “multistate” kinetics in protein folding: Transient aggregation and changing transition-state ensembles. Acc. Chem. Res. 31 765–772. [Google Scholar]
- Sanchez, I.E. and Kiefhaber, T. 2003a. Hammond behavior versus ground state effects in protein folding: Evidence for narrow free energy barriers and residual structure in unfolded states. J. Mol. Biol. 327 867–884. [DOI] [PubMed] [Google Scholar]
- ———. 2003b. Origin of unusual Φ-values in protein folding: Evidence against specific nucleation sites. J. Mol. Biol. 334 1077–1085. [DOI] [PubMed] [Google Scholar]
- Sandberg, A., Leckner, J., Shi, Y., Schwarz, F.P., and Karlsson, B.G. 2002. Effects of metal ligation and oxygen on the reversibility of the thermal denaturation of Pseudomonas aeruginosa azurin. Biochemistry 41 1060–1069. [DOI] [PubMed] [Google Scholar]
- Sandberg, A., Leckner, J., and Karlsson, B.G. 2004. Apo-azurin folds via an intermediate that resembles the molten-globule. Protein Sci. (this issue). [DOI] [PMC free article] [PubMed]
- Santoro, M.M. and Bolen, D.W. 1988. Unfolding free energy changes determined by the linear extrapolation method. 1. Unfolding of phenylmethanesulfonyl α-chymotrypsin using different denaturants. Biochemistry 27 8063–8068. [DOI] [PubMed] [Google Scholar]
- Silow, M. and Oliveberg, M. 1997. Transient aggregates in protein folding are easily mistaken for folding intermediates. Proc. Natl. Acad. Sci. 94 6084–6086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefani, M. and Dobson, C.M. 2003. Protein aggregation and aggregate toxicity: New insights into protein folding, misfolding diseases and biological evolution. J. Mol. Med. 81 678–699. [DOI] [PubMed] [Google Scholar]
- van den Berg, B., Ellis, R.J., and Dobson, C.M. 1999. Effects of macromolecular crowding on protein folding and aggregation. EMBO J. 18 6927–6933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vivian, J.T. and Callis, P.R. 2001. Mechanisms of tryptophan fluorescence shifts in proteins. Biophys. J. 80 2093–2109. [DOI] [PMC free article] [PubMed] [Google Scholar]