

Abstract
Utilizing concepts of protein building blocks, we propose a de novo computational algorithm that is similar to combinatorial shuffling experiments. Our goal is to engineer new naturally occurring folds with low homology to existing proteins. A selected protein is first partitioned into its building blocks based on their compactness, degree of isolation from the rest of the structure, and hydrophobicity. Next, the protein building blocks are substituted by fragments taken from other proteins with overall low sequence identity, but with a similar hydrophobic/hydrophilic pattern and a high structural similarity. These criteria ensure that the designed protein has a similar fold, low sequence identity, and a good hydrophobic core compared with its native counterpart. Here, we have selected two proteins for engineering, protein G B1 domain and ubiquitin. The two engineered proteins share ~20% and ~25% amino acid sequence identities with their native counterparts, respectively. The stabilities of the engineered proteins are tested by explicit water molecular dynamics simulations. The algorithm implements a strategy of designing a protein using relatively stable fragments, with a high population time. Here, we have selected the fragments by searching for local minima along the polypeptide chain using the protein building block model. Such an approach provides a new method for engineering new proteins with similar folds and low homology.
Keywords: protein building block, computational protein design, combinatorial assembly, protein G, ubiquitin, molecular dynamics simulation
Protein folding is not a random search process (Levinthal 1968; Wolynes et al. 1995; Dill and Chan 1997; Dobson et al. 1998). Currently, the new view of protein folding with a funnel shape energy landscape (Wolynes et al. 1995; Dill and Chan 1997; Onuchic et al. 1997; Brooks et al. 1998) appears to most appropriately describe the observed protein folding processes. Nevertheless, some experiments (Bai et al. 1995) have shown that folding can be considered to occur as a sequential process rather than in numerous different pathways. The building block folding model (Lesk and Rose 1981; Baldwin and Rose 1999a,b; Tsai and Nussinov 2001b; Tsai et al. 2002), which states that protein folding is a process of combinational assembly of building blocks, is a “practical” folding model along the guidelines of the views of funnel energy landscape. An arbitrary fragment in a protein is considered as a building block if one or some preferred conformations are more stable (or with a higher population time) than other alternative conformations (Tsai et al. 2000, 2002; Tsai and Nussinov 2001a). Based on concepts of hierarchical protein folding, the building block model defines a protein building block by means of compactness, degree of isolation, and hydrophobicity of candidate building blocks (Tsai et al. 2000). The results are consistent with limited proteolysis experiments (Tsai et al. 2002). In this model, proteins in the same family yield very similar building blocks. However, because a building block fragment is a conformationally independent entity, building blocks from different protein families can also share similar building block structures (Haspel et al. 2003a). This suggests that building blocks may be useful in designing proteins. To test this idea, we use a new computational algorithm to engineer proteins with naturally occurring folds; however, with low sequence homology. The sequence identity is kept as low as possible to avoid a homology-based bias.
The computational procedures are outlined in Figure 1 ▶: Briefly,
Figure 1.
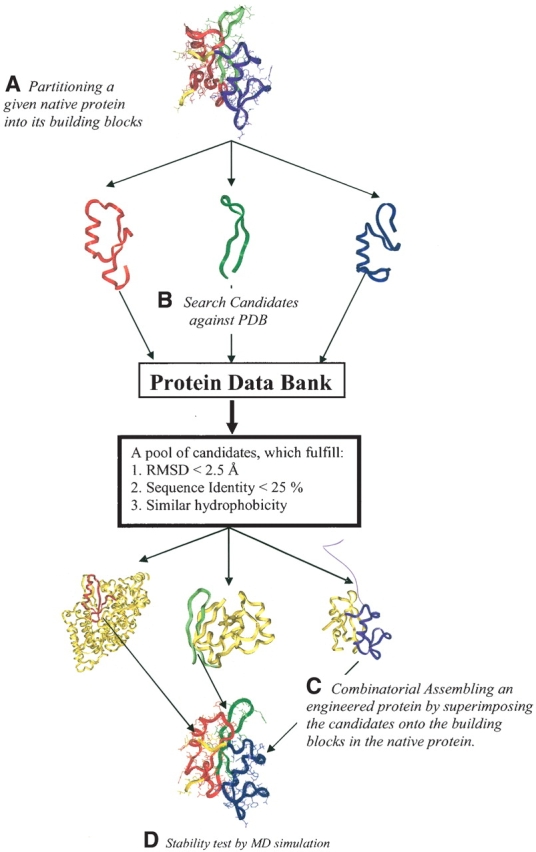
Graphic outline of the computational protein engineering algorithm. (A) A given native protein 3D structure is partitioned into building blocks using a computational cutting algorithm and a fragment length independent scoring function (Tsai et al. 2000). (B) Using the structure and sequence of building blocks from step A, candidate substitution fragments are searched against the PDB. The goal is a Cα-RMSD smaller than 2.5 Å and sequence identity lower than 25%. At the same time, candidate building blocks should have a similar hydrophobic/hydrophilic pattern. (C) By superimposing the selected substitution fragments onto the native protein, the new engineered protein is constructed by combinatorial assembly. (D) Finally, the stability of the engineered proteins are examined by explicit water MD simulations. This algorithm ensures the engineered protein has the similar fold as its parent protein, but with low sequence identity.
In a given protein, its 3D topology is partitioned into building blocks based on a building block cutting algorithm and a scoring function (Tsai et al. 2000).
Each building block is searched against the Protein Data Bank to find candidates of substitute fragments. A candidate should have low root-mean-squared deviation (RMSD), here <2.5 Å), similar hydrophobicity and low sequence identity (<25%). Several candidates can be found, depending on the topology and sequence of each building block.
The “best” candidate is selected from the pool. Its topology is superimposed onto the building block in the original native protein. These procedures (A, B, and C) are repeated for each building block. Then, the engineered protein is built by combinatorial assembly.
Finally, the stability of the engineered protein is examined by explicit water molecular dynamics simulations.
In our algorithm, the criterion of hydrophobicity will ensure that the candidates will have similar hydrophobic/hydrophilic pattern as the original building blocks. On the other hand, the small RMSD criterion constrains the candidates to those with similar topology as the original building blocks. In the combinatorial assembly procedure, candidates are superimposed onto their corresponding building blocks in the native protein. Thus, this procedure ensures that the engineered protein will have a fold similar to the original native protein, similar hydrophobic and hydrophilic pattern, but low sequence identity. The minor nonequilibrium energy, which may exist in the original engineered proteins, is removed by force field energy minimization.
The algorithm proposed here is very similar to experiments of protein domain swapping and combinatorial shuffling of polypeptide segments except that the “domain” is defined by building blocks in our algorithm. In the computational and experimental domain swapping design study, Voigt et al. (2002) defined the protein building blocks by minimum disturbance of the integrity of the protein 3D structure using concepts of schema theory of genetic algorithms. Building blocks defined either by minimum disturbance or by fold independence can be regarded as relatively stable protein fragments in a given protein. Mayo and Arnold (Meyer et al. 2003) have further constructed a combinatorial library to estimate the disruption caused upon substitution of schemas due to altered interactions in the 3D structures upon schema shuffling. Other fragment-based approaches include protein design by phage display libraries. This strategy has been employed to computationally and experimentally design a four-helix bundle protein (Chu et al. 2002), coupling phage display and proteolysis. Interestingly, the authors find that the positions of the cutting sites of the protease may significantly influence the selection of structures. Pioneering studies of limited proteolysis by Fontana et al. (1997, 1999) have long shown that fragments obtained through a limited proteolysis strategy can be combined to yield the native protein. This suggests that fragments obtained through such applications can be used both for studies of protein folding pathways and for protein design. The number of potential combinations in protein design is huge, as shown in the first pioneering completely automated zinc finger redesign by Mayo and his colleagues (Dahiyat and Mayo 1997; Dahiyat et al. 1997). Fragment-based approaches reduce the number of combinations in a designed protein. An alternate algorithm to reduce the huge number of degrees of freedom involves a statistical computationally assisted design strategy. This method has recently successfully designed water-soluble analogs of a potassium channel (Slovic et al. 2004) and a monomeric helical dinuclear metalloprotein (Calhoun et al. 2003). Still another promising strategy involves an application of the Rosetta Design algorithm (Dantas et al. 2003). Additionally, new protein engineering techniques using multiple stabilizing substitutions were recently employed by Peng and coworkers (Cammett et al. 2003). These techniques were shown to yield remarkable results, enhancing the stability of cyclin-dependent kinase inhibitor and renovating Cdk4 binding activity of several flawed cancer-associated mutant proteins.
Recombination is a powerful tool for the engineering and optimization of proteins in vitro (Crameri et al. 1998; Riechmann and Winter 2000). It enhances design through combination of fragments from different proteins to form a new protein with a potential new function. Here, rather than substituting a single residue at each location, our approach substitutes fragments. Importantly the fragment size varies, depending on its identification as a local minimum along the polypeptide chain. The minimum size is 15 amino acids, and the maximum can be any size. A fragment-based approach reduces the computational cost dramatically. At the same time, criteria such as those defined above ensure that the topology and hydrophobic/hydrophilic patterns of engineered protein are similar to the native protein. The similarity between an engineered protein and its parent native protein will likely ensure that the engineered protein has good opportunity to be stable.
Two proteins, protein G B1 domain (PDB code: 2gb1) and ubiquitin (PDB code: 1ubq), were selected for engineering. These two engineered proteins share ~20% and ~25% amino acid identity, respectively. Like native proteins, the engineered proteins also have a hydrophobic core and the hydrophilic side chains are exposed to the protein surface. In addition, the engineered proteins have similar folds as their corresponding native proteins. On the other hand, two “nonproteins” with inverted polar/nonpolar residue patterns (with no or poor hydrophobic cores) based on the topologies of protein G B1 domain and ubiquitin were also engineered for control. The stabilities of the engineered and control proteins were tested by explicit water molecular dynamics simulations. Employing this computational algorithm, we are able to engineer new, similar fold, low homology proteins based on a selected native protein, and to examine the idea whether the building blocks are stand-alone fragments. The computational methods developed here may assist in combinatorial design of new functional proteins.
Computational algorithm
There are three major procedures in computational algorithm of protein engineering: (1) building block (BB) cutting algorithm, (2) candidate BB searching and in silico protein engineering algorithms, and (3) stability tests by molecular dynamics simulations. The tertiary structure of selected native protein is partitioned into a set of building blocks by estimating their compactness, degree of isolation, and hydrophobilicity. The building blocks are regarded as relatively stable and highly populated fragments. Based on the structure, sequence, and the hydrophobilicity pattern of the building blocks, candidate BBs with similar structure, low sequence identity, and similar hydrophobic/hydrophilic pattern are searched against the Protein Data Bank (Berman et al. 2000). The best candidate BBs are superimposed onto the corresponding BBs’ Cα architectures. Finally, the stability of the engineered protein is examined by molecular simulations. The three procedures are described as below.
Building block cutting algorithm
The detailed description of the building block cutting algorithm has been published elsewhere (Tsai et al. 2000), and is only briefly outlined here. A scoring function estimates the relative stability of a candidate building block. The scoring function is expressed as:
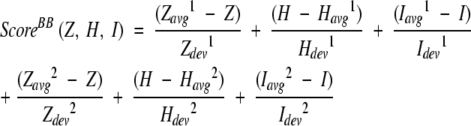 |
(1) |
where Z, H, and I are the compactness, hydrophobicity, and degree of isolation, respectively. The hydrophobicity score (H) is defined as the fraction of the buried nonpolar surface area over the total nonpolar surface area,
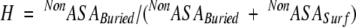 |
(2) |
where NonASABuried and NonASASurf are the buried and the exposed nonpolar surface area (Tsai et al. 2000). The subscripts avg and dev are the arithmetic average and the standard deviation, respectively, obtained from a nonredundant data set of 930 representative single chain proteins. Quantities with superscripts 1 and 2 are calculated with respect to fragment size and a function of the fraction of fragment size to the entire protein, respectively. The selected candidate BB has a high stability score as estimated from equation 1, which represents the minimum deviation from the averaged values. Fragments with various lengths (minimum 15) are estimated for their stability scores. The procedure is carried out iteratively until the building blocks can no longer be cut. The resulting spanning tree delineates the most likely protein folding pathways.
Candidate BB search and in silico protein engineering algorithms
Once a native target protein has been cut into its building blocks, the structures and sequences of its BBs are used to search the PDB for substitute fragments. Four criteria are used in the candidate BBs search:
Low Cα-RMSD: The Cα-RMSD (original vs. candidate BB) is expected to be as small as possible (<2.5 Å).
Low sequence identity: To avoid selection of a homologous protein, the candidate BB should have sequence identity lower than 25%. No residue insertion or deletion is considered.
-
Similar hydrophobic/hydrophilic pattern: Binary and hydrophobilicity patterns are used in the candidate BB search. The binary pattern is calculated by comparing the sequence hydrophobic and hydrophibilic similarities. Candidate BB with a higher binary pattern is selected (usually higher than 70%). In contrast to the binary pattern, we introduce another quantity called hydrophobilicity pattern, calculated from the experimental hydrophobicity scale (EHS; Fauchere and Pliska 1983) difference between the original and candidate building blocks. This criterion selects candidates with similar side-chain environments as the original building blocks. The hydrophobilicity pattern is defined as:

(3) where N is the number of residues and the 〈expectation value〉 is the expected value of the experimental hydrophobicity scale (EHS) difference between the 20 amino acids. The experimental hydrophobicity scales are taken from Fauchere and Pliska’s work, and the expectation value is 1.151 based on this scale (Fauchere and Pliska 1983). Therefore, for a candidate without any similarity to the original BB, we expect its hydrophobilicity pattern to be equal to a unit. A selected candidate has a smaller hydrophobilicity pattern.
No disulfide bond or cofactor is considered. Either disulfide bond or cofactor can stabilize the proteins, which may not allow us to examine the performance of our algorithm fairly. Thus, fragments with disulfide bond or cofactor are excluded.
In this study, 19,294 protein structures with a total of 36,653 chains (when chain length >15) deposited in the Protein Data Bank were searched. Finally, the engineered protein is assembled by superimposing the candidate BBs onto the native protein architecture. To ensure that two connected BBs are covalently joined properly, larger (10 times) weighting factors are used for the N- and C-terminal Cα atoms of each candidate BB in the superimposition and assembly procedures. The unassigned fragments (i.e., those between BBs) are kept in the engineered protein. These criteria and procedures ensure that the engineered protein will have a similar fold as the native protein. On the other hand, it will have low sequence identity. Additionally, it will also own a good hydrophobic core.
Stability test by MD simulations
The stability of the engineered proteins is tested by molecular dynamics (MD) simulations. To assess whether a protein is stable and folded by computer simulations is a challenging task. It is not only limited by the accuracy of the theory (e.g., force field), but also restricted by the computer power (i.e., simulation time). Protein folding is on the milliseconds to microseconds to seconds time scale. Current computers are incapable of routinely offering such long time simulations. The engineered proteins constructed based on the algorithms proposed above are assumed to have structures similar to their native structures. Namely, the original engineered protein may have a structure similar to its native one. Therefore, explicit water MD simulation on an order of nanosecond simulation time might be long enough to serve as a first test in examining the stability of the engineered proteins.
All simulations were performed with CHARMM (Brooks et al. 1983). The system was treated explicitly with the all atom model using CHARMM-22 force field (MacKerell et al. 1998). A series of MD simulations were performed for the native, engineered, and nonproteins at room temperature with the explicit water TIP3P model (Jorgensen et al. 1983). The proteins were solvated with explicit water molecules in a cubic box. The size of box depends on the size of the protein to preserve infinite dilution. All simulations were performed using the NVT ensemble under periodic boundary conditions with the minimum image convention. The systems were energy-minimized by the Adopted Basis Newton-Raphson (ABNR) prior to the MD simulations. A group based distance cutoff was applied at 12 Å and 13 Å when generating the list of pairs. The force switching function was used to smooth the electrostatic potential energy (pair-wise distances between 8–12 Å), whereas the van der Waals shift function was used to smooth the van der Waals potential energy (Steinbach and Brooks 1994). The non-bonded neighboring list was updated every 20 steps. In the simulations, the Cα-RMSD of the native proteins was expected to be lower than that of the engineered proteins, which was used as the low bound reference. In contrast, in the absence of compact hydrophobic core, the Cα-RMSD of nonproteins was expected to be higher. Thus, the Cα-RMSD of a nonprotein was employed as the upper bound reference.
Results
Two proteins are engineered. Their corresponding parent native proteins are protein G B1 domain (PDB code: 2gb1) and ubiquitin (PDB code: 1ubq). For convenience, these two native proteins are abbreviated as nat-2gb1 and nat-1ubq. The proteins engineered based on nat-2gb1 and nat-1ubq have good hydrophobic cores are denoted as eng-2gb1 and eng-1ubq, respectively. In contrast to the engineered proteins, two proteins are also assembled with small or inverted polar/nonpolar residue patterns (called nonproteins). The candidate building blocks selected for assembling the nonproteins own a larger hydrophobilicity pattern (>1.00). They are labeled as non-2gb1 and non-1ubq, respectively.
Protein G B1 domain
Protein G B1 domain consists of 56 residues with two building blocks (Fig. 2A ▶). Building block-I (BB-I) has 38 residues (residues 2–39) and building block-II (BB-II) has 20 residues (residues 37–56). Residue 1 in the N terminus is unassigned and is kept in the engineered protein. For convenience, the three overlapped residues (37–39) between building blocks-I and -II are assigned to BB-II only. Therefore, the adjusted BB-I has 35 residues (from 2 to 36) and BB-II has its original 20 residues (from 37 to 56). The sequence of native-2gb1 is shown in Table 1.
Figure 2.
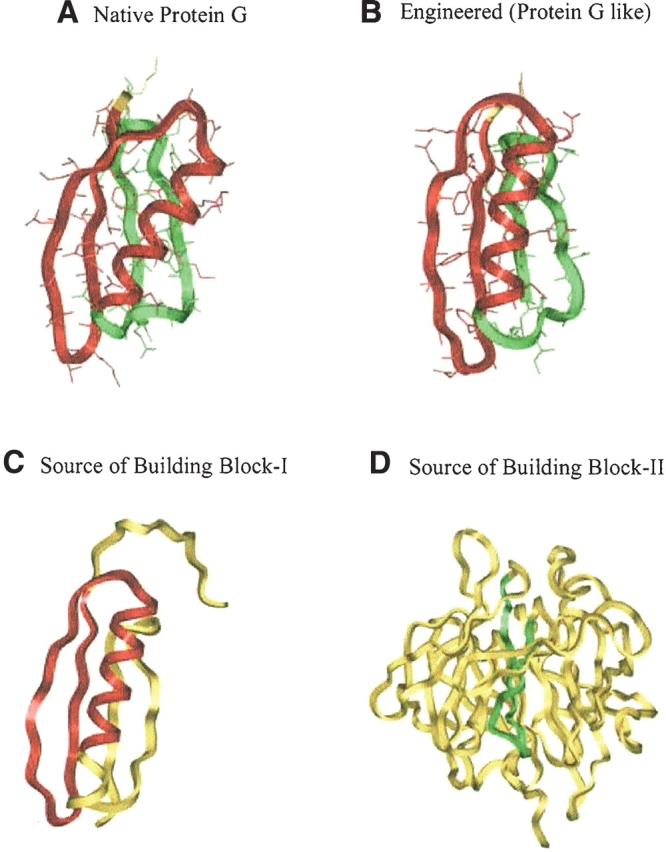
The structures and building blocks of the native protein G B1 domain and the engineered protein. The sources used in engineering protein are also shown. Building block-I (BB-I) is shown in red and building blocks-II (BB-II) is highlighted in green. The unassigned and unused residues are in yellow. (A) The two building blocks of the native protein G B1 domain. Building block-I (BB-I, residues 2–36) consists of a β-hairpin and an α-helix. Building block-II (BB-II, residues 37–56) is a β-hairpin. (B) The two building blocks of the engineered protein (protein G-like; eng-2gb1). Similar to BB-I in nat-2gb1, BB-I of eng-2gb1 also consists of a β-hairpin and an α-helix, whereas BB-II is a β-hairpin. (C) The structure of the Protein L B1 Domain, the source protein used for the engineered protein. The fragment used for engineering eng-2gb1 is highlighted in red. (D) The structure of diisopropylfluorophosphatase, the source protein of BB-II in eng-2gb1. The fragment used in engineering eng-2gb1 is denoted in green.
Table 1.
This table lists the detailed information of engineered protein (eng-2gb1) and native protein G B1 domain (nat-2gb1)
| Source | PDB code | Residue numbera | RMSD (Å)b | Sequence identity | Binary pattern | Hydrophobicity patternc | |
| BB-I | Protein L B1 domain | 1k53(A) | 5–39 | 2.43 | 20.0% | 74.3% | 0.83 |
| BB-II | Diisopropylfluorophosphatase | 1e1a(A) | 70–89 | 1.74 | 15.0% | 75.0% | 0.41 |
| eng-2gb1d | — | — | — | 2.21 | 19.6% | 75.0% | 0.67 |
| Amino acid sequence information | |||||||
| | – – – – – – – – – – – – – – – B B – I – – – – – – – – – – – – – – | – – – – – – – B B – I I – – – – – – – | | |||||||
| Residue #: | ——— • ——— 1 ——— • ——— 2 ——— • ——— 3 ——— • ——— 4 ——— · ——— 5 ——— • — | ||||||
| Binary pattern: | * * * * * –* –* – * * * * * * –* – – * * * –* * * * – * * – * * * * * * * – * * * * * * * – – * * * – *– * | ||||||
| nat-2gb1: | M T Y K L I L N G K T L K G E T T T E A V D A A T A E K V F K Q Y A N D N G V D G E W T Y D D A T K T F T V T E | ||||||
| eng-2gb1: | M T I K A N L I F A N A S T Q T A E F K G T F E K A T S E A Y A Y A D T G G I P A G C Q C D R D A N Q L F V A D | ||||||
| Sequence identity: | * *– * – –* – – – – – – – – * – – – – – – – – –* – – – – – – * * – – –* – – – – – – –* – – – – – – – *– – | ||||||
aThe numbers shown here are the residue numbers in their original corresponding PDB files.
bThe RMSDs are calculated against native structure and are based on Cαatoms only.
cHydrophobicity patterns are calculated from equation 3.
dThe unassigned fragment (residue 1) is included in computing the information of engineered protein (eng-2gb1). The RMSDs are calculated based on the
Cα atoms of initial assembled structure against nat-2gb1. Other information of eng-2gb1, sequence identity, binary pattern, and hydrophobicity pattern, is calculated against nat-2gb1.
To engineer a new protein based on our criteria and procedures, the BBs in the native protein are searched against the PDB. More than one candidate is usually found. The candidate distribution for the nat-2gb1 BBs is shown in Figure 3 ▶. The distribution is separated into two groups: one with higher sequence identity, and a smaller hydrophobicity scale, the second one with lower sequence identity, but with a higher hydrophobicity scale. Candidates with low sequence identity and low hydrophobicity scale are selected. Even though there is usually more than one candidate that fulfills these criteria, only the best one is selected for the assembly. For the engineered protein based on protein G (Fig. 2B ▶; eng-2gb1), BB-I is adopted from protein L B1 domain (Fig. 2C ▶, PDB code: 1k53[A]) with an RMSD of 2.4 Å compared to the native BB-I (in nat-2gb1). BB-II is tailored from diisorpropylfluorophosphatase (Fig. 2D ▶; PDB code: 1e1a[A]) with an RMSD of 1.7 Å compared to the BB-II in the native protein. With respect to the whole native protein, the initial structure of the engineered eng-2gb1 has an overall RMSD of 2.2 Å, sequence identity of 19.6%, binary pattern of 73.2%, and hydrophobicity pattern of 0.67. Therefore, eng-2gb1 has similar fold and low homology to nat-2gb1. Sequence and structural information of eng-2gb1 and nat-2gb1 are given in Table 1.
Figure 3.
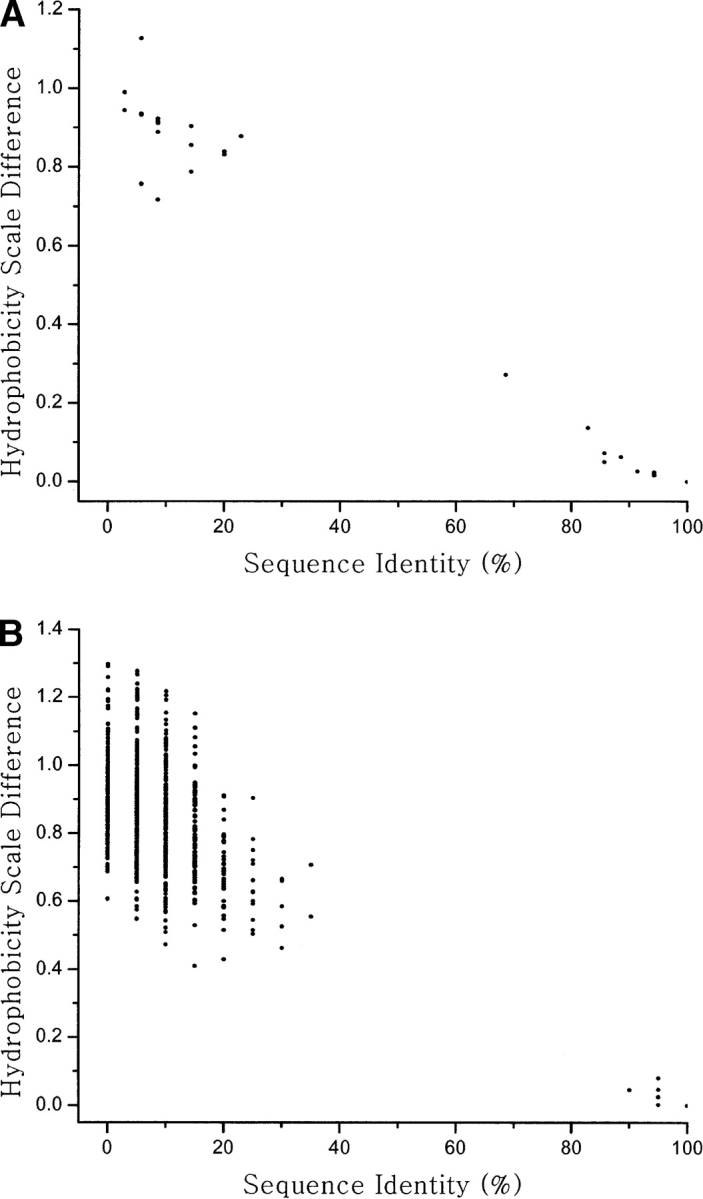
The distribution of candidate fragments for engineering of the native protein G. The distribution of candidate fragments are projected on two coordinates, sequence identity, and hydrophobicity scale difference when their Cα-RMSD is smaller than 2.5 Å. (A) The candidate fragment distribution for building block-I of the native protein G. (B) The candidate fragment distribution for building block-II of the native protein G. Fragments with lower sequence identity and smaller hydrophobicity scale difference are used for engineering protein G.
The nonprotein (non-2gb1) is assembled with inverted polar/nonpolar residue pattern. Its BB-I is adopted from acetate kinase (PDB code: 1g99[A]) and BB-II is from thioredoxin reductase (PDB code: 1tdf). In contrast to eng-2gb1, the building blocks of non-2gb1 have a larger hydrophobicity pattern. Overall, due to the inverted pattern requirement, the nonprotein is less similar to nat-2gb1. The RMSD of the initial structure of the non-2gb1 is 2.3 Å; the sequence identity is only 5.3%; the binary pattern is 50%; and the hydrophobicity pattern is 1.17. The non-2gb1 information is in Table 2.
Table 2.
This table lists the detailed information of nonprotein (non-2gb1)
| Source | PDB code | Residue numbera | RMSD (Å)b | Sequence identity | Binary pattern | Hydrophobicity patternc | |
| BB-I | Acetate kinase | 1g99(A) | 38–72 | 2.46 | 5.7% | 48.6% | 1.13 |
| BB-II | Thioredoxin reductase | 1tdf | 80–99 | 2.09 | 0.0% | 50.0% | 1.29 |
| non-2gb1d | — | — | — | 2.34 | 5.4% | 50.0% | 1.17 |
| Amino acid sequence information | |||||||
| | – – – – – – – – – – – – – – B B – I – – – – – – – – – – – – – – | – – – – – – – – B B – I I – – – – – – – | | |||||||
| Residue #: | ——— • ——— 1 ——— • ——— 2 ——— • ——— 3 ——— • ——— 4 ——— • ——— 5 ——— • — | ||||||
| Binary pattern: | * * * –* – – * * – * * * * –* * – * – * – – – * – – – – – – – * * – * –* – * * * * –* – * –* – – – – – * * | ||||||
| native-2gb1: | M T Y K L I L N G K T L K G E T T T E A V D A A T A E K V F K Q Y A N D N G V D G E W T Y D D A T K T F T V T E | ||||||
| non-2gb1: | M N S I I T Q K K F | D G K K L E K L T D L P T H K D A L E E V V K A L T I F D H I N K V D L Q N R P F R L N G D | |||||
| Sequence identity: | * – – – – – – – – – – – * – – – – – – – – – – – – – – – – – – – – * – – – – – – – – – – – – – – – – – – – – – – | ||||||
aThe numbers shown here are the residue numbers in their original corresponding PDB files.
bThe RMSDs are calculated against native structure and are based on Cα atoms only.
cHydrophobicity patterns are calculated from equation 3.
dThe unassigned fragment (residue 1) is included in computing the information of nonprotein (non-2gb1). The RMSDs are calculated based on the Cα atoms of initial assembled structure against nat-2gb1. Other information of non-2gb1, sequence identity, binary pattern, and hydrophobicity pattern, is calculated against nat-2gb1.
The binary pattern of nat-2gb1, eng-2gb1, and non-2gb1 in their 3D structures are shown in Figure S1 (in the Supplemental Material). Clearly, nat-2gb1 and eng-2gb1 have larger hydrophobic cores, and the hydrophilic residues are located on the surface. In contrast, the hydrophobic core of the nonprotein is relatively smaller. The computed hydrophobicity also shows that nat-2gb1 and eng-2gb1 have higher hydrophobicity scores (H; equation 2). In contrast, the hydrophobicity score of non-2gb1 is low. The hydrophobicity scores of nat-2gb1, eng-2gb1, and non-2gb1 are summarized in Table S (in the Supplemental Material).
Ubiquitin
Ubiquitin has 76 residues with three building blocks, BB-I (residues 1–21), BB-II (residues 21–41), and BB-III (residues 42–68). For simplicity, the overlapped residue 21 is assigned to BB-II. Residues 69–76 in the C terminus are unassigned. BB-I is a β-hairpin; BB-II is an α-helix–rich fragment; BB-III is a large loop. The structure of native ubiquitin (nat-1ubq) along with its three building block sources is shown in Figure 4 ▶.
Figure 4.

The structure and three building blocks of the native ubiquitin and the engineered ubiquitin. The sources used for engineering eng-1ubq are also shown. BB-I is shown in red, BB-II is shown in green, and BB-III is highlighted in blue. The unassigned and unused fragments are shown in yellow. (A) The native ubiquitin: Building block-I (BB-I, residues 1–20) consists of a β-hairpin. Building block-II (BB-II, residues 21–41) is an α-helix rich fragment. Building block-III (BB-III, residues 42–68) is a loop. (B) The structure and building blocks of eng-1ubq. Similar to the BB-I in nat-1ubq, BB-I of eng-1ubq is a β-hairpin. BB-II is an α-helix rich fragment. BB-III is a loop. (C) The structure of Escherichia coli topoisomerase I, the source protein of BB-I of eng-1ubq. The residues used as BB-I of eng-1ubq are shown in red. (D) The structure of urinate isomerase, the source protein of BB-II of eng-1ubq. The residues used for BB-II of eng-2gb1 are shown in green. (E) The structure of Sumo-I, the source protein of BB-III of eng-1ubq. The residues used as BB-III of eng-1ubq are shown in blue.
The candidate distribution of nat-1ubq’s BBs is shown in Figure 5 ▶. The candidates of BB-I and BB-II of nat-1ubq are shown when their RMSDs are smaller than 2.5 Å. The RMSD criterion used for BB-III is 2.8 Å. Only one candidate has an RMSD smaller than 2.5 Å for the BB-III loop. The best candidate for each building block that fulfills the criteria of low sequence identity and small hydrophobicity scale difference, is used for engineering the protein. For eng-1ubq (Fig. 4B ▶), BB-I is taken from L-Amino Acid Oxi-dase (Fig. 4C ▶, PDB code: 1f8r[A]); BB-II from 2c-Methyl-D-Erythritol 2,4-Cyclodiphosphate Synthase (Fig. 4D ▶, PDB code: 1jn1[A]); and BB-III is adopted from the ubiquitin-like protein SMT3 (Fig. 4E ▶, PDB code: 1euv[B]). Although BB-III is adopted from an ubiquitin-like protein, the sequence identity is still under 25%. Overall, the sequence identity of eng-1ubq is 26.3%. The RMSD, binary pattern, and hydrophobilicity pattern of entire eng-1ubq are 1.51 Å, 85.5%, and 0.41, respectively. The sequence information of eng-1ubq along with nat-1ubq is given in Table 3.
Figure 5.
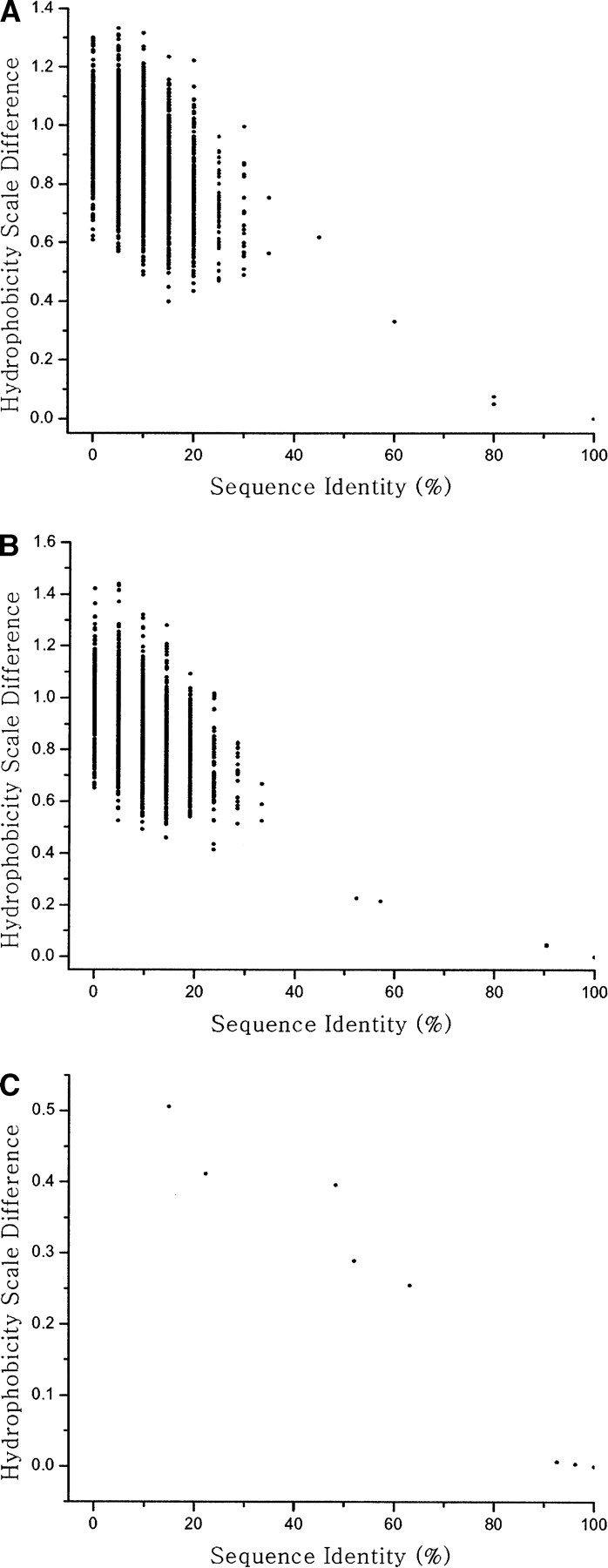
The distribution of candidate fragments for engineering of native ubiquitin. The distribution of candidate fragments is projected in two coordinates, sequence identity, and hydrophobicity scale difference when their Cα-RNSDs are smaller than 2.5 Å (2.8 Å for BB-III). (A) The candidate fragment distribution for building block-I, (B) for building block-II, and (C) for building block-III of native ubiquitin. Fragments with lower sequence identity as well as the smaller hydrophobicity scale difference are used for engineering ubiquitin.
Table 3.
This table shows the detailed information of engineered ubiquitin (eng-1ubq) and native ubiquitin (nat-1ubq)
| Source | PDB code | Residue numbera | RMSD (Å)b | Sequence identity | Binary pattern | Hydrophobicity patternc | |
| BB-I | L-Amino acid oxidase | 1f8r(A) | 271–290 | 1.11 | 15.0% | 95.0% | 0.40 |
| BB-II | 2c-Methyl-D-erythritol 2,4-Cyclodiphosphate synthase | 1jn1(A) | 108–128 | 1.53 | 23.8% | 71.4% | 0.44 |
| BB-III | Ubiquitin-like protein SMT3 | 1euv(B) | 64–90 | 0.86 | 14.8% | 85.2% | 0.51 |
| eng-1ubqd | — | — | — | 1.51 | 26.3% | 85.5% | 0.41 |
| Amino acid sequence information | |||||||
| | – – – – – – – B B – I – – – – – – – | – – – – – – – – B B – I I – – – – – – – | | |||||||
| Residue #: | ——— • ——— 1 ——— • ——— 2 ——— • ——— 3 ——— • ——— 4 ——— • — | ||||||
| Binary pattern: | * * * * * * * * * * * * * * * * * * * – –* * * – * * * * * – * * –* * – * * * – | ||||||
| nat-1ubq: | M Q I F V K T L T G K T I T L E V E P S D T I E N V K A K I Q D K E G I P P D Q Q | ||||||
| eng-1ubq: | V T V V Y E T L S K E T P S V T A D Y V P H I D A M R A K I A E D L Q C D I E Q V | ||||||
| Sequence identity: | − − − − − − + + − − − + − − − − − − − − − − + − − − − + + + − − − − − − − − − + − | ||||||
| | – – – – – – – – – – B B – I I I – – – – – – – – – – | | |||||||
| Residue #: | ——— • ——— 5 ——— • ——— 6 ——— • ——— 7 ——— • — | ||||||
| Binary pattern: | * * * * –* –* * * – * * * * * * * * ** * * * – * * * * * * * * * * | ||||||
| nat-1ubq: | R L I F A G K Q L E D G R T L S D Y N I Q K E S T L H L V L R L R G G | ||||||
| eng-1ubq: | R F L Y D G I R I Q A D Q T P E D L D M E D N D I I E L V L R L R G G | ||||||
| Sequence identity: | + − − − − + − − − − − − − + − − + − − − − − − − − − − * * * * * * * * * | ||||||
aThe numbers shown here are the residue numbers in their original corresponding PDB files.
bThe RMSDs are calculated against native structure and are based on Cα atoms only.
cHydrophobicity patterns are calculated from equation 3.
dThe unassigned fragment (residues 69–76) is included in computing the information of engineered protein (eng-1ubq). The RMSDs are calculated based
on the Cα atoms of initial assembled structure against nat-1ubq. Other information of eng-1ubq, sequence identity, binary pattern, and hydrophobicity
pattern, is calculated against nat-1ubq.
The nonprotein (non-1ubq) is assembled with an inverted polar/nonpolar residue pattern. Its BB-I is adopted from horse plasma gelsolin (PDB code: 1d0n[A]; BB-II from thymidylate synthase (PDB code: 1vza); BB-III is from phosphotransferase (PDB code: 2hid). The RMSD of BB-III is relatively higher (3.4 Å). Because BB-III is a larger loop, there is no good candidate with RMSD under 2.5 Å. In contrast to the eng-1ubq, the hydrophobicity patterns of building blocks of non-1ubq are larger than 1.00. In non-1ubq, some residues in the inner protein core are hydrophilic. The RMSD of non-1ubq is 2.9 Å, the sequence identity is only 14.7%, the binary pattern is 42.1%, and the hydrophobicity pattern is 1.19. Details of non-1ubq are summarized in Table 4.
Table 4.
This table shows the detailed information of non-ubiquitin (non-1ubq)
| Source | PDB code | Residue numbera | RMSD (Å)b | Sequence identity | Binary pattern | Hydrophobicity patternc | |
| BB-I | Horse plasma gelsolin | 1d0n(A) | 70–89 | 2.47 | 10.0% | 35.0% | 1.32 |
| BB-II | Thymidylate synthase | 1vza | 53–73 | 2.38 | 4.8% | 33.3% | 1.44 |
| BB-III | Phosphotransferase | 2hid | 35–61 | 3.43 | 0.0% | 37.0% | 1.25 |
| non-1ubqd | — | — | — | 2.89 | 14.7% | 42.1% | 1.19 |
| Amino acid sequence information | |||||||
| | – – – – – – – B B – I – – – – – – – | – – – – – – – – B B – I I – – – – – – – | | |||||||
| Residue #: | ——— • ——— 1 ——— • ——— 2 ——— • ——— 3 ——— • ——— 4 ——— • — | ||||||
| Binary pattern: | * – – –* * – –* * – – –* – – –* – – – –* – – –* – – * * – –* * – – – – * – | ||||||
| nat-1ubq: | M Q I F V K T L T G K T I T L E V E P S D T I E N V K A K I Q D K E G I P P D Q Q | ||||||
| non-1ubq: | I L K T V Q L R N G I L Q Y D L H Y W L P F G L I K S D L L W F L H G D T N I R F | ||||||
| Sequence identity: | – – – –* – – – – * – – – – – – – – – – – – – – – – – – – – – – – –* – – – – – – | ||||||
| | – – – – – – – – – – B B – I I I – – – – – – – – – – | | |||||||
| Residue #: | ——— • ——— 5 ——— • ——— 6 ——— • ——— 7 ——— • — | ||||||
| Binary pattern: | – – – –* * * – – – * * – –* – –* – * – – * * – – – * * * * * * * * | ||||||
| nat-1ubq: | R L I F A G K Q L E D G R T L S D Y N I Q K E S T L H L V L R L R G G | ||||||
| non-1ubq: | L E Y N G K T V N L K S I M G V V S L G I A K G A E I L V L R L R G G | ||||||
| Sequence identity: | – – – – – – – – – – – – – – – – – – – – – – – – – – – * * * * * * * * | ||||||
aThe numbers shown here are the residue numbers in their original corresponding PDB files.
bThe RMSDs are calculated against native structure and are based on Cα atoms only.
cHydrophobicity patterns are calculated from equation 3.
dThe unassigned fragment (residues 69–76) is included in computing the information of nonprotein (non-1ubq). The RMSDs are calculated based on the
Cα atoms of initial assembled structure against nat-1ubq. Other information of non-1ubq, sequence identity, binary pattern, and hydrophobicity pattern, is calculated against nat-1ubq.
The binary patterns of the nat-1ubq, eng-1ubq, and non-1ubq are shown in Figure S2, with 3D structures (in the Supplemental Material). Nat-1ubq and eng-1ubq have large hydrophobic cores and the hydrophilic residues are exposed on the surface, unlike non-1ubq. The calculations also show the nat-1ubq and eng-1ubq have higher hydrophobicity score (0.79 and 0.75, respectively). In contrast, the hydrophobicity score of non-1ubq is low (0.69) (see Table S in the Supplemental Material).
Discussion
Here, we propose a de novo protein engineering method based on substitution of stand-alone protein fragments. We largely focus on whether the engineered proteins are stable and folded. To computationally test these questions are still out of current computational power. The ideal strategy involves iterative modifications of the computationally engineered proteins and their experimental stability tests (D. Raleigh, pers. comm.). Here, explicit water MD simulations are employed to examine the stability of the engineered proteins and to evaluate the advantages and disadvantages of this engineering method. To examine the stability of the engineered proteins, the RMSDs of the native proteins and of “nonproteins” are used as lower and upper bound references. We assume that during the simulations, the native protein will have smaller and the nonproteins larger Cα-RMSDs versus their energy minimized structures.
The Cα-RMSDs of nat-2gb1, eng-2gb1, and non-2gb1 in 8.0-nsec explicit water MD simulations are shown in Figure 6 ▶. The Cα-RMSD of nat-2gb1 fluctuates around 1.0 Å during the entire course of the simulation (Fig. 6A ▶). In contrast, the Cα-RMSD of non-2gb1 with an inverted hydrophobic core increases with the simulation time indicating that its energy-minimized structure cannot be maintained. For the engineered protein (eng-2gb1), its structure fluctuates around its energy-minimized structure (with a compact core) with a Cα-RMSD of ~2.5 Å during the simulation. As expected, the Cα-RMSD of the engineered protein (eng-2gb1) locates between the low bound Cα-RMSD of the native protein (nat-2gb1) and the upper bound Cα-RMSD of the nonprotein (non-2gb1), suggesting that the engineered protein is potentially stable in vitro. Figure 6B ▶ shows the averaged Cα-RMSD of nat-2gb1, eng-2gb1, and non-2gb1 as a function of their residue position. Again, the Cα-RMSD of eng-2gb1 lies between those of nat-2gb1 and non-2gb1. To further analyze the stabilities of each building block in individual proteins, their Cα-RMSDs as a function of time are calculated (Fig. 6C,D ▶). The Cα-RMSDs of building block-I of nonprotein increases with simulation time, whereas the others are stable. Surprisingly, building block-II of nonprotein is also stable in the simulation, indicating that this fragment can be a stand-alone building block, and the mutual stabilization from other fragments may not be important.
Figure 6.

The RMSDs of nat-2gb1, eng-2gb1, and non-2gb1 in 8.0-nsec explicit water MD simulations. The units of RMSDs are shown in Å. (A) The RMSDs of the whole proteins (nat-2gb1, eng-2gb1, and non-2gb1) as a function of simulation time. (B) The RMSDs of nat-2gb1, eng-2gb1, and non-2gb1 as a function of Cα atoms. (C) The RMSDs of building block-I of nat-2gb1, eng-2gb1, and non-2gb1 as a function of simulation time. (D) The RMSDs of building block-II of nat-2gb1, eng-2gb1, and non-2gb1 as a function of simulation time.
Figure 7 ▶ shows the Cα-RMSDs of nat-1ubq, eng-1ubq, and non-1ubq in 9-nsec explicit water MD simulations. Similar to the behavior of eng-2gb1, the Cα-RMSDs of eng-1ubq lies between the non-1ubq and nat-1ubq. Nevertheless, its Cα-RMSD is only slightly lower than that of non-1ubq, indicating that the engineered protein cannot be very stable. To further investigate why the eng-1ubq is not very stable, the Cα-RMSDs of each building block as a function of time were calculated. The Cα-RMSDs of the whole proteins as a function of the residue number were also computed. The results are as expected: The Cα-RMSD of building block-II of eng-1ubq is stable, nearly overlapping that of nat-1ubq. The Cα-RMSD of building block-I of eng-1ubq fluctuates, but with relatively low magnitude. In contrast, the Cα-RMSD of building block-III of eng-1ubq increases rapidly with simulation time. The BB-III large loop is much more flexible than the helical and β-stranded structures. In addition, few qualified candidates can be found for this loop building block (see Fig. 5 ▶) resulting in a less stable structure.
Figure 7.

The RMSDs of nat-1ubq, eng-1ubq, and non-1ubq in 9.0-nsec explicit water MD simulations. The units of RMSDs are shown in Å. (A) The RMSDs of the whole proteins (nat-1ubq, eng-1ubq, and non-1ubq) as a function of simulation time. (B) The RMSDs of nat-1ubq, eng-1ubq, and non-1ubq atoms. (C) The RMSDs of building block-I of nat-1ubq, eng-1ubq, and non-1ubq as a function of simulation time. (D) The RMSDs of building block-II of nat-1ubq, eng-1ubq, and non-1ubq as a function of simulation time. (E) The RMSDs of building block-III of nat-1ubq, eng-1ubq, and non-1ubq as a function of simulation time.
Hence, the fold of eng-2gb1 is maintained during the MD simulations. The RMSD of eng-2gb1 is larger than the lower bound RMSD of nat-2gb1 and smaller than the upper bound RMSD of nonproteins (non-2gb1). In contrast, the engineered ubiquitin is less stable. This is due to the flexible loop (BB-III), suggesting that it is not easy to engineer long loop structures. Such a conclusion is consistent with insights obtained from limited proteolysis experiments (Fontana et al. 1997, 1999).
Conclusions and future work
In this study, a de novo computational algorithm is proposed to engineer proteins in terms of protein building blocks. This approach is similar to combinatorial experiments, where protein building blocks are used as “shuffling domains.” Here, BBs are defined as fragments that form local minima along the polypeptide chain. As such, they have relatively high population times. Because protein building blocks are conformationally independent entities (Haspel et al. 2003a,b), we test the feasibility of partitioning proteins into building blocks and exchanging between BBs with similar conformations and hydrophobic/hydrophilic patterns taken from different proteins. This approach is similar to combinatorial experiments, where protein building blocks are used as “shuffling domains.” The sequence identities of the selected fragments are chosen to be as low as possible (<25%) to avoid a homology bias. Based on these criteria, a new protein can be assembled with a similar fold and low sequence identity compared to the selected native protein. Two proteins (protein G B1 domain and ubiquitin) are selected to illustrate this engineering algorithm. The stability of engineered proteins is tested by simulations. The MD simulations show that the fold of one engineered protein (protein G B1 domain denoted as eng-2gb1) is kept during the 8-nsec explicit water simulations. The RMSD of eng-2gb1 is in between the lower bound RMSD of the native protein and the upper bound RMSD of the “nonprotein” (with inverted hydrophobic core). However, the newly engineered ubiquitin is much less stable because BB-III contains a large flexible loop. Our searches of the PDB found only a few candidate large loop BBs with a similar static conformations. Because a crystal structure is an average structure and large loops are particularly flexible, it is quite possible that the structure we have captured by picking the crystal coordinates does not represent the optimal conformation for this building block. We conclude that in a fragment-based engineering strategy, engineering large loops is very challenging. Overall, our study suggests that it is potentially feasible to engineer proteins in terms of protein building blocks.
Here, we have demonstrated that proteins can be engineered in terms of protein building blocks in silico. The next essential step is to experimentally synthesize the engineered proteins and validate their stability by in vitro experiments. The scoring function used to select the candidate fragments should also be improved to enhance the stability of engineered proteins. For example, deletion and insertion of residues might be included in the candidate fragment search in an attempt to find additional, possibly better candidates. The volume of amino acids may further be considered in the matching, even though the volume of amino acids has been implemented in the scoring function by hydrophobicity scale difference. Moreover, the packing of hydrophobic core may be further optimized (Lazar and Handel 1998; Malakauskas and Mayo 1998). Computationally engineered proteins and their experimental stability tests should best be performed iteratively.
Acknowledgments
We thank our other group members, Drs. Gunasekaran, Pan, and Zanuy, for helpful discussion. In particular, we also thank Dr. Jacob V. Maizel for encouragement. This study utilized the high-performance computational capabilities of the Biowulf PC/Linux cluster at the National Institutes of Health, Bethesda, MD (http://biowulf.nih.gov). The research of R.N. in Israel has been supported in part by the Ministry of Science grant, and by the “Center of Excellence in Geometric Computing and its Applications,” funded by the Israel Science Foundation (administered by the Israel Academy of Sciences). This project has been funded in whole or in part with federal funds from the National Cancer Institute, NIH, under contract number NO1-CO-12400. The content of this publication does not necessarily reflect the view or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organization imply endorsement by the U.S. Government.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.04774004.
Supplemental material: see www.proteinscience.org
References
- Bai, Y.W., Sosnick, T.R., Mayne, L., and Englander, S.W. 1995. Protein-folding intermediates—Native-state hydrogen-exchange. Science 269 192–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldwin, R.L. and Rose, G.D. 1999a. Is protein folding hierarchic? I. Local structure and peptide folding. Trends Biochem. Sci. 24 26–33. [DOI] [PubMed] [Google Scholar]
- ———. 1999b. Is protein folding hierarchic? II. Folding intermediates and transition states. Trends Biochem. Sci. 24 77–83. [DOI] [PubMed] [Google Scholar]
- Berman, H.M., Westbrook, J., Feng, Z., Gilliland, G., Bhat, T.N., Weissig, H., Shindyalov, I.N., and Bourne, P.E. 2000. The Protein Data Bank. Nucleic Acids Res. 28 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks, B.R., Bruccoleri, R.E., Olafson, B.D., States, D.J., Swaminathan, S., and Karplus, M. 1983. Charmm—A program for macromolecular energy, minimization, and dynamics calculations. J. Comput. Chem. 4 187–217. [Google Scholar]
- Brooks, C.L., Gruebele, M., Onuchic, J.N., and Wolynes, P.G. 1998. Chemical physics of protein folding. Proc. Natl. Acad. Sci. 95 11037–11038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calhoun, J.R., Kono, H., Lahr, S., Wang, W., DeGrado, W.F., and Saven, J.G. 2003. Computational design and characterization of a monomeric helical dinuclear metalloprotein. J. Mol. Biol. 334 1101–1115. [DOI] [PubMed] [Google Scholar]
- Cammett, T.J., Luo, L., and Peng, Z.Y. 2003. Design and characterization of a hyperstable p16(INK4a) that restores Cdk4 binding activity when combined with oncogenic mutations. J. Mol. Biol. 327 285–297. [DOI] [PubMed] [Google Scholar]
- Chu, R., Takei, J., Knowlton, J.R., Andrykovitch, M., Pei, W.H., Kajava, A.V., Steinbach, P.J., Ji, X.H., and Bai, Y.W. 2002. Redesign of a four-helix bundle protein by phage display coupled with proteolysis and structural characterization by NMR and X-ray crystallography. J. Mol. Biol. 323 253–262. [DOI] [PubMed] [Google Scholar]
- Crameri, A., Raillard, S.A., Bermudez, E., and Stemmer, W.P.C. 1998. DNA shuffling of a family of genes from diverse species accelerates directed evolution. Nature 391 288–291. [DOI] [PubMed] [Google Scholar]
- Dahiyat, B.I. and Mayo, S.L. 1997. De novo protein design: Fully automated sequence selection. Science 278 82–87. [DOI] [PubMed] [Google Scholar]
- Dahiyat, B.I., Sarisky, C.A., and Mayo, S.L. 1997. De novo protein design: Towards fully automated sequence selection. J. Mol. Biol. 273 789–796. [DOI] [PubMed] [Google Scholar]
- Dantas, G., Kuhlman, B., Callender, D., Wong, M., and Baker, D. 2003. A large scale test of computational protein design: Folding and stability of nine completely redesigned globular proteins. J. Mol. Biol. 332 449–460. [DOI] [PubMed] [Google Scholar]
- Dill, K.A. and Chan, H.S. 1997. From Levinthal to pathways to funnels. Nat. Struct. Biol. 4 10–19. [DOI] [PubMed] [Google Scholar]
- Dobson, C.M., Sali, A., and Karplus, M. 1998. Protein folding: A perspective from theory and experiment. Angew. Chem. (Intl. Ed.) 37 868–893. [DOI] [PubMed] [Google Scholar]
- Fauchere, J.L. and Pliska, V. 1983. Hydrophobic parameters-Pi of amino-acid side-chains from the partitioning of N-acetyl-amino-acid amides. Eur. J. Med. Chem. 18 369–375. [Google Scholar]
- Fontana, A., Polverino deLaureto, P., DeFilippis, V., Scaramella, E., and Zam-bonin, M. 1997. Probing the partly folded states of proteins by limited proteolysis. Fold. Des. 2 R17–R26. [DOI] [PubMed] [Google Scholar]
- ———. 1999. Limited proteolysis in the study of protein conformation. In Proteolytic enzymes: Tools and targets (eds. E.E. Sterchi and W. Stocker), pp. 257–284. Springer Verlag, Heidelberg, Germany.
- Haspel, N., Tsai, C.J., Wolfson, H., and Nussinov, R. 2003a. Hierarchical protein folding pathways: A computational study of protein fragments. Proteins 51 203–215. [DOI] [PubMed] [Google Scholar]
- ———. 2003b. Reducing the computational complexity of protein folding via fragment folding and assembly. Protein Sci. 12 1177–1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen, W.L., Chandrasekhar, J., Madura, J.D., Impey, R.W., and Klein, M.L. 1983. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79 926–935. [Google Scholar]
- Lazar, G.A. and Handel, T.M. 1998. Hydrophobic core packing and protein design. Curr. Opin. Chem. Biol. 2 675–679. [DOI] [PubMed] [Google Scholar]
- Lesk, A.M. and Rose, G.D. 1981. Folding units in globular–proteins. Proc. Natl. Acad. Sci. 78 4304–4308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levinthal, C. 1968. Are there pathways for protein folding? J. Chim. Phys. 65 44–45. [Google Scholar]
- MacKerell, A.D., Bashford, D., Bellott, M., Dunbrack, R.L., Evanseck, J.D., Field, M.J., Fischer, S., Gao, J., Guo, H., Ha, S., et al. 1998. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B 102 3586–3616. [DOI] [PubMed] [Google Scholar]
- Malakauskas, S.M. and Mayo, S.L. 1998. Design, structure and stability of a hyperthermophilic protein variant. Nat. Struct. Biol. 5 470–475. [DOI] [PubMed] [Google Scholar]
- Meyer, M.M., Silberg, J.J., Voigt, C.A., Endelman, J.B., Mayo, S.L., Wang, Z.G., and Arnold, F.H. 2003. Library analysis of SCHEMA-guided protein recombination. Protein Sci. 12 1686–1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onuchic, J.N., LutheySchulten, Z., and Wolynes, P.G. 1997. Theory of protein folding: The energy landscape perspective. Annu. Rev. Phys. Chem. 48 545–600. [DOI] [PubMed] [Google Scholar]
- Riechmann, L. and Winter, G. 2000. Novel folded protein domains generated by combinatorial shuffling of polypeptide segments. Proc. Natl. Acad. Sci. 97 10068–10073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slovic, A.M., Kono, H., Lear, J.D., Saven, J.G., and DeGrado, W.F. 2004. Computational design of water-soluble analogues of the potassium channel KcsA. Proc. Natl. Acad. Sci. 101 1828–1833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinbach, P.J. and Brooks, B.R. 1994. New spherical-cutoff methods for long-range forces in macromolecular simulation. J. Comput. Chem. 15 667–683. [Google Scholar]
- Tsai, C.J. and Nussinov, R. 2001a. The building block folding model and the kinetics of protein folding. Protein Eng. 14 723–733. [DOI] [PubMed] [Google Scholar]
- ———. 2001b. Transient, highly populated, building blocks folding model. Cell Biochem. Biophys. 34 209–235. [DOI] [PubMed] [Google Scholar]
- Tsai, C.J., Maizel, J.V., and Nussinov, R. 2000. Anatomy of protein structures: Visualizing how a one-dimensional protein chain folds into a three-dimensional shape. Proc. Natl. Acad. Sci. 97 12038–12043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai, C.J., de Laureto, P.P., Fontana, A., and Nussinov, R. 2002. Comparison of protein fragments identified by limited proteolysis and by computational cutting of proteins. Protein Sci. 11 1753–1770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voigt, C.A., Martinez, C., Wang, Z.G., Mayo, S.L., and Arnold, F.H. 2002. Protein building blocks preserved by recombination. Nat. Struct. Biol. 9 553–558. [DOI] [PubMed] [Google Scholar]
- Wolynes, P.G., Onuchic, J.N., and Thirumalai, D. 1995. Navigating the folding routes. Science 267 1619–1620. [DOI] [PubMed] [Google Scholar]