

Abstract
A number of computational tools are available for detecting signal peptides, but their abilities to locate the signal peptide cleavage sites vary significantly and are often less than satisfactory. We characterized a set of 270 secreted recombinant human proteins by automated Edman analysis and used the verified cleavage sites to evaluate the success rate of a number of computational prediction programs. An examination of the frequency of amino acid in the N-terminal region of the data set showed a preference of proline and glutamine but a bias against tyrosine. The data set was compared to the SWISS-PROT database and revealed a high percentage of discrepancies with cleavage site annotations that were computationally generated. The best program for predicting signal sequences was found to be SignalP 2.0-NN with an accuracy of 78.1% for cleavage site recognition. The new data set can be utilized for refining prediction algorithms, and we have built an improved version of profile hidden Markov model for signal peptides based on the new data.
Keywords: signal peptide, cleavage site, experimental verification, SWISS-PROT annotation, computational prediction
Secreted and cell-surface proteins are fundamental to inter-cellular communications for multicellular organisms. The extracellular accessibility of these proteins makes them ideal targets for protein therapeutics. In fact, virtually all protein-based therapeutic drugs on the market target these secreted and cell-surface proteins or are secreted proteins themselves. Secreted proteins and a majority of cell-surface proteins possess an N-terminal signal peptide. The signal peptide is typically between 15 and 40 amino acids long and is essential for protein secretion, and is then subsequently cleaved from the mature protein (Nakai 2000).
The importance of signal peptide-containing proteins has motivated the development of several computational methods for predicting signal peptides and determining the signal cleavage sites. These include SigCleave, based on the SigPep data set (von Heijne 1986, 1987), SignalP 2.0-NN, which utilizes a neural network method (Nielsen et al. 1997a,b), SignalP 2.0-HMM, based on a hidden Markov model (Nielsen and Krogh 1998), SigPfam, based on a Pfam-compatible profile hidden Markov model (Zhang and Wood 2003), and a few other methods (Chou 2001a,b,c; Vert 2002; Cai et al. 2003; Chen et al. 2003). More recently, an updated version of SignalP (3.0) was reported that showed performance improvement (Dyrlov Bendtsen et al. 2004). All of these methods rely on protein annotations from publicly available databases. The SWISS-PROT database (Bairoch and Apweiler 2000) is the most commonly used and arguably the best annotated protein sequence database. Although many of the available prediction methods reportedly perform well in distinguishing signal peptides from nonsignal sequences, the recurrent use of the SWISS-PROT data sets for training and validating raises concerns over the true prediction accuracies. In particular, it is critical to realistically assess the cleavage site prediction accuracy, because it is often desirable to produce hybrid, functional secreted proteins with tags linked precisely to the N termini of mature proteins for scientific and commercial purposes.
The performance of computational prediction methods should be ultimately evaluated by an independent data set that is experimentally determined. Our large-scale efforts in identifying human secreted and transmembrane proteins (Clark et al. 2003) provided an opportunity for producing such a data set for signal peptide studies. We expressed and purified 270 proteins and experimentally determined the N-terminal sequences of the mature proteins, and used the validated data for evaluating various computational methods for predicting cleavage sites. This data should also be valuable for improving some of the SWISS-PROT annotations as well as refining existing prediction tools.
Materials and methods
Protein expression, purification, and sequence determination
Secreted and cell-surface proteins were identified from the SPDI efforts (Clark et al. 2003). Proteins were expressed in CHO cells (Lucas et al. 1996) and 293 and Sf9 cells (Lee et al. 2001). Fusion proteins were made using a C-terminal tagged 8Xhis tag and purified on nickel affinity columns. Proteins were also expressed with a C-terminal tag of the Fc region of human IgG1 and purified over a protein A column. The first 15 residues of the purified proteins were determined using automated Edman degradation. No special selection criteria were applied to pick the 270 proteins being reported in this article.
High throughput automated protein sequencing was performed on PE-Applied Biosystems Procise 494 HT protein sequencers using 20-min Edman cycles (Henzel et al. 1999; Pham et al. 2003).
The SWISS-PROT (Release 42) protein sequences were downloaded from ftp://us.expasy.org/databases/swiss-prot/release/.
Signal peptide predictions
The signal peptide potential for each protein sequence was analyzed using several commonly used prediction algorithms. SigCleave is the EMBOSS implementation of the weight matrix method (von Heijne 1986) and is, in principle, identical to the SigSeq program (Popowicz and Dash 1988). The default cutoff value of 3.5 was used for predicting signal peptide potential, and the highest scoring cleavage site was assumed to be the correct prediction. SigPfam is based on a Pfam-compatible profile hidden Markov model (Zhang and Wood 2003) we previously developed. Using the hmmpfam program from the HMMER package (Eddy 1998) to evaluate the first 70-amino-acid region, we set −0.5 as the cutoff score for signal potential and derived the cleavage site from the alignment coordinates. The SignalP V2.0- and SignalP V3.0-based predictions were performed via their web interfaces (http://www.cbs.dtu.dk/services/SignalP-2.0/ and http://www.cbs.dtu.dk/services/SignalP/) with default settings. The SignalP 2.0-NN is a neural network method trained on a data set derived from SWISS-PROT release 35 (Nielsen et al. 1997a,b), whereas SignalP 2.0-HMM is the implementation of a hidden Markov model. The SignalP V3.0 was an improved version of SignalP that was recently released (Dyrlov Bendtsen et al. 2004).
Building an improved profile HMM model for signal peptides
The Pfam-compatible signal peptide model was built using the HMMER 2.3.2 package following a procedure previously described (Zhang and Wood 2003). The optimal “architecture prior” parameter was determined to be 0.90. All the components associated with this model (HMM model, data sets, prediction programs) are available online at http://share.gene.com/share/cleavagesite.
Results
Comparison of cleavage site prediction accuracies
We experimentally determined the N-terminal sequences of 270 mature secreted and cell-surface proteins. The N termini of mature proteins were recorded as signal peptide cleavage sites, unless evidence existed for any further post-translational cleavage of protein precursors. This data set (Supplementary Table 1) was used for signal cleavage site studies. The performance of signal peptide prediction algorithms is usually evaluated in two areas: the ability to distinguish signal peptides from nonsignal sequences and the ability to locate the signal cleavage sites. Usually, only the percentages of correctly predicted sites among positively predicted sequences are reported. However, we find it more helpful to also include the overall percentage based on all test sequences.
Benchmarked by our confirmed signal peptide sequences, all six programs gave high sensitivities of detecting signal-containing proteins, with SignalP 2.0-HMM and SignalP 3.0-NN being the highest, both at 98.5%. However, these programs showed much greater variation of accuracies in predicting the cleavage sites (Fig. 1A ▶). The best program appeared to be SignalP 2.0-NN, which precisely predicted 78.1% of sites. In contrast, both SigCleave and SigPfam yielded markedly lower accuracies. Our analysis indicates SignalP 2.0-NN is the best overall prediction program, consist with a previous observation (Menne et al. 2000). It should be noted that SignalP 2.0-HMM is reported to be better at distinguishing cleavable signal peptides from non-cleavable signal anchors (Nielsen and Krogh 1998), which we did not test. We did not observe major performance differences between the two versions of SignalP-NN, but the new version of SignalP-HMM (3.0) appeared to be better at recognizing signal cleavage sites than SignalP 2.0-HMM.
Figure 1.
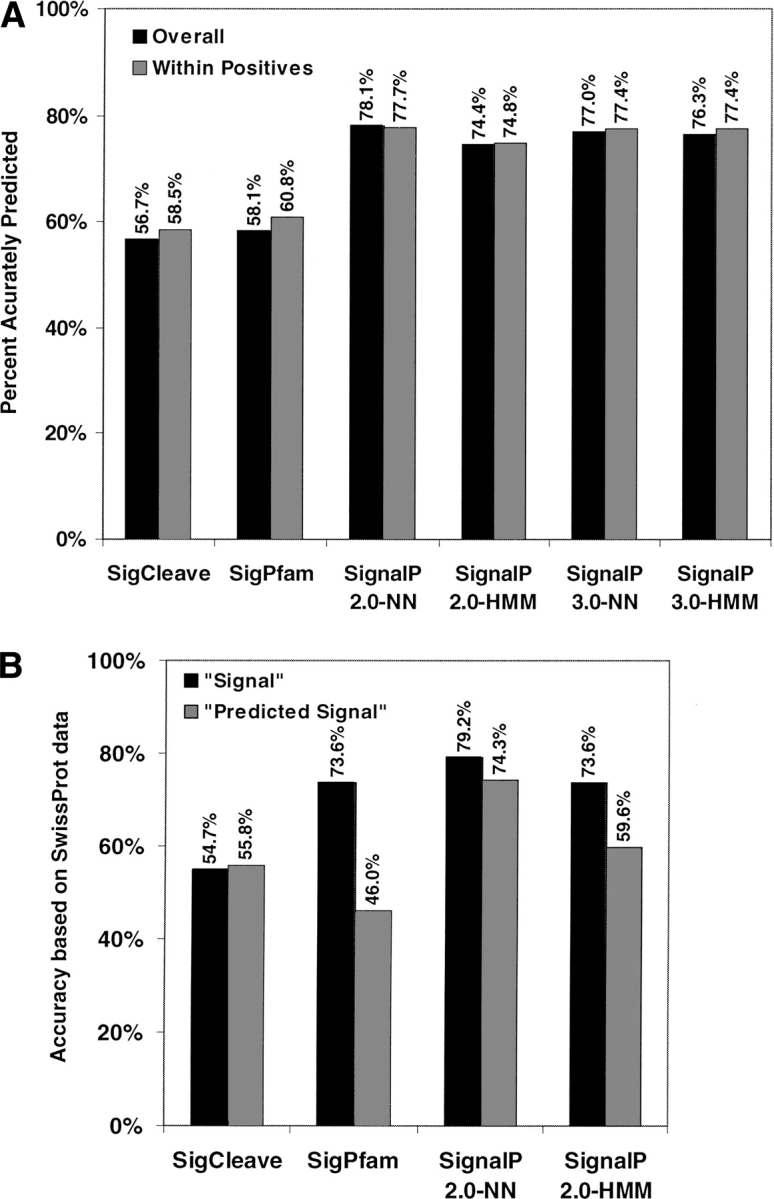
The cleavage sites of signal peptides are recognized at varying degrees of accuracy by six different programs. The Y-axis indicates the percentage of signal peptide sequences where the cleavage site is placed correctly. (A) The prediction results based on the experimentally verified cleavage sites. The solid bars represent overall percentages that are measured using the entire 270 protein sequences. The gray bars represent the percentages of correctly predicted sites among positively predicted sequences by each program. (B) The perceived site prediction accuracies when benchmarked by the subset of SWISS-PROT sequences that overlap with our validated sequence set. The solid bars show results that are based on 52 SWISS-PROT sequences with annotated “Signal” sites (with experimental evidence). The gray bars show results that are based on 106 SWISS-PROT sequences with computationally predicted signal cleavage sites (without experimental evidence).
Analysis of matched SWISS-PROT data set
One of the intriguing aspects of developing and evaluating prediction algorithms is that both training and testing data sets are typically collected from the SWISS-PROT database. Although SWISS-PROT is arguably the best annotated protein sequence database, any incorrectly annotated entries will likely propagate into various prediction tools and results. It is therefore useful to evaluate the reliability of signal peptide annotation in the SWISS-PROT database. Of all the human protein sequences with annotated signal sequences in the current release of SWISS-PROT (Release 42), only 33.6% are marked to contain “Signal” under the feature table, which implies that there are experimental data for the presence and location of the signal peptides. The remaining 66.4% protein entries are marked to contain “Signal By Similarity,” “Signal Potential,” or “Signal Probable,” which implies that these are derived computationally, either by a signal peptide prediction program or by sequence similarity.
Of the 270 total protein sequences, 169 are represented by SWISS-PROT Release 42. Three of the SWISS-PROT entries, KAC_HUMAN, CK15_HUMAN, and CRI1_HUMAN, do not have the annotation of signal peptide even though they are either secreted or transmembrane proteins. Of the remaining 166 proteins marked to have signal peptide, 70.5% of the annotated cleavage sites are consistent with our verified sites. However, different types of signal annotation gave different results. Of the 113 protein entries marked to contain predicted signal peptides, only 63.7% of the predicted cleavage sites agree with our data. The remaining 53 are marked to contain “Signal” under the feature table and are therefore expected to be supported by experimental evidence. Forty-five of these annotated sites, or 85.0%, are identical to our verified sites. Those eight discrepancies were investigated further based on cited literature.
Surprisingly, five of the SWISS-PROT entries with discrepancies, AXO1_HUMAN, FCG1_HUMAN, HGFA_HUMAN, T10C_HUMAN, and TRLT_HUMAN, contain signal annotations based on prediction rather than experimental data (Allen and Seed 1989; Miyazawa et al. 1993; Tsiotra et al. 1993; Pan et al. 1997; Sica et al. 2001). In the case of T10C_HUMAN, both predicted (Pan et al. 1997) and experimental (Sheridan et al. 1997) data are available, but SWISS-PROT contains the predicted data. The experimental data are consistent with our results. For the remaining two proteins with discrepancies, INA5_HUMAN and INA6_HUMAN, it is not clear to us whether their signal annotations were based on prediction or experimental data. Therefore, although references are provided that support a “Signal” annotation, the papers themselves may not contain experimental data to support the claim. The remaining discrepancy associated with SWISS-PROT, PTHY_HUMAN, is caused by postcleavage modification of preProPTH (Vasicek et al. 1983). In this case we observed the site for postsignal cleavage sites. We estimate that such a mistake due to postcleavage modification has a very low occurrence and will not influence the overall quality of our data set. Regardless, the cleavage site for PTHY_HUMAN has been corrected.
To understand how previously reported prediction accuracies were achieved, we estimated the perceived accuracies for site prediction when benchmarked against SWISS-PROT annotation, as the SWISS-PROT data are usually used for validation. As shown in Figure 1B ▶, when comparing with the SWISS-PROT entries that have “Signal” annotations, the perceived prediction accuracies for SignalP 2.0-NN and SignalP 2.0-HMM are similar to those observed by us (Fig. 1A ▶). A much higher perceived accuracy (73.6%) was observed with the SigPfam program, which was originally trained with similar sets of SWISS-PROT human sequences (Zhang and Wood 2003). This discrepancy suggests that HMM models could be overtrained or that the SWISS-PROT data might not always be the best test data. As expected, when analyzing the SWISS-PROT entries with computationally predicted signal peptides, the perceived accuracies are mostly lower. Interestingly, none of the prediction programs tested here matches with the SWISS-PROT annotation extremely well, indicating the SWISS-PROT annotations were historically based on multiple prediction algorithms before recently settled down to the SignalP program.
Excluding the 45 proteins that are already correctly annotated in SWISS-PROT regarding the signal cleavage sites, the availability of our verified data for 225 proteins would represent a significant increase (32%) of human protein sequences with annotations of verified cleavage sites in the current SWISS-PROT database.
Improving signal peptide prediction using verified cleavage sites
The confirmed N-terminal sequences of mature proteins provide a reliable data source for studying preferential amino acid usage after the signal peptide cleavage sites. We first determined the expected frequencies of amino acid usage by sampling the entire mature proteins, and then compared the usage at each of the positions after the cleavage sites with expected frequencies. The log ratios of the observed and expected frequencies are plotted to reveal any usage preferences or antipreferences (Fig. 2 ▶). It is apparent that whereas some of the residues such as glycine are not preferentially used in any of the positions, several residues show biased usage. Tyrosine, for example, is obviously discriminated against in the first few positions after cleavage sites. Proline, on the other hand, is preferred in many of the sites with the exception of the +1 position, where it is almost never found. Glutamine was found at the +1 residue in 10.7% of the 270 proteins in the data set. Because N-terminal glutamine is cyclized to pyroglutamic acid by glutamine cyclase during protein synthesis (Kamp et al. 1998), the N-terminal pyroglutamic acid may serve to protect these secreted proteins from degradation by extracellular amino-peptidase.
Figure 2.
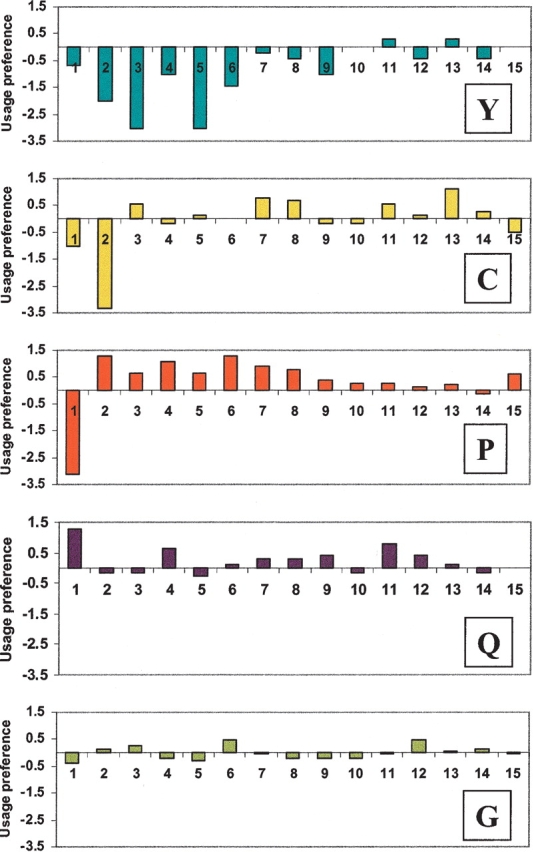
Amino acid usage patterns at the N-terminal positions of mature proteins. The X-axis indicates the amino acid positions relative to the signal peptide cleavage site. The Y-axis indicates the usage preference, measured by the base 2 log ratios of observed and expected frequencies, where the expected frequencies were determined by examining all residues of all the mature proteins in the data set.
The availability of our confirmed signal cleavage sites also provides new opportunities for refining existing prediction programs. By removing incorrectly aligned signal peptide sequences and adding new ones that were confirmed experimentally, we believe that the alignment-based models for signal peptides could be improved. As a test, we built a new Pfam-compatible HMM model for signal peptides based on these confirmed sequences. The sensitivity of the SigPfam program based on the new model increased from 91.5% to 93.5% based on a leave-one-out validation test. (The specificity was not tested for lack of negative data set for this study.) In addition, the overall cleavage site prediction accuracy increased from 58.1% to 73.7%. Despite this performance improvement, SignalP 2.0-NN remains the best signal prediction program. It is possible that the SignalP package can be improved further with the assistance of our data set. Nevertheless, the improved SigPfam would be a useful tool as it can be easily implemented, configured, and tuned. All components of SigPfam are freely available at http://share.gene.com/share/cleavagesite.
Discussion
It is critical to accurately locate signal peptide cleavage sites when making constructs for producing recombinant secreted proteins or receptors. At present, this process is usually accomplished either by checking annotations from protein sequence databases or by running one of the existing computer programs for predicting signal peptides. Although many of the computer programs are extremely powerful in distinguishing signal peptides from nonsignal sequences (as high as 99% accuracy), the best program, SignalP 2.0-NN, localizes only 78% of the signal cleavage sites accurately. The suboptimal performance in this regard can be partially explained by insufficient experimental data available for modeling the cleavage sites. In fact, a majority of the annotated cleavage sites in protein sequence databases, such as SWISS-PROT, are based on sequence similarities or computational predictions. It is alarming that over one-third of the computationally derived cleavage sites are incorrect according to our assessment.
To improve the accuracy of signal cleavage site prediction, it is necessary to continue to accumulate cleavage site data that are experimentally validated. Our work to determine the N-terminal amino acid sequences of human mature proteins should help assess the performance of existing computational tools, improve the cleavage site annotation in protein sequence databases, and perhaps help improve existing programs for predicting signal peptides. Indeed, we noticed improvement of the SigPfam program when retrained with our new data set. Other programs, for example, SignalP 2.0, could perhaps also benefit from the improved training data. Furthermore, the precise localization of the signal cleavage sites also prompted us to revisit the amino acid usage at the N-terminal regions of mature proteins. Early work described amino acid patterns in this region (von Heijne 1983, 1984), but most prediction programs do not weigh this region heavily. The differential amino acid usage patterns (Fig. 2 ▶) based on our data set should be considered when developing cleavage site prediction tools to improve the overall performance. The development of signal peptide analysis tools should perhaps be a continual process to take advantage of new and improved data, as even the best tool currently available, SignalP 2.0-NN, has room for improvement.
Acknowledgments
We thank Colin Watanabe, Mike Ward, Hilary Clark, and William Wood for discussion and comments. We also thank members of the Bill Henzel, Craig Crowley, Dan Yansura, and Austin Gurney laboratories at Genentech for reagents and data.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Article published online ahead of print. Article and publication date are at http://www.proteinscience.org/cgi/doi/10.1110/ps.04682504.
Supplemental material: see www.proteinscience.org
References
- Allen, J.M. and Seed, B. 1989. Isolation and expression of functional high-affinity Fc receptor complementary DNAs. Science 243 378–381. [DOI] [PubMed] [Google Scholar]
- Bairoch, A. and Apweiler, R. 2000. The SWISS-PROT protein sequence database and its supplement TrEMBL in 2000. Nucleic Acids Res. 28 45–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai, Y.D., Lin, S., and Chou, K.C. 2003. Support vector machines for prediction of protein signal sequences and their cleavage sites. Peptides 24 159–161. [DOI] [PubMed] [Google Scholar]
- Chen, Y., Yu, P., Luo, J., and Jiang, Y. 2003. Secreted protein prediction system combining CJ-SPHMM, TMHMM, and PSORT. Mamm. Genome 14 859–865. [DOI] [PubMed] [Google Scholar]
- Chou, K.C. 2001a. Prediction of protein signal sequences and their cleavage sites. Proteins 42 136–139. [DOI] [PubMed] [Google Scholar]
- ———. 2001b. Prediction of signal peptides using scaled window. Peptides 22 1973–1979. [DOI] [PubMed] [Google Scholar]
- ———. 2001c. Using subsite coupling to predict signal peptides. Protein Eng. 14 75–79. [DOI] [PubMed] [Google Scholar]
- Clark, H.F., Gurney, A.L., Abaya, E., Baker, K., Baldwin, D., Brush, J., Chen, J., Chow, B., Chui, C., Crowley, C., et al. 2003. The Secreted Protein Discovery Initiative (SPDI), a large-scale effort to identify novel human secreted and transmembrane proteins: A bioinformatics assessment. Genome Res. 13 2265–2270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyrlov Bendtsen, J., Nielsen, H., Von Heijne, G., and Brunak, S. 2004. Improved prediction of signal peptides: SignalP 3.0. J. Mol. Biol. 340 783–795. [DOI] [PubMed] [Google Scholar]
- Eddy, S.R. 1998. Profile hidden Markov models. Bioinformatics 14 755–763. [DOI] [PubMed] [Google Scholar]
- Henzel, W.J., Tropea, J., and Dupont, D. 1999. Protein identification using 20-minute Edman cycles and sequence mixture analysis. Anal. Biochem. 267 148–160. [DOI] [PubMed] [Google Scholar]
- Kamp, R.M., Tsunasawa, S., and Hirano, H. 1998. Application of new deblocking aminopeptidase from Pyrococcus furiosis for microsequence analysis of blocked proteins. J. Protein Chem. 17 512–513. [PubMed] [Google Scholar]
- Lee, J., Ho, W.H., Maruoka, M., Corpuz, R.T., Baldwin, D.T., Foster, J.S., Goddard, A.D., Yansura, D.G., Vandlen, R.L., Wood, W.I., et al. 2001. IL-17E, a novel proinflammatory ligand for the IL-17 receptor homolog IL-17Rh1. J. Biol. Chem. 276 1660–1664. [DOI] [PubMed] [Google Scholar]
- Lucas, B.K., Giere, L.M., DeMarco, R.A., Shen, A., Chisholm, V., and Crowley, C.W. 1996. High-level production of recombinant proteins in CHO cells using a dicistronic DHFR intron expression vector. Nucleic Acids Res. 24 1774–1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menne, K.M., Hermjakob, H., and Apweiler, R. 2000. A comparison of signal sequence prediction methods using a test set of signal peptides. Bioinformatics 16 741–742. [DOI] [PubMed] [Google Scholar]
- Miyazawa, K., Shimomura, T., Kitamura, A., Kondo, J., Morimoto, Y., and Kitamura, N. 1993. Molecular cloning and sequence analysis of the cDNA for a human serine protease reponsible for activation of hepatocyte growth factor. Structural similarity of the protease precursor to blood coagulation factor XII. J. Biol. Chem. 268 10024–10028. [PubMed] [Google Scholar]
- Nakai, K. 2000. Protein sorting signals and prediction of subcellular localization. Adv. Protein Chem. 54 277–344. [DOI] [PubMed] [Google Scholar]
- Nielsen, H. and Krogh, A. 1998. Prediction of signal peptides and signal anchors by a hidden Markov model. Proc. Int. Conf. Intell. Syst. Mol. Biol. 6 122–130. [PubMed] [Google Scholar]
- Nielsen, H., Engelbrecht, J., Brunak, S., and von Heijne, G. 1997a. Identification of prokaryotic and eukaryotic signal peptides and prediction of their cleavage sites. Protein Eng. 10 1–6. [DOI] [PubMed] [Google Scholar]
- ———. 1997b. A neural network method for identification of prokaryotic and eukaryotic signal peptides and prediction of their cleavage sites. Int. J. Neural Syst. 8 581–599. [DOI] [PubMed] [Google Scholar]
- Pan, G., Ni, J., Wei, Y.F., Yu, G., Gentz, R., and Dixit, V.M. 1997. An antagonist decoy receptor and a death domain-containing receptor for TRAIL. Science 277 815–818. [DOI] [PubMed] [Google Scholar]
- Pham, V., Tropea, J., Wong, S., Quach, J., and Henzel, W.J. 2003. High-throughput protein sequencing. Anal. Chem. 75 875–882. [DOI] [PubMed] [Google Scholar]
- Popowicz, A.M. and Dash, P.F. 1988. SIGSEQ: A computer program for predicting signal sequence cleavage sites. Comput. Appl. Biosci. 4 405–406. [DOI] [PubMed] [Google Scholar]
- Sheridan, J.P., Marsters, S.A., Pitti, R.M., Gurney, A., Skubatch, M., Baldwin, D., Ramakrishnan, L., Gray, C.L., Baker, K., Wood, W.I., et al. 1997. Control of TRAIL-induced apoptosis by a family of signaling and decoy receptors. Science 277 818–821. [DOI] [PubMed] [Google Scholar]
- Sica, G.L., Zhu, G., Tamada, K., Liu, D., Ni, J., and Chen, L. 2001. RELT, a new member of the tumor necrosis factor receptor superfamily, is selectively expressed in hematopoietic tissues and activates transcription factor NF-κB. Blood 97 2702–2707. [DOI] [PubMed] [Google Scholar]
- Tsiotra, P.C., Karagogeos, D., Theodorakis, K., Michaelidis, T.M., Modi, W.S., Furley, A.J., Jessell, T.M., and Papamatheakis, J. 1993. Isolation of the cDNA and chromosomal localization of the gene (TAX1) encoding the human axonal glycoprotein TAG-1. Genomics 18 562–567. [DOI] [PubMed] [Google Scholar]
- Vasicek, T.J., McDevitt, B.E., Freeman, M.W., Fennick, B.J., Hendy, G.N., Potts Jr., J.T., Rich, A., and Kronenberg, H.M. 1983. Nucleotide sequence of the human parathyroid hormone gene. Proc. Natl. Acad. Sci. 80 2127–2131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vert, J.P. 2002. Support vector machine prediction of signal peptide cleavage site using a new class of kernels for strings. Pac. Symp. Biocomput. 7 649–660. [DOI] [PubMed] [Google Scholar]
- von Heijne, G. 1983. Patterns of amino acids near signal-sequence cleavage sites. Eur. J. Biochem. 133 17–21. [DOI] [PubMed] [Google Scholar]
- ———. 1984. Analysis of the distribution of charged residues in the N-terminal region of signal sequences: Implications for protein export in prokaryotic and eukaryotic cells. EMBO J. 3 2315–2318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ———. 1986. A new method for predicting signal sequence cleavage sites. Nucleic Acids Res. 14 4683–4690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ———. 1987. SIGPEP: A sequence database for secretory signal peptides. Protein Seq. Data Anal. 1 41–42. [PubMed] [Google Scholar]
- Zhang, Z. and Wood, W.I. 2003. A profile hidden Markov model for signal peptides generated by HMMER. Bioinformatics 19 307–308. [DOI] [PubMed] [Google Scholar]