

Abstract
In general, α-helical conformations in proteins depend in large part on the amino acid residues within the helix and their proximal interactions. For example, an alanine residue has a high propensity to adopt an α-helical conformation, whereas that of a glycine residue is low. The sequence preferences for β-sheet formation are less obvious. To identify the factors that influence β-sheet conformation, a series of scanning polyalanine mutations were made within the strands and associated turns of the β-sheet region in T4 lysozyme. For each construct the stability of the folded protein was reduced substantially, consistent with removal of native packing interactions. However, the crystal structures showed that each of the mutants retained the β-sheet conformation. These results suggest that the structure of the β-sheet region of T4 lysozyme is maintained to a substantial extent by tertiary interactions with the surrounding parts of the protein. Such tertiary interactions may be important in determining the structures of β-sheets in general.
Keywords: β-sheet, T4 lysozyme, secondary structure, tertiary interactions, alanine mutagenesis
The formation and packing of secondary structure elements in proteins guides folding and determines the final tertiary structure. Still, the factors that dictate whether a sequence forms a helix, strand, turn, or coil in the folded protein remain poorly understood. A number of studies suggest that the formation and stability of α-helices depend on proximal interactions within the helix, with some residues having a higher propensity than others to adopt helical conformations (e.g., Chakrabartty and Baldwin 1995; Muñoz and Serrano 1995; Frishman and Argos 1997; Sagermann et al. 1999). Whereas helical conformations depend on interactions within the helical region, β-sheets depend on global interactions in the folded or nearly folded protein. Residues within a β-strand have extended conformations, thus the hydrogen-bonding needs of the main chain are satisfied by residues that are in general distant in sequence. The influence of local amino acid sequence on β-sheet formation is not well understood.
Bacteriophage T4 lysozyme is made up of an α-helical domain and a smaller domain that contains a four-stranded β-sheet (Fig. 1A ▶; Remington et al. 1978). It was shown previously (Heinz et al. 1992; Blaber et al. 1995; Zhang et al. 2002) that the protein is highly tolerant of polyalanine substitutions within α-helices, and in many cases these are actually stabilizing. However, the extent to which such substitutions could be tolerated in the β-sheet region was not investigated. Here we address this question by replacing residues within the β-sheet region of T4 lysozyme (Fig. 1 ▶) with alanine and determining the structure and stability of the resulting mutants.
Figure 1.

Organization of β-sheet in bacteriophage T4 lysozyme. (A) Ribbons diagram of T4 lysozyme with the three β-sheet strands—β1, β2, and β3—shown in red. The three α-helices—αA, αB, and αC—are also identified. The three β-sheet strands are labeled, as are the A, B, and C helices. In this view both the amino and carboxyl termini of the molecule are at the “back” of the upper domain. (B) Schematic of the β-sheet region showing hydrogen-bonding interactions (after Remington et al. 1978). Shaded segments correspond to each of the four mutants constructed and red lettering identifies the residues changed to alanine. Arrowheads denote the direction of the main chain.
Results
We constructed four polyalanine mutants within strands β1, β2, and β3 and the associated connecting loops in the β-sheet region of bacteriophage T4 lysozyme. The mutants are defined in Table 1 and illustrated in Figure 1B ▶. Mutant β1-A6, for example, has six alanine substitutions within the first portion of the 3-sheet. All studies were carried out in the cysteine-free pseudo-wild-type variant C54T/C97A, designated WT*.
Table 1.
Thermal stabilities of mutant lysozymes
| Name of mutant | Mutations included | pH of measurement | Tm(°C) | ΔH(kcal/mole) | ΔΔG(kcal/mole) |
| WT*a | C54T/C97A | 3.02 | 51.6 | 128 | — |
| β1-A6b | R14A/K16A/I17A/K19A/T21A/E22A/WT* | 3.02 | 44 | 60 | −3 |
| β2-A4b | Y24A/Y25A/T26A/I27A/WT* | 3.05 | 35 | 55 | −5.5 |
| β2,3-A3b | G28A/I29A/G30A/WT* | 3.05 | 36 | 61 | −5.5 |
| β3-A3b | L32A/L33A/T34A/E108V/WT* | 3.05 | 44 | 62 | −3 |
| E108Va | E108V/WT* | 3.02 | 55.2 | 134 | +1.4 |
| G28Aa | G28A/WT* | 3.02 | 46.5 | 109 | −2.0 |
a Data are well modeled by two-state analysis. ΔG° for unfolding was determined at the Tm of WT* and ΔΔG [ΔG°(mut) – ΔG°(WT*)] is expected to have an uncertainty of ±0.15 kcal/mole.
b Apparent enthalpies of unfolding for these mutants were too low to have resulted from strictly two-state transitions. The corresponding ΔΔG values are expected to be minimum estimates of destabilization and are uncertain to at least ± 0.5 kcal/mole.
All four of the constructs were significantly destabilized compared to WT* (Table 1). The second β-strand is the most buried in the structure, and mutations within this region (β2-A4 and β2,3-A3) were the most destabilizing, consistent with disruption of the native packing interactions. Polyalanine mutations within the most solvent-exposed β-strand (β1-A6) had the least effect on protein stability.
Each of the mutants crystallized in a different space group (Table 2). The resulting crystal structures show that despite the mutations, the β-sheet structure was well-conserved (Fig. 2 ▶). The largest differences between the crystal structures were seen in the solvent-exposed loop between β-strand 1 and 2 (loop 2) and the exposed loop after β-strand 3 (loop 3). In addition, the loop between helices B and C (loop 4) also shifts among some of the structures. A number of these differences are in regions of the structure that are involved in crystal contacts. Because the crystal contacts involving these loops differ among the mutants, we cannot rule out crystal packing interactions as a contributing factor to the different conformations we observe. Still, the β-sheet is highly similar among all of the mutants and WT* (Fig. 2E ▶; Table 3). The following sections provide a more detailed analysis of the properties of each of the mutants.
Table 2.
Crystallographic data for mutant lysozymes
| Mutant | β1-A6 | β2-A4 | β2,3-A3 | β3-A3 |
| Data collection | ||||
| Space group | P6222 | P3221 | C2 | I422 |
| Cell dimensions | ||||
| a (Å) | 120.5 | 60.8 | 123.0 | 96.0 |
| b (Å) | 120.5 | 60.8 | 53.7 | 96.0 |
| c (Å) | 50.3 | 96.1 | 60.5 | 77.3 |
| β (°) | B1A6 | B3A3 | 102.3 | — |
| Resolution (Å) | 39-2.15(2.2–2.15)a | 20-2.1(2.8–2.1) | 20-2.4(2.53–2.4) | 30-1.8(1.85–1.8) |
| Wavelength (Å) | 1.0 | 1.54 | 1.54 | 1.0 |
| Number of unique reflections | 11,852 (718) | 10,422 (5252) | 15,444 (1756) | 16,663 (1083) |
| Multiplicity | 5.2 (3.8) | 3.3 (2.9) | 2.7 (2.6) | 8.3 (7.7) |
| Completeness (%) | 97.6 (83.3) | 99.6 (99.5) | 88.3 (82.5) | 97.7 (97.7) |
| Rmerge (%) | 12.5 (46.8) | 10.5 (34.7) | 6.7 (21.1) | 3.4 (8.3) |
| I/σ(I) | 5.1 (1.6) | 5.2 (1.8) | 12.4 (2.8) | 15.1 (8.7) |
| Refinement | ||||
| Rcryst(%) | 22.2 | 18.7 | 17.5 | 18.4 |
| Rfree(%) | 27.4 | 21.5 | 23.6 | 21.5 |
| Number of protein atoms | 1269 | 1290 | 2614 | 1256 |
| Number of waters | 99 | 83 | 76 | 189 |
| Average protein B-value (Å2) | 36.0 | 28.0 | 32.6 | 20.6 |
| Average solvent B-value (Å2) | 37.4 | 62.5 | 43.9 | 46.2 |
| Rms deviation from ideal | ||||
| Bond lengths (Å) | 0.008 | 0.008 | 0.008 | 0.007 |
| Bond angles (°) | 1.2 | 1.2 | 1.2 | 1.4 |
| Ramachandran plt | ||||
| Most favored (%) | 94.5 | 92.6 | 92.3 | 94.3 |
| Additionally allowed (%) | 5.5 | 7.4 | 6.7 | 5.7 |
| Generally allowed (%) | 0 | 0 | 0.7 | 0 |
| Protein Data Bank (PDB) code | 1T8F | 1SSW | 1SSY | 1T8G |
a Numbers in parentheses refer to the highest resolution shell. Rmerge gives the agreement between repeated intensity measurements and Rcryst the agreement between the observed and calculated structure amplitudes.
Figure 2.

Superposition of the amino-terminal region (residues 11–65) of each of the mutant lysozyme structures (red) on that of WT* (cyan; PDB ID 1L63). In all comparisons, the β-sheet is conserved. Three of the mutants crystallized in space groups different than WT* (space group P3221) and the resulting crystal contacts in this region differ. (A) Mutant β1-A6 (red), with its β-strands and α-helices labeled, as well as its four solvent-exposed loops (thick black line). Loops 1–4 and α-helix B all have differences in crystal packing relative to WT* (cyan). (B) Mutant β2-A4 (red) crystallized in the same space group as WT* (cyan), conserving both the β-sheet structure and surface loops. © Mutant β2,3-A3 (red) crystallized in space group C2 with two molecules in the asymmetric unit, although for simplicity only molecule A is shown. Despite differences in crystal contacts, this mutant has loop conformations similar to WT* (cyan). (D) Mutant β3-A3 (space group I422) conserves the β-sheet structure despite large differences in regions involved in crystal contacts (loop 2, 3, and α-helix B). Loops 2 and 4 differ in crystal contacts but conserve the structure seen in WT* (cyan). Loop 3 is disordered in the crystal, and is not modeled (red asterisks). (E) Stereo diagram superposition of only the β-sheet region for all mutants (including both molecules A and B in mutant β2,3-A3) with WT* (cyan) to show the conservation of the hydrogen bonding network.
Table 3.
Structural changes within the β-sheet region
| Proteins compared | Shift (Å) |
| WT* vs. β1-A6 | 0.29 |
| WT* vs. β2-A4 | 0.18 |
| WT* vs. β2,3-A3 (molecule A) | 0.28 |
| WT* vs. β2,3-A3 (molecule B) | 0.31 |
| B2,3-A3 (molecule A vs. molecule B) | 0.16 |
| WT* vs. β3-A3 | 0.24 |
The values quoted are the r.m.s.d. derived from the superposition of the backbone atoms of residues 14–17 plus 25–32, i.e., the residues within strands β1, β2, and β3, plus the buried turn between β2 and β3 (Fig. 1B ▶).
Mutant β1-A6
In mutant β1-A6 six residues were replaced with alanine (β1 strand, loop 2; Fig. 1B ▶). The protein is substantially destabilized with the melting temperature at pH 3.0 reduced by 7.6°C (Table 1). The unfolding transition is also broader than WT* with the enthalpy of unfolding being reduced from 128 kcal/mole for WT* to 60 kcal/mole for the mutant. Thermal unfolding transitions of this breadth are characteristic of multistate unfolding, suggesting that the calculated loss in free energy (Table 1) is a minimum estimate.
In the 2.1-Å resolution structure the overall structure of the β-sheet is very similar to WT* with an r.m.s.d. of 0.29 Å for the main chain atoms (Fig. 2A ▶; Table 3). The largest shifts within the β-sheet (~0.5 Å) occur in the vicinity of residues 17–20 at the end of β1, with larger deviations (up to 3 Å) in loop 2 (Fig. 2A ▶). Three of the mutations—K19A, T21A, and E22A—are in loop 2 and are involved in crystal contacts that are distinct from WT*. It may be that these mutations stabilize an alternate conformation of the loop that allows for different crystal packing and the new space group. The largest shifts (~4.3 Å) in the amino-terminal domain are in loop 3, which forms part of the same crystal contact as loop 2 (Fig. 2A ▶). Mutations R14A, K16A, K19A, and T21A replace largely solvent-exposed side chains, and therefore do not affect internal packing. E22A disrupts a salt bridge with R137, with the latter adopting a new rotomer and participating in a crystal contact in the new space group. Only I17A affects intramolecular packing, and removal of this partially buried, bulky side chain would be expected to be destabilizing (Fig. 3A ▶). The loss in stability of the single mutant I17A (ΔΔG = −2.7 kcal/mole)(Xu et al. 1998) accounts for a substantial part of the overall destabilization ofβ1-A6.
Figure 3.
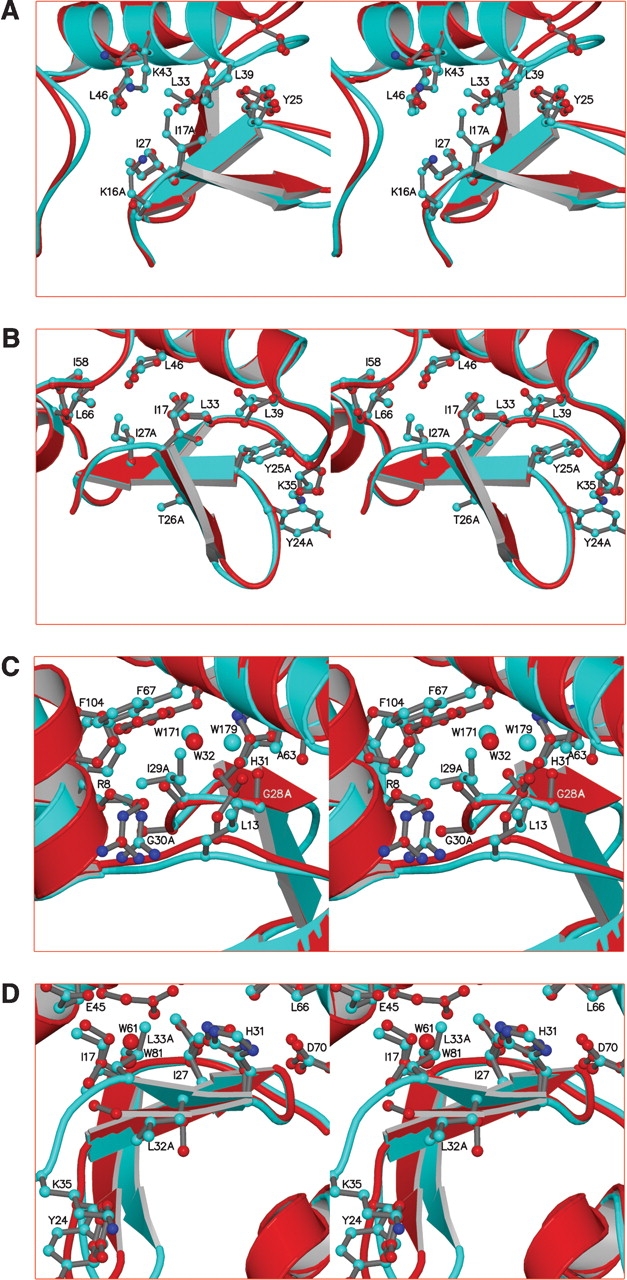
Stereo diagrams superimposing the β-sheet region of the polyalanine mutants (red) on WT* (cyan) to show detailed changes that occur in response to the mutations. The amino acids that were substituted with alanine are identified. The superpositions are based on the backbone atoms of residues 14–17 plus 25–32. (A) Mutant β1-A6. (B) Mutant β2-A4. (C) Mutant β2,3-A3. (D) Mutant β3-A3. The chain was disordered past Ala 33.
Mutant β2-A4
In mutant β2-A4 the four consecutive residues 24–27, which include strand β2, were replaced with alanine (Fig. 1B ▶). Although these residues form the core of the β-sheet, these potentially disruptive mutations have surprisingly modest effects on the overall structure of the β-sheet (Table 3). Mutations Y24A and Y25A remove packing interactions with side chains in loop 3 and α-helix B (Figs. 2B ▶, 3B ▶). As a result, Ala 24 in loop 2 and Lys 35 of loop 3 shift by ~0.4 Å and ~0.8 Å, respectively (Fig. 3B ▶). These shifts reduce the distance between the Cα atoms of residues 24 and 35 from 5.4 Å to 4.5 Å and minimize the void due to the loss of the two tyrosines. Site 26 is partly solvent-exposed and the mutation T26A would not be expected to adversely affect protein stability. The single mutation I27A, however, is known to be destabilizing (ΔΔG = −3.1 kcal/mole)(Xu et al. 1998). Ile 27 in WT* is involved in packing interactions with several residues, including Ile 17, Leu 46, and Ile 58 (Fig. 3B ▶). In the mutant structure, all of these side chains shift toward the cavity left by the I27A mutation, with the main chain of L46 shifting the most (0.46 Å). Given the potential large cavities formed by these substitutions, it is not surprising that the β2-A4 mutant is severely destabilized. Again, the unfolding transition is very broad (ΔH ~55 kcal/mole) and the apparent melting temperature drops about 17°C relative to WT* (Table 1).
Mutant β2,3-A3
Gly 28, Ile 29, and Gly 30 form a tight turn between the β2 and β3 strands in the native structure and are buried in the junction between the amino- and carboxy-terminal domains. This junction acts as a hinge, allowing the angle between the two domains to vary over a range of 50° in crystal structures of different mutants (Zhang et al. 1995). In mutant β2,3-A3, residues 28–30 were substituted to give G28A/I29A/G30A (Fig. 1B ▶). The structure was determined in a space group different than that of WT*, and contained two molecules, A and B, in the asymmetric unit (Fig. 4 ▶). Although the β-sheet structure in mutant β2,3-A3 is highly similar to WT* (Fig. 2C ▶), the hinge-bending angle between the amino- and car-boxy-terminal domains increases by 14.1° and 14.7° for molecule A and B, respectively. Changes in hinge-bending angles can occur when lysozyme variants are crystallized in different space groups, or have different environments in the same crystal (Zhang et al. 1995). It is not possible to attribute these changes in angle to the mutations alone, but given their nature and location they could be expected to have a significant effect on the junction. The G28A inserts a β-carbon into the junction, and if the hinge-bending angle were the same as WT*, the Cβ would be close (ca. 3.5 Å) to the β-carbon of Ala 63 (Fig. 3C ▶). With the increase in hinge-bending angles, the distances have increased to 4.4 Å in both molecule A and B.
Figure 4.

Similarity of the amino-terminal domains of molecule A (red) and molecule B (green) of mutant β2,3-A3.
In addition, WT* buries two water molecules in this junction (W171 and W179). If the structure were unchanged, the Cβ of Ala 28 would be unfavorably close (~2 Å) to W179 (Fig. 3C ▶). Presumably as a result of this steric clash, the water structure has changed, leaving only a single buried water molecule (W32) at this site (Fig. 3C ▶).
The backbone conformation of G28A is very similar to WT* (Table 4). For an alanine residue, this represents a strained, although not unobserved, conformation (Karplus 1996). Theoretical calculations based on bond stretching and angle bending estimate this conformation to be unfavorable by ~2 kcal/mole, although this calculation neglects stabilizing hydrogen bonding and packing interactions in the folded protein (Karplus 1996). The single mutant, G28A/WT*, showed a destabilization of 2 kcal/mole (Table 1). Significant destabilization is also expected for the I29A mutation. Ile 29 is a core residue, and removal of its bulky side chain is accompanied by shifts in nearby hydrophobic residues as they attempt to fill the newly formed void. The single mutant I29A is known to cause a 2.6-kcal/mole loss of stability (Xu et al. 1998). G30A would seem to have the least effect on the structure, as it is relatively solvent-accessible and only requires the solvent-exposed carboxylate of E11 to shift. In fact, the single mutant G30A has been found to have no effect on stability (Shoichet et al. 1995).
Table 4.
Backbone conformation of mutant β2,3-A3
| WT* | Mutant β2,3-A3 | ||||||
| Molecule A | Molecule B | ||||||
| φ (°) | ψ (°) | φ (°) | ψ (°) | φ (°) | ψ (°) | ||
| Ile27 | −149 | 166 | Ile27 | −140 | 169 | −141 | 172 |
| Gly28 | 70 | −131 | Ala28 | 56 | −129 | 56 | −128 |
| Ile29 | −103 | 72 | Ala29 | −104 | 84 | −98 | 85 |
| Gly30 | 63 | 34 | Ala30 | 56 | 39 | 52 | 37 |
| His31 | −82 | 103 | His31 | −91 | 105 | −88 | 108 |
Mutant β3-A3
In mutant β3-A3, residues Leu 32, Leu 33, and Thr 34 were mutated to alanine (Fig. 1B ▶). Because it was known that the single mutant L33A decreased the stability of WT* lysozyme by 3.6 kcal/mole (Xu et al. 1998), it was decided to at least partially offset this by including the mutation E108V in β3-A3. E108V is distal to the β-sheet and stabilizes the WT* protein structure (Table 1; Wray et al. 1999). In the crystal structure the β-sheet is intact, although residues 34–37 are largely disordered and are not included in the model (Fig. 5 ▶). In WT* the side chain of Leu 32 is partially buried in the protein surface and along with the side chain of Tyr 24 buttresses the main chain of loop 3 (Fig. 3D ▶). In the absence of packing interactions from the side chain of Leu 32, loop 3 becomes disordered. The electron density for Ala 33 is very weak, and its Cα moves ~2 Å into the space left by the Leu 32 side chain. Despite this large main chain displacement all of the hydrogen bonds between the β2 and β3 strands seem to be maintained (Figs. 1B ▶, 2E ▶). The side chain of Leu 33 in WT* is largely buried. Its replacement with alanine, coupled with the main chain displacement leaves a declivity exposed to the surface. Glu 45 adopts a new rotomer that places its carboxylate near the opening of the declivity and two water molecules (W61 and W81) occupy the position of the Leu 33 side chain (Fig. 3D ▶). ΔΔG for β3-A3 is estimated to be at least −4.4 kcal/mole with respect to E108V/WT* (Table 1).
Figure 5.

Electron density for mutant β3-A3 showing the quality of the fit of L33A in a simulated-annealing omit map in which Ala 33 was left out of the molecular dynamics simulation. The coefficients are 2Fo-Fc and the map is contoured at 0.9 σ. Beyond Ala 33, the electron density becomes uninterpretable.
Discussion
Perhaps the most striking feature of the results described here is the ability of the protein to maintain an essentially wild-type structure notwithstanding very substantial changes in the amino acid sequence. An example is provided by the mutant β1-A6, R14A/K16A/I17A/K19A/T21A/E22A, in which six residues are mutated to alanine (Fig. 1B ▶). Five of the six substitutions are what might be described as “large-to-small,” and four of them involve alterations in charge. None of these substitutions would be considered as “conservative.” Nevertheless, the conformation of the β1 strand is well maintained (Figs. 2A ▶, 3A ▶). In general terms it provides further evidence of the ability of protein structures to tolerate substitutions in the amino acid sequence, even some that may substantially destabilize the folded protein (Matthews 1987, 1996).
It is particularly striking that in all of the mutants the core region of the β-sheet is especially well conserved. If each of the structures is superimposed on WT* based on residues 14–17 plus 25–32 (Fig. 2E ▶), that is, the residues within strands β1, β2, and β3 plus the buried turn connecting β2 to β3, then the discrepancy in no case exceeds 0.31 Å (Table 3). Furthermore, the hydrogen-bonding network between the β-sheet strands is well conserved (Fig. 2E ▶).
Some of the structural differences that are seen in the mutant structures occur in regions of crystal contact (Fig. 2A ▶). It is possible that differences in such crystal contacts may contribute to the structural changes, especially within the solvent-exposed loop regions. On the other hand, the two independent molecules in the crystals of mutant β2,3-A3 have different crystallographic environments, but very similar crystal structures. This suggests that structural changes due to differences in crystal contacts are probably modest.
The results described here make it possible to compare the consequences of multiple alanine substitutions in β-sheet strands with those in α-helices. Previous studies with T4 lysozyme showed that the protein was, in general, very tolerant of alanine substitutions within α-helices (Heinz et al. 1992; Zhang et al. 1992; Blaber et al. 1995). For example, within the α-helix that includes residues 40–50, Asn 40, Ser 44, Glu 45, Asp 47, and Lys 48 can be replaced with alanine, yet the stability actually increases by 3.1°C at pH 3.0 (Heinz et al. 1992). Even if alanines are placed at all 10 consecutive positions 40–49, the protein folds and has a structure very similar to WT*. To some extent these results may be facilitated by the fact that the substituting residue, alanine, is helix-favoring (Lyu et al. 1990; O’Neil and DeGrado 1990; Blaber et al. 1993b; Chakrabartty and Baldwin 1995). If an alanine substitution is made at a fully solvent-exposed site in an α-helix, the stability of the protein can often be increased by 0.2–0.4 kcal/mole (Blaber et al. 1993a, 1995). On the other hand, if the alanine substitution is made for a fully buried bulky residue such as leucine, there can be substantial loss of hydrophobic stabilization in the range 2–5 kcal/mole (Eriksson et al. 1992; Blaber et al. 1995). Thus, if multiple alanine substitutions are made in a single helix, it is to be expected that some of these may be seriously destabilizing. At the same time, this loss of stability can to some degree be offset by the modest, but cumulative, stabilization arising from favorable helix propensity.
For alanine substitutions in a β-sheet, however, the situation is different. The numerical energy values for different β-sheet propensity scales differ (e.g., Kim and Berg 1993; Minor Jr. and Kim 1994; Smith et al. 1994), but it is generally agreed that alanine is one of the poorest β-sheet-forming residues (Smith et al. 1994). Thus it is perhaps not surprising that all four of the multiple alanine constructs within the β-sheet region of T4 lysozyme are destabilizing (Table 1). Still, each of these constructs retains its β-sheet conformation despite alanine’s poor β-sheet propensity. This supports the idea that, at least in T4 lysozyme, the conformation of the β-sheet region is determined to a substantial degree by the structural environment provided by the surrounding parts of the protein and less so by the amino acid sequence within the individual β-strands.
Related findings were reported by Sagermann and Matthews (2002). They took a peptide corresponding to the β2 and β3 strands as well as the turn that connects them (residues 24–35; Fig. 1 ▶) and appended it to the carboxyl terminus of T4 lysozyme. The structure was ambivalent. In one crystal form it appeared to be largely α-helical and in another crystal it appeared to be weakly ordered, extending through the active site cleft. In neither case did the appended peptide have a structure similar to a strand-turn-strand. Therefore, there was no suggestion that the amino acid sequence from residue 24 to residue 35 was of itself sufficient to cause this region to adopt the strand-turn-strand conformation seen in the WT* structure.
In contrast to the situation in the β-sheet region of T4 lysozyme, there is evidence that the conformations of at least some of the α-helices are determined by the amino acid sequence within, or adjacent to, the helix. This evidence does not come from polyalanine substitutions (which would be expected to be helix-favoring), but from sequence duplication experiments. One such experiment focused on residues 40–50, which form α-helix B (Fig. 1 ▶). If the amino acid sequence corresponding to this helix is duplicated in tandem, the duplicated region is also largely α-helical (Sagermann et al. 1999). This was taken as evidence that the helical structure was determined predominantly by amino acid sequence within the α-helix.
There are also parallels between the results obtained here and those of Minor Jr. and Kim (1994), who showed that an 11-amino-acid sequence (the “chameleon” sequence) could adopt either an α-helical or a β-sheet conformation depending on its location within the IgG-binding domain of protein G. These results illustrate the importance of the structural context, together with the local sequence, in defining the final conformation of structural elements in proteins.
It can be asked whether the mutants described here define the limit to which T4 lysozyme can be destabilized yet still fold. For β2-A4 and β2,3-A3 (Table 1), the reduction in ΔΔG is 5.5 kcal/mole and, as is typically the case for such unstable mutants, the enthalpies of unfolding are also substantially reduced relative to WT*. There are a few other mutant lysozymes that are somewhat less stable (ΔΔG ~8 kcal/mole), and for which crystal structures have been determined. These include mutant L99A/F153A that introduces a large cavity (Eriksson et al. 1992), mutant M102K that introduces a charged group into the core (Dao-pin et al. 1991), and some mutants that introduce up to nine methionines in the carboxy-terminal domain (Gassner et al. 2003). Thus, the mutants described in this article do not strictly reach the accessible lower limits of lysozyme stability and crystallizability, but they do approach that regime.
Materials and methods
Mutagenesis and protein purification
Mutant β1-A6 was constructed by the method of Kunkel (1985). The other variants were constructed by two-stage PCR (overlap-extension; Ho et al. 1989) using the unique restriction sites BamHI and HindIII. The two-stage PCR products were digested with BamHI and HindIII and were then ligated into the template vector that was digested with the same enzymes. The mutants were constructed in a cysteine-free version of the T4 lysozyme gene designated as WT* (Matsumura and Matthews 1989).
Mutations were confirmed by DNA sequencing. Proteins were expressed and purified as described (Muchmore et al. 1989; Poteete et al. 1991). The mutant proteins listed in Table 1 remained soluble and monomeric as judged by elusion profiles from sizing columns (data not shown). In vivo halo assays indicated that the mutant proteins in Table 1 remained catalytically active albeit to less extent than wild type (Poteete et al. 1991).
Thermal stability
Thermal unfolding was monitored by the circular dichroism (CD) change at 223 nm as a function of temperature (Eriksson et al. 1993) using a protein concentration of ~0.015 mg/mL in 20 mM of glycine-HCl, 1 mM of EDTA at pH 3.0. For the multiple mutant data presented here the van’t Hoff enthalpies of unfolding were too low to have resulted from two-state transitions (Carra et al. 1996). For those cases, calculation of the differential free energy of unfolding was done at the apparent melting temperature of the mutants assuming two-state unfolding for WT* and a ΔCp of 2.5 kcal/mole-K. The ΔΔG values derived in this manner should be minimum estimates of the degree of destabilization resulting from the mutational changes.
Crystallographic analysis
Mutant proteins β1-A6, β2-A4, and β2,3-A3 were crystallized by using the hanging drop method under “standard” conditions for WT* lysozyme (Eriksson et al. 1993). Typically, concentrated protein solutions (15–20 mg/mL) were mixed 1:1 with precipitant solutions containing approximately 50 mM of oxidized β-mercaptoethanol, 50 mM of reduced β-mercaptoethanol, 1.8–2.5 M of sodium/potassium phosphate at pH 6.5–7.1 (Lipscomb et al. 1998). The 1:1 mixture was then suspended over a well containing the undiluted precipitant solution. For mutant β3-A3, the well solution contained 200–300 mM of NaCl, 20 mM of HEPES at pH 7.0, 12%–16% PEG8000, 10% 2-propanol, and 2 mM of NiCl2. Hanging drops were made by mixing 5 μL of well solution with an equal volume of 1 mM protein solution in 20 mM of HEPES at pH 7.0 (Wray et al. 2000). The crystals of β2-A4 were isomorphous with WT* with the space group of P3221 and one molecule per asymmetric unit. Although similar conditions were used, mutant β2,3-A3 crystallized in space group C2 with two molecules per asymmetric unit and mutant β1-A6 crystallized in space group P6222. Mutant protein β3-A3 crystallized in space group I422 with one molecule per asymmetric unit.
X-ray diffraction data for β2-A4 and β2,3-A3 were collected under cryo conditions (Mooers et al. 2003) on a Rigaku R-AXIS imaging plate mounted on a rotating anode source (Table 2). The diffraction data for the crystal of β3-A3 were collected on beam-line 9–1 at the Stanford Synchrotron Radiation Laboratory. All data sets were integrated and scaled using the MOSFLM and SCALA within the CCP4 suite (Dodson et al. 1997).
Structures of mutants with space group P3221 were determined and refined by standard methods (Table 2; Eriksson et al. 1993). The structures of β2,3-A3 and β3-A3 were solved by molecular replacement using AMoRe (Navaza 1994). CNS (Brünger and Rice 1997) and TNT (Tronrud 1996) were used for model refinement (Table 2). Model building was performed by using the program O (Jones and Kjeldgaard 1997). Figures were prepared by using BOBSCRIPT (Esnouf 1997), MOLSCRIPT (Kraulis 1991), and RASTER3D (Merritt and Murphy 1994).
Coordinates and structure factors have been deposited in the PDB and will be released on publication of the article. Access codes are given in Table 2.
Acknowledgments
We are most grateful to Dr. Dirk Heinz for constructing mutant β1-A6; Leslie Gay and Joel Lindstrom for help in purifying and characterizing some of the mutant lysozymes; the Stanford Synchrotron Radiation Laboratory (SSRL) for data collection of mutant β3-A3; and Drs. Larry Weaver, Dale Tronrud, and Martin Sagermann for help with X-ray crystallography. This work was supported in part by NIH grant GM21967 to B.W.M.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Article published online ahead of print. Article and publication date are at http://www.proteinscience.org/cgi/doi/10.1110/ps.04875504.
References
- Blaber, M., Lindstrom, J.D., Gassner, N., Xu, J., Heinz, D.W., and Matthews, B.W. 1993a. Energetic cost and structural consequences of burying a hydroxyl group within the core of a protein determined from Ala → Ser and Val → Thr substitutions in T4 lysozyme. Biochemistry 32 11363–11373. [DOI] [PubMed] [Google Scholar]
- Blaber, M., Zhang, X-J., and Matthews, B.W. 1993b. Structural basis of amino acid α-helix propensity. Science 260 1637–1640. [DOI] [PubMed] [Google Scholar]
- Blaber, M., Baase, W.A., Gassner, N., and Matthews, B.W. 1995. Alanine scanning mutagenesis of the α-helix 115–123 of phage T4 lysozyme: Effects on structure, stability and the binding of solvent. J. Mol. Biol. 246 317–330. [DOI] [PubMed] [Google Scholar]
- Brünger, A.T. and Rice, L.M. 1997. Crystallographic refinement by simulated annealing: Methods and applications. Methods Enzymol. 277 243–269. [DOI] [PubMed] [Google Scholar]
- Carra, J.H., Murphy, E.C., and Privalov, P.L. 1996. Thermodynamic effects of mutations on the denaturation of T4 lysozyme. Biophys. J. 71 1994–2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakrabartty, A. and Baldwin, R.L. 1995. Stability of α-helices. Adv. Prot. Chem. 46 141–176. [PubMed] [Google Scholar]
- Dao-pin, S., Anderson, D.E., Baase, W.A., Dahlquist, F.W., and Matthews, B.W. 1991. Structural and thermodynamic consequences of burying a charged residue within the hydrophobic core of T4 lysozyme. Biochemistry 30 11521–11529. [DOI] [PubMed] [Google Scholar]
- Dodson, E.J., Winn, M., and Ralph, A. 1997. Collaborative Computational Project, Number 4: Providing programs for protein crystallography. Methods Enzymol. 277 620–633. [DOI] [PubMed] [Google Scholar]
- Eriksson, A.E., Baase, W.A., Zhang, X.-J., Heinz, D.W., Blaber, M., Baldwin, E.P., and Matthews, B.W. 1992. Response of a protein structure to cavity-creating mutations and its relation to the hydrophobic effect. Science 255 178–183. [DOI] [PubMed] [Google Scholar]
- Eriksson, A.E., Baase, W.A., and Matthews, B.W. 1993. Similar hydrophobic replacements of Leu 99 and Phe 153 within the core of T4 lysozyme have different structural and thermodynamic consequences. J. Mol. Biol. 229 747–769. [DOI] [PubMed] [Google Scholar]
- Esnouf, R.M. 1997. An extensively modified version of MolScript that includes greatly enhanced coloring capabilities. J. Mol. Graphics 15 133–138. [DOI] [PubMed] [Google Scholar]
- Frishman, D. and Argos, P. 1997. Seventy-five percent accuracy in protein secondary structure prediction. Proteins 27 329–335. [DOI] [PubMed] [Google Scholar]
- Gassner, N.C., Baase, W.A., Mooers, B.H.M., Busam, R.D., Weaver, L.H., Lindstrom, J.D., Quillin, M.L., and Matthews, B.W. 2003. Multiple methionine substitutions are tolerated in T4 lysozyme and have coupled effects on folding and stability. Biophys. Chem. 100 325–340. [DOI] [PubMed] [Google Scholar]
- Heinz, D.W., Baase, W.A., and Matthews, B.W. 1992. Folding and function of a T4 lysozyme containing 10 consecutive alanines illustrate the redundancy of information in an amino acid sequence. Proc. Natl. Acad. Sci. 89 3751–3755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho, S.N., Hunt, H.D., Horton, R.M., Pullen, J.K., and Pease, L.R. 1989. Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene 77 51–59. [DOI] [PubMed] [Google Scholar]
- Jones, T.A. and Kjeldgaard, M. 1997. Electron-density map interpretation. Methods Enzymol. 277 173–208. [DOI] [PubMed] [Google Scholar]
- Karplus, P.A. 1996. Experimentally observed conformation-dependent geometry and hidden strain in proteins. Protein Sci. 5 1406–1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, C.A. and Berg, J.M. 1993. Thermodynamic β-sheet propensities measured using a zinc-finger host peptide. Nature 362 267–270. [DOI] [PubMed] [Google Scholar]
- Kraulis, P.J. 1991. MOLSCRIPT: A program to produce both detailed and schematic plots of protein structures. J. Appl. Cryst. 24 946–950. [Google Scholar]
- Kunkel, T.A. 1985. Rapid and efficient site-specific mutagenesis without phenotypic selection. Proc. Natl. Acad. Sci. 82 488–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipscomb, L.A., Gassner, N.C., Snow, S.D., Eldridge, A.M., Baase, W.A., Drew, D.L., and Matthews, B.W. 1998. Context-dependent protein stabilization by methionine-to-leucine substitution shown in T4 lysozyme. Protein Sci. 7 765–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyu, P.C., Liff, M.I., Marky, L.A., and Kallenbach, N.R. 1990. Side-chain contributions to the stability of α-helical structure in peptides. Science 250 669–673. [DOI] [PubMed] [Google Scholar]
- Matsumura, M. and Matthews, B.W. 1989. Control of enzyme activity by an engineered disulfide bond. Science 243 792–794. [DOI] [PubMed] [Google Scholar]
- Matthews, B.W. 1987. Genetic and structural analysis of the protein stability problem. Biochemistry 26 6885–6888. [DOI] [PubMed] [Google Scholar]
- ———. 1996. Structural and genetic analysis of the folding and function of T4 lysozyme. FASEB J. 10 35–41. [DOI] [PubMed] [Google Scholar]
- Merritt, E.A. and Murphy, M.E.P. 1994. Raster3D version 2.0. A program for photorealistic molecular graphics. Acta Crystallogr. D 50 869–873. [DOI] [PubMed] [Google Scholar]
- Minor Jr., D.L. and Kim, P.S. 1994. Context is a major determinant of β-sheet propensity. Nature 371 264–268. [DOI] [PubMed] [Google Scholar]
- Mooers, B.H.M., Datta, D., Baase, W.A., Zollars, E.S., Mayo, S.L., and Matthews, B.W. 2003. Repacking the core of T4 lysozyme by automated design. J. Mol. Biol. 332 741–756. [DOI] [PubMed] [Google Scholar]
- Muchmore, D.C., McIntosh, L.P., Russell, C.B., Anderson, D.E., and Dahlquist, F.W. 1989. Expression and 15N labelling of proteins for proton and nitrogen-15 NMR. Methods Enzymol. 177 44–73. [DOI] [PubMed] [Google Scholar]
- Muñoz, V. and Serrano, L. 1995. Elucidating the folding problem of helical peptides using empirical parameters. II. Helix macrodipole effects and rational modification of the helical content of natural peptides. J. Mol. Biol. 245 275–296. [DOI] [PubMed] [Google Scholar]
- O’Neil, K.T. and DeGrado, W.F. 1990. A thermodynamic scale for the helix-forming tendencies of the commonly occurring amino acids. Science 250 646–651. [DOI] [PubMed] [Google Scholar]
- Poteete, A.R., Dao-pin, S., Nicholson, H., and Matthews, B.W. 1991. Second-site revertants of an inactive T4 lysozyme mutant restore activity structuring the active site cleft. Biochemistry 30 1425–1432. [DOI] [PubMed] [Google Scholar]
- Remington, S.J., Anderson, W.F., Owen, J., Ten Eyck, L.F., Grainger, C.T., and Matthews, B.W. 1978. Structure of the lysozyme from bacteriophage T4: An electron density map at 2.4 Å resolution. J. Mol. Biol. 118 81–98. [DOI] [PubMed] [Google Scholar]
- Sagermann, M. and Matthews, B.W. 2002. Crystal structures of a T4-lysozyme-duplication extension mutant demonstrates that the highly conserved β-sheet region in T4 has low intrinsic folding propensity. J. Mol. Biol. 316 931–940. [DOI] [PubMed] [Google Scholar]
- Sagermann, M., Baase, W.A., and Matthews, B.W. 1999. Structural characterization of an engineered tandem repeat contrasts the importance of context and sequence in protein folding. Proc. Natl. Acad. Sci. 96 6078–6083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoichet, B.K., Baase, W.A., Kuroki, R., and Matthews, B.W. 1995. A relationship between protein stability and protein function. Proc. Natl. Acad. Sci. 92 452–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith, C.K., Withka, J.M., and Regan, L. 1994. A thermodynamic scale for the β-sheet forming tendencies of the amino acids. Biochemistry 33 5510–5517. [DOI] [PubMed] [Google Scholar]
- Tronrud, D.E. 1996. Knowledge-based B-factor restraints for the refinement of proteins. J. Appl. Cryst. 29 100–104. [Google Scholar]
- Wray, J.W., Baase, W.A., Lindstrom, J.D., Weaver, L.H., Poteete, A.R., and Matthews, B.W. 1999. Structural analysis of a non-contiguous second-site revertant in T4 lysozyme shows that increasing the rigidity of a protein can enhance its stability. J. Mol. Biol. 292 1111–1120. [DOI] [PubMed] [Google Scholar]
- Wray, J.W., Baase, W.A., Ostheimer, G.J., Zhang, X.-J., and Matthews, B.W. 2000. Use of a non-rigid region in T4 lysozyme to design an adequate metal-binding site. Protein Eng. 13 313–321. [DOI] [PubMed] [Google Scholar]
- Xu, J., Baase, W.A., Baldwin, E., and Matthews, B.W. 1998. The response of T4 lysozyme to large-to-small substitutions within the core and its relation to the hydrophobic effect. Protein Sci. 7 158–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, X.-J., Baase, W.A., and Matthews, B.W. 1992. Multiple alanine replacements within α-helix 126–134 of T4 lysozyme have independent, additive effects on both structure and stability. Protein Sci. 1 761–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, X.-J., Wozniak, J.A., and Matthews, B.W. 1995. Protein flexibility and adaptability seen in 25 crystal forms of T4 lysozyme. J. Mol. Biol. 250 527–552. [DOI] [PubMed] [Google Scholar]
- Zhang, X.-J., Baase, W.A., and Matthews, B.W. 2002. A helix initiation signal in T4 lysozyme identified by polyalanine mutagenesis. Biophys. Chem. 101–102: 43–56. [DOI] [PubMed] [Google Scholar]