

Abstract
Aggregation of β-amyloid (Aβ) into fibrillar deposits is widely believed to initiate a cascade of adverse biological responses associated with Alzheimer’s disease. Although it was once assumed that the mature fibril was the toxic form of Aβ, recent evidence supports the hypothesis that Aβ oligomers, intermediates in the fibrillogenic pathway, are the dominant toxic species. In this work we used urea to reduce the driving force for Aβ aggregation, in an effort to isolate stable intermediate species. The effect of urea on secondary structure, size distribution, aggregation kinetics, and aggregate morphology was examined. With increasing urea concentration, β-sheet content and the fraction of aggregated peptide decreased, the average size of aggregates was reduced, and the morphology of aggregates changed from linear to a globular/linear mixture and then to globular. The data were analyzed using a previously published model of Aβ aggregation kinetics. The model and data were consistent with the hypothesis that the globular aggregates were intermediates in the amyloidogenesis pathway rather than alternatively aggregated species. Increasing the urea concentration from 0.4 M to 2 M decreased the rate of filament initiation the most; between 2 M and 4 M urea the largest change was in partitioning between the nonamyloid and amyloid pathways, and between 4 M and 6 M urea, the most significant change was a reduction in the rate of filament elongation.
Keywords: amyloid, β-amyloid, light scattering, atomic force microscopy, peptide aggregation
Alzheimer’s disease (AD) is an age-associated neurodegenerative disease characterized by loss of memory, language skill, and cognitive function. One of the defining characteristics of AD is the formation in the brain of extracellular amyloid senile plaques. The major protein constituent of amyloid plaques is the 4-kDa polypeptide β-amyloid (Aβ). Aβ undergoes spontaneous aggregation into β-sheet structured fibrils (Serpell 2000). In vitro, aggregation of Aβ is linked to cellular dysfunction and death (Pike et al. 1993; Seilheimer et al. 1997). Genetic and transgenic animal studies support the “amyloid cascade” hypothesis: that Aβ fibrillogenesis is required to initiate AD pathology (Selkoe 1997).
It was once assumed that the fully mature fibrillar amyloid deposits were toxic. More recently, it was proposed that small Aβ oligomeric species, intermediates in the fibrillogenesis pathway, are responsible for cell death, and that Aβ toxicity is caused by the aggregation process rather than the final product of aggregation (Lansbury 1999; Kirkitadze et al. 2002). If this hypothesis is true, then characterization of transient intermediates in the aggregation pathway is essential for a molecular-level description of Alzheimer’s disease pathology, and for rational design of Aβ toxicity inhibitors. This line of research would benefit by establishing solution conditions that arrest fibrillogenesis at these intermediate stages.
Urea and other chemical denaturants at moderate concentrations have been used extensively in protein-folding studies to trap folding intermediates (Cymes et al. 1996; Ayed and Duckworth 1999). Urea affects protein stability by hydrogen-bonding to the peptide backbone, thus destabilizing secondary structure, and by allowing greater solvation of hydrophobic side chains. Because amyloid assembly involves the same forces as those driving protein folding—that is, establishment of secondary structure through backbone hydrogen bonding, and burial of hydrophobic side chains—we hypothesized that urea could be used to trap amyloid “misfolding” intermediates.
In previous work, we showed that dissolution of Aβ in 8 M urea (pH 10) produced monomeric and denatured peptide (Pallitto and Murphy 2001). Subsequent dilution into phosphate-buffered saline with azide (PBSA) provided conditions for reproducibly generating Aβ fibrils. At Aβ concentrations of ~100 μM, in solvents at physiological pH and ionic strength, initial Aβ assembly is rapid (Pallitto and Murphy 2001), making elucidation of early events difficult. Retardation of Aβ aggregation by manipulating solvent conditions may provide a method to isolate early precursors in the fibrillogenesis pathway. In the work reported here, we tested whether increasing the urea concentration could slow down Aβ aggregation kinetics sufficiently to lead to isolation of intermediates, by reducing the thermodynamic driving force for aggregation (Wang and Bolen 1997). Solutions of Aβ in PBSA containing from 0.4 M to 6 M urea were assayed using several biophysical techniques: circular dichroism, size exclusion chromatography, dynamic and static light scattering, and atomic force microscopy. The data were analyzed in light of a previously published kinetic model.
Results
Secondary structure
Aβ in 8 M urea (pH 10) is believed to be monomeric and unstructured (Pallitto and Murphy 2001). This condition served as the starting point for dilution of Aβ into PBSA at varying urea concentrations. Circular dichroic spectra of Aβ in 0.4 M, 2 M, 4 M, and 6 M urea (with PBSA) are presented in Figure 1 ▶. In 0.4 M urea, Aβ contained a mix of random coil and β-sheet structure. Both θ217 and θ222 increased monotonically with increasing urea, indicating that there was a continuous increase in the unstructured content of Aβ as the urea concentration was increased (Creighton 1993).
Figure 1.

CD spectra of Aβ in 0.4 M, 2 M, 4 M, and 6 M urea (from bottom to top). All the samples contained 140 μM of Aβ and were incubated for ~22–24 h before CD measurements. Due to residual urea, the minimum wavelength was limited to 210–214 nm.
Monomer/dimer/aggregate distribution
The distribution of Aβ between monomeric and oligomeric species was determined by size exclusion chromatography (SEC). Aβ in 8 M urea (pH 10) elutes as a monomer, with full recovery of injected material (Pallitto and Murphy 2001). At 0.4 M and 2 M urea, SEC elution profiles were very similar, displaying two peaks with elution times corresponding to molecular weights of monomer and dimer, with the dimer by far the dominant species (Fig. 2 ▶). Little or no material was detected in the void, because Aβ aggregates tend to stick to the column under these conditions. To determine the fraction of Aβ in aggregated versus nonaggregated (monomer/dimer) form, we compared peak areas for identical samples injected with and without the column in place. In both 0.4 M and in 2 M urea, just over one-third of the peptide was incorporated into large aggregates (Table 1); this fraction was stable for at least ~22 h (data not shown).
Figure 2.

Representative size exclusion chromatograms of 140 μM of Aβ in 0.4 M, 2 M, 4 M, and 6 M urea (solid line, from top to bottom). The mobile phase was matched to that of the sample buffer. Molecular weight of Aβ peak was determined by calibration of column at each urea concentration using insulin chain B (3.5 kDa), ubiquitin (8.5 kDa), ribonuclease A (13.7 kDa), ovalbumin (43 kDa), and BSA (67 kDa). The letters M and D represent monomer and dimer, respectively. Arrows represent elution times of ubiquitin at different urea concentrations. The shift in elution times with different urea concentrations in mobile phase is likely due to urea-induced volume expansion. In 4 M urea, dimeric species were observed in four of six replicates; in the remaining two samples, initially only monomer was observed, but there was subsequently complete conversion to dimer within ~2–3 h. In 6 M urea, seven of nine replicates showed a purely monomeric peak initially, shown as a dotted line, followed by a complete switch to purely dimer within ~2–3 h. Only dimer was detected in remaining 2/9 replicates.
Table 1.
Effect of urea on distribution of Aβ between aggregates and nonaggregated populations
| Urea concentration | ||||
| 0.4 M (n = 6) | 2 M (n = 7) | 4 M (n = 6) | 6 M (n = 9) | |
| % Nonaggregates (M + D)a | 63 ± 8 | 63 ± 6 | 80 ± 8 | 78 ± 8 |
| % Aggregatesa | 37 ± 8 | 37 ± 6 | 20 ± 8 | 22 ± 8 |
a Identical samples were injected using the same sample loop and detector with and without the column in place. To calculate the mass fraction of Aβ in monomer and dimer populations, the individual peak areas (obtained with the column in place) were divided by the peak area without the column. The mass fraction of Aβ as aggregates (>70 kDa) was calculated by difference.
Injection of Aβ in PBSA containing 4 M and 6 M urea produced modestly different SEC elution profiles (Fig. 2 ▶). In 4 M urea, dimeric species were observed in four of six replicates. In the remaining two samples, initially only monomer was observed, but there was subsequently complete conversion to dimer within ~2–3 h (data not shown). In 6 M urea, seven of nine replicate preparations showed a purely monomeric peak initially, followed by a complete switch to purely dimer within ~2–3 h. In the remaining replicates, only dimer was detected. At both 4 M and 6 M urea, peak areas and elution times were stable from ~4–5 h to > 48 h after dilution. In these samples, only about one-fifth of the peptide was aggregated (Table 1). The elution time of the putative dimer shifted from ~30 min to ~27 min with a urea concentration change from 0.4 M to 6 M. This suggests volume expansion of the dimer, similar to that observed for ubiquitin (data not shown).
Aggregation kinetics
We measured the rate of increase in size of Aβ aggregates using dynamic light scattering (DLS). Autocorrelation data were taken at a 90° scattering angle repeatedly over a 30-h time period. The data were analyzed using the cumulants method to obtain the z-average hydrodynamic diameter dsph. The average scattered intensity at the 90° scattering angle, I(90°), was measured over the same 30-h period. dsph is indicative of the average molecular size, whereas I(90°) is related to average molecular mass. Because of the way that data from a heterogeneous mixture of particles are weighted, dsph is more sensitive to larger particles than is I(90°).
At the earliest time point measured, dsph decreased with increasing urea concentration, from ~24 nm at 0.4 M urea to ~10 nm at 6 M urea (Fig. 3A ▶). At 0.4 M and 2 M urea, dsph increased over time, with a much greater rate of increase at 0.4 M urea. In contrast, for samples in 4 M and 6 M urea, dsph was independent of time. I(90°) decreased dramatically as the urea concentration increased from 0.4 M to 2 M (Fig. 3B ▶). At both urea concentrations, a temporal increase in I(90°) was observed, with the rate of increase much greater at 0.4 M urea. Initial I(90°) at 4 M and 6 M urea was slightly lower than the value at 2 M urea; I(90°) did not change with time for these samples.
Figure 3.

(A) Hydrodynamic diameter dsph and (B) scattering intensity at 90° angle I(90°) as determined by dynamic light scattering for Aβ in 0.4 M (○), 2 M (□), 4 M (♦), and 6 M (×) urea after sample preparation. Light scattering data are shown along with the fits to the kinetic model (see text).
Autocorrelation data were further analyzed using CONTIN, a constrained regularization method useful for determining size distributions, especially for broad or polydisperse populations (Provencher 1982). These distributions are intensity-averaged and are therefore heavily weighted toward the largest particles. Representative size distributions for samples early (~0.5–3 h) and late (22–24 h) in the aggregation process are presented in Figure 4 ▶. At the lowest and highest urea concentrations, unimodal distributions were observed, whereas at the two intermediate urea concentrations, multimodal distributions were detected. At early times, the most-populated size range was 20–30 nm, regardless of urea concentration. Additionally, small (<10 nm) particles were detected at the highest urea concentration. Interestingly, a population of much larger particles was detected at intermediate (2 M and 4 M), but not at the lowest or highest, urea concentrations. With time, the size distribution shifted toward larger particles for the 0.4 M and 2 M urea samples. On closer examination, a difference emerges: At 0.4 M urea, there is a shift in the entire distribution toward larger particles, whereas at 2 M urea the population of aggregates centered at ~20 nm remains stable but the larger-sized particles grow with time. At 4 M or 6 M urea, no change in size distribution is observed, consistent with the lack of temporal effect on dsph.
Figure 4.

Size distribution of Aβ aggregates as determined by CONTIN analysis of DLS data. Urea concentration is 0.4 M, 2 M, 4 M, and 6 M reading from top to bottom. Left and right panels for each urea concentration showed Aβ distributions with detectable intensities after ~0.5–3 and ~22–24 h, respectively. Distributions from at least seven data points were collected and averaged for each panel.
Interestingly, the time to formation of visible precipitates was shortest at an intermediate urea concentration: at 0.4 M urea, precipitates were observed at ~50 h; at 2 M urea, at ~30 h; and at 4 M and 6 M urea, at > ~70 h. This is likely due to the greater insolubility of the long aggregates formed at 2 M urea.
Size and shape of Aβ aggregates
To obtain detailed information about the morphology and characteristic dimensions of Aβ aggregates, static light scattering measurements were collected after incubating samples for ~22–24 h. Weight-averaged molecular weight <M>w and z-averaged square radius of gyration 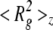 for each sample were calculated from linear regression fits to the data (Table 2). <M>wincludes contributions from monomers, dimers, and aggregates. The average molecular weight of aggregates, Magg, was calculated by dividing <M>w by the mass fraction of aggregated peptide (Table 1). <M>w (and Magg) decreased dramatically with an increase in urea concentration from 0.4 M to 2 M, with smaller decreases as the urea concentration rose further to 4 M or 6 M. Surprisingly, <Rg2>1/2z was greatest at the intermediate (2 M) urea concentration. (It should be noted that <Rg2>1/2z is heavily weighted toward the largest aggregates and is affected by volume expansion at high urea concentrations.)
for each sample were calculated from linear regression fits to the data (Table 2). <M>wincludes contributions from monomers, dimers, and aggregates. The average molecular weight of aggregates, Magg, was calculated by dividing <M>w by the mass fraction of aggregated peptide (Table 1). <M>w (and Magg) decreased dramatically with an increase in urea concentration from 0.4 M to 2 M, with smaller decreases as the urea concentration rose further to 4 M or 6 M. Surprisingly, <Rg2>1/2z was greatest at the intermediate (2 M) urea concentration. (It should be noted that <Rg2>1/2z is heavily weighted toward the largest aggregates and is affected by volume expansion at high urea concentrations.)
Table 2.
Effect of urea on Aβ aggregate size
| Urea concentration | ||||
| 0.4 M | 2 M | 4 M | 6 M | |
| 〈M〉w (kDa) | 1100 ± 110 | 100 ± 20 | 32 ± 2 | 14 ± 2 |
| Magg(kDa)a | 3000 ± 300 | 270 ± 40 | 160 ± 10 | 63 ± 9 |
| 〈Rg2〉1/2z (nm) | 56 ± 8 | 96 ± 22 | 50 ± 12 | 37 ± 12 |
| Lc(nm) | 150 ± 5 | 290 ± 30 | NDc | ND |
| dagg(nm)b | 6.8 | ND | ND | ND |
a We assumed only monomer, dimer, and large aggregates to be present. Then, Magg = (〈Μ〉w− WmonomerMmonomer − WdimerMdimer)/wagg.
b Calculated from Magg, Lc, and assumed specific volume of hydrated peptide.
c ND, not determined.
SLS data for 0.4 and 2 M urea samples were further analyzed to ascertain particle shape (e.g., rod vs. globular). (Particle sizes in 4 M and 6 M urea samples were too small to discriminate among different particle shapes.) The data were accurately described assuming a particle structure factor P(q) for rod-like particles (data not shown). Using an equation for P(q) of rod-like particles, the average length Lc of the particles was determined by nonlinear regression fit to the data; results are summarized in Table 2. Increasing the urea concentration from 0.4 to 2 M led to a sharp (~10-fold) decrease in <M>w but, surprisingly, a twofold increase in Lc. This is consistent with the observation by DLS of a small population of very long aggregates in the 2 M urea sample.
The diameter of the rodlike particles at 0.4 M urea was estimated, from Magg, Lc, and an assumed partial specific volume for the hydrated peptide vh of 1.1 cm3/g, to be 6.8 nm. The mass per unit length, Magg /Lc, of 20 kDa/nm, is in close agreement with that of the “protofilament” proposed by Tycko and colleagues (Petkova et al. 2002). At 2 M urea, the heterogeneity of the sample morphology (see CONTIN analysis and data below) precludes a simple calculation of the mass per unit length of these aggregates.
To provide visual confirmation of the particle morphologies and sizes, Aβ aggregates were imaged using atomic force microscopy (AFM). Representative images are shown in Figure 5 ▶. At 0.4 M urea, the distribution was unimodal, with most aggregates appearing as short rod-like particles about 6–7 nm in height and 50–150 nm in length. Similar structures have been observed by others using AFM (Harper et al. 1997; Kowalewski and Holtzman 1999). These AFM results are quantitatively consistent with analysis of static light scattering data (Table 2), recalling that light scattering is biased toward larger particles. At 2 M urea, two distinct populations were observed. Most of the structures were globular blobs of ~6–7 nm height (Fig. 5C,D ▶). Assuming a spherical geometry and vh of 1.1 cm3/g produces an estimate of the molecular mass of these blobs of ~60–90 kDa (~15–20 mer). Nonfibrillar globular aggregates of similar dimension have been observed by others (Kowalewski and Holtzman 1999). A few definite fibrillar aggregates were readily observed, some with a helical or twisted appearance (Fig. 5C ▶). These fibrillar aggregates were generally ~8–12 nm in height and roughly 200–700 nm in length. This height is the same as the diameter of mature Aβ fibrils (Merz et al. 1983). The AFM images are quite consistent with the bimodal distribution detected by analysis of DLS data (Fig. 4 ▶), as well as the presence of large linear aggregates suggested by Kratky analysis of SLS data. The small globular blobs contribute to <M>w and Magg but not to the determination of Lc.
Figure 5.

AFM images of Aβ in (A) 0.4 M urea, fourfold diluted; (B) 0.4 M urea, 10-fold diluted; (C) 2 M urea, fourfold diluted; (D) 2 M urea, undiluted; (E) 4 M urea, fourfold diluted; and (F) 6 M urea, fourfold diluted. Aβ in each urea concentration was incubated for 20–22 h before taking AFM images in solution tapping mode. Samples were diluted as indicated just before imaging. Scale bar represents 100 nm.
AFM images taken of Aβ in 4 M and 6 M urea revealed only small spherical globules ~1–2 nm in height, a size consistent with Aβ monomers or dimers (Walsh et al. 1997; Stine et al. 2003). No fibrils or larger globular blobs were observed, even though light scattering and SEC data indicated the presence of some oligomers in these samples. There are two possible reasons for this apparent inconsistency: (1) Binding of these oligomers to the mica surface may be inhibited by the higher urea concentrations, and (2) the oligomers may be a very small number fraction of all species. Indeed, we estimate from the combined light scattering and SEC data at 6 M urea that, although the oligomers contain ~20% of the total mass of peptide, on a number basis only ~2% of all particles are oligomers.
Summary of experimental results
Experimental results are summarized in Table 3. Briefly, we observed that increasing the urea concentration in PBSA (1) decreased the quantity of β-sheet, (2) decreased the amount of aggregated material, (3) drastically reduced the average size of aggregates, (4) inhibited the slow growth of aggregates, and (5) changed morphology from fibrillar to globular/fibrillar mixture to globular. At 0.4 M urea in PBSA, Aβ contained a mix of β-sheet and random coil structure, with about 37 wt% highly aggregated. Aβ aggregates were rodlike particles with a unimodal distribution, 50–150 nm in length and 6–7 nm in diameter. Aggregates grew smoothly over time. Increasing urea to 2 M did not change the mass fraction of aggregated peptide, but slightly reduced the β-sheet content. There was a dramatic shift in the particle size distribution toward a bimodal distribution consisting of numerous small stable globular blobs (6–7 nm) and very few large and slow-growing fibrillar aggregates, 8–12 nm in diameter and 200–700 nm in length. At 4 M urea, the fraction of aggregated material dropped significantly, and the β-sheet content decreased further. Primarily 1–2-nm spheres, likely monomers/dimers, were observed by AFM imaging. Some aggregates were still detectable by light scattering, a technique that is particularly sensitive to larger particles. We surmise that these aggregates are distributed between small globules and a few large aggregates. Finally, at 6 M urea, there was a further drop in β-sheet content and the sample contained mainly 1–2-nm particles. Still, a small fraction of material was aggregated; by light scattering analysis these aggregates were predominantly ~15-mers, similar to the estimated size of the globules detected by AFM at 2 M urea.
Table 3.
Summary of experimental results
| Urea conc. | θ217 (CD) | % Aggregate (SEC) | Average no. of monomers in aggregates (LS) | Growth over time? (LS) | Aggregate morphology (AFM) |
| 0.4 M | −2058 | 37 | 700 | Yes | Fibrillar: 50–150 nm long, 6–7 nm high |
| 2 M | −1674 | 37 | 65 | Slight | Mixed: |
| Globular: 6–7 nm high | |||||
| Fibrillar: 200–700 nm long, 8–12 nm high | |||||
| 4 M | −1419 | 20 | 35 | No | Globular: 1–2 nm high |
| 6 M | −1135 | 22 | 15 | No | Globular: 1–2 nm high |
Kinetic modeling
We previously developed a detailed mathematical model describing the kinetics of Aβ aggregation (Pallitto and Murphy 2001). A schematic of the model is shown in Figure 6 ▶. Urea-denatured Aβ is postulated to undergo four major steps upon dilution into PBSA: (1) rapid refolding into either a nonfibrillogenic monomer/dimer M/D or a fibrillogenic dimeric intermediate I, (2) cooperative association of I into a nucleus N, (3) elongation of N by addition of I to form thin filaments f of variable length, and (4) slow lateral association of f into fibrils F. The model successfully recapitulated multiple experimental observations. We initially postulated that the same model structure would describe Aβ self-association in the presence of increasing urea, with model parameter values that would change with urea concentration. Initial efforts at fitting the data at higher urea concentrations to the original model were unsuccessful, however.
Figure 6.

Aβ aggregation model schematic.
To proceed, therefore, we needed to consider the nature of the globular oligomeric assemblies. Two possibilities were considered: (1) The globules are off-pathway aggregates, unrelated to fibril formation and growth, or (2) the globules are intermediates in the fibrillogenesis pathway, arrested in their growth by the higher urea concentrations. Several pieces of evidence support the latter hypothesis, that the globular oligomers are indeed trapped fibril precursors. First, at 2 M urea, a mixture of globular assemblies and larger fibrils was observed. Heights of elongated structures observed at 0.4 M urea were identical to those of globular structures at 2 M urea (6–7 nm). Second, in all four samples, an initial population of aggregates with dsph ~10 nm was observed (Fig. 4 ▶); at 0.4 M urea this population disappeared as larger linear aggregates were formed, but at the higher urea concentrations this population remained stable. These data are inconsistent with irreversible formation of off-pathway globules. Third, the average number of monomers per globular blob at 2 M urea estimated from AFM (~15), and the average molecular weight of aggregates Magg in 6 M urea estimated from light scattering (63 kDa) are both very similar to that of the nucleus proposed in previous work (Pallitto and Murphy 2001). Fourth, if globular structures were a dead-end product, insufficient amyloidogenic Aβ would remain at 2 M urea to lead to the formation of long fibrils. We attempted to develop kinetic models where the globules were dead-end off-pathway products rather than fibrillar precursors. However, we were unable to fit the data to these models despite significant effort. Most problematic was the data at 2 M urea: If the globules were off-pathway and unable to participate in fibril formation, then the concentration of I was driven too low for any significant fibril growth, in contrast to our observations. For these reasons, we hypothesized that the observed globular aggregates of 6–7 nm diameter and containing ~15 monomers were, indeed, trapped nuclei N, on the amyloid pathway.
The model as originally proposed (Pallitto and Murphy 2001) assumed that the rate constants for filament initiation (N + I→fn+1) and for filament elongation (fi + I →fi+1) were identical. However, this assumption precludes the possibility of simultaneous appearance of small oligomeric assemblies (e.g., substantial concentration of N) and large fibrillar aggregates. We relaxed this assumption and allowed the rate constants for filament initiation and filament elongation to be distinct.
We proceeded to evaluate model parameters as follows: First we examined the parameters related to the distribution between monomer, dimer, and larger aggregates. SEC data indicated that monomer/dimer/aggregate distributions at 0.4 M and 2 M urea were identical; therefore, values of kM/kI, kD/kI, and KMD were kept identical for these two conditions. There was a stepwise decrease in fraction of aggregated material as the urea concentration increased from 2 M to 4 M. At both 4 and 6M urea, a transient monomer switched to dimer within 2–3 h (Fig. 2 ▶). We postulated that the transient monomer is the fully denatured monomer Mu present in 8 M urea (pH 10), and is slower to refold to a stable dimer D upon dilution because of the large urea concentration (e.g., kD is slower). (This was a simpler and more attractive hypothesis than that an alternatively folded unstable monomer was formed.) We eliminated stable M formation (and hence kM/kI and KMD) for these samples. kD/kI values at 4 M and 6 M urea were evaluated by integration of second-order rate equations of D and I with respect to fully denatured monomer Mu, using the information on Aβ distribution given in Table 1. Fitted values are listed in Table 4. These results show that nonamyloidogenic D formation is favored 4:1 over amyloidogenic I at higher (4 M and 6 M) urea.
Table 4.
Effect of urea on model parameters
| Urea concentration | ||||
| 0.4 M | 2 M | 4 M | 6 M | |
| kM /kI (μM) | 80 ± 30 | 80 ± 30 | 0 | 0 |
| kD/kI | 0.65 ± 0.15 | 0.65 ± 0.15 | 4 ± 0.5 | 3.6 ± 0.4 |
| KMD (μM−1) | 0.64 ± 0.08 | 0.64 ± 0.08 | ND | ND |
| kpn /kn (μM4) | 6 ± 1 × 108a | 1.1 ± 0.6 | 10 ± 9 | 3.4 ± 2.7 |
| kpf/kn (μM4) | 6 ± 1 × 108a | 50 ± 20 × 103 | 40 ± 20 × 103 | 2 ± 2 × 103 |
| kla (μM−2 h−1) | 0.047 ± 0.003 | 2 ± 1 | ND | ND |
Number of filaments in fibril, p, number of intermediates I in nucleus N, n, and reaction order of filaments association into fibrils, q were assumed to be independent of urea concentration.
a From Pallitto and Murphy (2001). ND, not determined. Fitted values of the model parameters are reported along with their 95% confidence intervals.
Next, we evaluated model parameters related to nucleus formation, filament initiation and elongation, and fibril growth. We replaced the single rate constant kp, used for both filament initiation and elongation by addition of I, with two constants: kpn, for filament initiation (N + I →fn+1), and kpf, for filament elongation (fi + I →fi+1). Mathematically, the original model equations (described in Materials and Methods) were modified as:
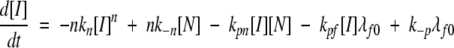 |
(5a) |
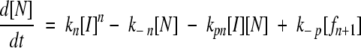 |
(6a) |
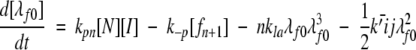 |
(7a) |
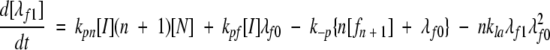 |
(8a) |
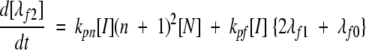 |
(9a) |
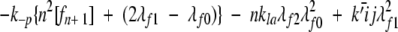 |
The experimental data are insufficient to determine unique values of kpn, kpf, and kn ; rather, we ascertain their ratio kpn/kn and kpf /kn. We used the methods developed previously (Pallitto and Murphy 2001) to relate the experimental light scattering data to the size distributions calculated from the model, with some minor modifications (Kim 2004).
The fitted model parameters are shown in Table 4, and the fitted curves are plotted in Figure 3 ▶. We used the fitted model parameters to calculate Magg and Lc in 2 M urea after 24 h; calculated values of ~260 kDa and ~260 nm, respectively, were close to experimental values (Table 2). The most striking feature of the 2 M urea sample was the coexistence of both small globules and long fibrillar aggregates (Fig. 5 ▶). The fibrillar aggregates were longer and thicker, but fewer in number, than those formed in 0.4 M urea. This implies that filament initiation is retarded more than filament elongation in this sample. The rate of aggregate growth was much slower at 2 M urea compared to 0.4 M urea. The fitted model parameters reflect these observations: a nearly 600-million-fold decrease in filament initiation (kpn/kn), an ~10-thousand-fold decrease in filament elongation (kpf /kn), and an ~40-fold increase in kla. At 4 and 6 M urea, the fraction of aggregated material decreased substantially, and AFM images indicated the presence of predominantly 1–2-nm particles, the expected size of Aβ monomers and dimers (Walsh et al. 1997; Stine et al. 2003). Light scattering, which is more sensitive to large particles, detected some oligomeric material. The average aggregate size was significantly smaller than that at the lower urea concentrations. These observations are reflected in the fitted model parameter values: There is an additional order-of-magnitude decrease in filament elongation (kpf /kn) at the highest urea concentration, and the rate constant for lateral aggregation, kla, could be reduced to zero.
The model results can be summarized succinctly as follows: Between 0.4 and 2 M urea, both filament initiation and elongation are reduced, and N is more stable than fn+1 in 2 M urea. Between 2 M and 4 M urea, the largest change is in the partitioning between nonamyloid and amyloidogenic pathways. Between 4 M and 6 M urea, the most significant change is a further reduction in filament elongation and a lack of any lateral association.
Discussion
Fibrillogenesis of Aβ is a likely causal factor in the onset of Alzheimer’s disease. Therefore, isolation of kinetic intermediates in the Aβ fibrillogenesis pathway is of great interest for the elucidation of a molecular-level description of AD pathology, and for rational design of Aβ toxicity inhibitors. During fibrillogenesis, an unfolded Aβ monomer converts to a β-sheet folded aggregate. We wondered whether moderate urea concentrations could trap intermediates in the Aβ fibrillogenesis pathway, akin to the common use of moderate denaturants to trap protein-folding intermediates.
Not surprisingly, increasing the urea concentration retarded Aβ aggregation kinetics significantly. Multiple effects were observed: a monotonic decrease in β-sheet content, a step decrease in the percent of aggregated material, and a change in aggregate morphology from shorter rod-like assemblies, to a mixture of globules and mature fibrils, to largely globules.
A key question is whether the globular assemblies are on or off the fibrillogenesis pathway. Although we are unable to provide definitive proof, the hypothesis that the globular assemblies are indeed fibrillar precursors was consistent with much of the experimental data. Furthermore, we were unable, despite numerous efforts, to develop a kinetic model based on the off-pathway hypothesis, whereas we were able to readily adapt our kinetic model of Aβ assembly to account for the effect of urea if we assumed that the globules are on-pathway.
Based on the experimental evidence and kinetic model combined, we propose that the observed globular oligomer is in fact the nucleus N. Further, we propose that N is ~6–7 nm in diameter and comprised of ~15 Aβ monomers, that N (and I) lack regular β-sheet structure, and that assembly of monomeric Aβ into I and N is driven primarily by hydrophobic forces. Thus, urea-driven solvation of hydrophobic side chains is likely the dominant mechanism behind the significant drop in the fraction of Aβ converted to I or N as the urea concentration is raised from 2 to 4 M. We further propose that conversion of nucleus to short filament (N + I →fn+1) and elongation of filaments by addition of amyloidogenic intermediate I (fi + I →fi+1), are accompanied by conversion to regular β-sheet structure, with its accompanying hydrogen bond formation. The large decrease in kpn/kn and kpf /kf observed when urea was raised from 0.4 M to 2 M is therefore likely due to disruption of hydrogen bonds by urea. We interpret the data to indicate that β-sheet formation occurs simultaneously with assembly of oligomers into filaments, and not with assembly of monomer into oligomers. This notion is particularly supported by the observation that, between 0.4 and 2 M urea, and again between 4 and 6 M urea, β-sheet content decreased whereas the mass fraction of aggregation material remained unchanged. Our results are in agreement with the work of Rosenberry and colleagues (Nichols et al. 2002), in which it was proposed that secondary structural changes occur upon elongation of filaments. Similarly, Yong et al. (2002) observed micelle-like Aβ assemblies at low temperature, and Stine et al. (2003) reported the presence of a small concentration of SDS-stable 60-kDa oligomers in a solution containing Aβ(1–40). Molecular dynamic simulations of aggregates of an Aβ fragment indicated that the aggregates were held together by a network of short-lived hydrogen bonds, and that aggregate interiors were molten globular or micellar in nature (Lakdawala et al. 2002). On the other hand, NMR studies indicate that mature fibrils do contain regular β-sheets and a core highly resistant to hydrogen exchange (Kheterpal et al. 2000; Antzutkin et al. 2002). These results draw a consistent picture of development of structure in stages, from unfolded to molten globular to β-sheet, as Aβ matures from monomer through nucleus to fibril.
There is a modest increase in the lateral association rate constant, kla, upon increasing the urea concentration from 0.4 M to 2 M. This result might be understood in light of the proposal that lateral association is driven primarily by electrostatic interactions (Petkova et al. 2002), given that urea reduces the dielectric constant of the solution. Perhaps a more likely explanation is that conversion of filaments to fibrils involves additional tertiary structural changes such as helical twisting, and that urea, by partially destabilizing filaments, actually reduces the energy barrier for conversion of filaments to fibrils. In line with this notion, mature fibrils under partially destabilizing solvent conditions have been observed by others (Booth et al. 1997; Kim et al. 2001).
Comparison with other amyloid-forming proteins may prove illuminating. Interestingly, moderate urea concentrations actually accelerated fibril formation with immunoglobulin light chain, by partially destabilizing the folded protein structure (Kim et al. 2001). With prion protein (PrP), increasing urea concentration led first to an increase in fibril formation kinetics followed by a decrease, with the maximum rate, at 2.4 M urea, corresponding to the midpoint of unfolding (Baskakov et al. 2004). PrP oligomers rich in β-sheet coexist with amyloid fibrils, with the oligomer favored at higher urea concentrations (Baskakov et al. 2002). However, several lines of evidence indicated that with PrP, the β-rich oligomer is not on the amyloid pathway. With Aβ, in contrast, we propose that the oligomer is not β-rich, but is on the amyloid pathway. Possibly, these differences are traceable to the starting conditions: PrP is natively folded, whereas monomeric Aβ is unfolded. Whether this pattern holds for a wider range of amyloid-forming proteins remains to be seen.
Materials and methods
Sample preparation
Lyophilized Aβ(1–40) was purchased from AnaSpec. Urea (electrophysiology/molecular biology grade) was purchased from Roche Biochemicals. All other chemicals were purchased from Sigma-Aldrich unless otherwise stated. Phosphate-buffered saline with azide (PBSA; 0.01 M Na2HPO4/NaH2PO4, 0.15 M NaCl, 0.02% (w/v) NaN3 [pH 7.4]) was double-filtered through 0.22-μm filters (Millex). 8 M urea was prepared in 10 mM glycine-NaOH buffer at pH 10, then filtered through 0.22-μm filters. Lyophilized Aβ(1–40) was solubilized using pre-filtered 8 M urea at a concentration of 2.8 mM for 10 min. These conditions were previously shown to produce monomeric and unfolded Aβ (Pallitto and Murphy 2001). Solutions were then diluted with filtered PBSA to 140 μM Aβ (equivalent monomer concentration), then quickly filtered through 0.45-μm filters directly into light scattering cuvettes (for light scattering) or microtubes (for size exclusion chromatography).
Circular dichroism spectroscopy
Circular dichroism (CD) spectra were obtained on Aβ samples prepared as described in urea/PBSA using an Aviv 62A DS Circular Spectrometer in the far-UV range with 0.1-cm pathlength cuvettes. The lower wavelength boundary was limited to 210–214 nm due to the residual urea in solution. The spectrum of the background was measured, followed by that of the sample. The background measurement was repeated to account for any machine drift and then subtracted from the average sample spectrum.
Size exclusion chromatography
The concentration of monomer and/or other small oligomers was determined by analysis of the samples by size exclusion chromatography (SEC), using a precision column pre-packed with Superdex 75 with a separation range of 3–70 kDa on a Pharmacia FPLC system. Briefly, the mobile phase flow rate was set at 0.05 mL/min and elution peaks were detected by UV absorbance at 280 nm. Mobile-phase solvent was matched to that used for sample preparation. The column was calibrated using insulin chain B (3.5 kDa), ubiquitin (8.5 kDa), ribonucleaseA (13.7 kDa), ovalbumin (43 kDa), and BSA (67 kDa). Urea caused a notable shift in the elution profiles of peptides and proteins used for calibration, presumably due to volume expansion. Therefore, the column was calibrated for each urea concentration investigated. Samples were also injected without the column in place; the peak area was used to calculate the total Aβ concentration of each sample. Samples were analyzed every hour for the first 5–7 h of aggregation, and then at least five more injections were completed after 24 h.
Laser light scattering
Samples were filtered through 0.45-μm filters directly into a cleaned light scattering cuvette placed in a bath of the index-matching solvent decahydronaphthalene, which was temperature-controlled to 25°C. A Coherent argon ion laser operated at 488 nm and a Malvern 4700c system were used to collect light scattering data. Autocorrelation data at 90° scattering angle were collected frequently (~10 times/h) for ~30 h, as described in more detail elsewhere (Shen et al. 1993). Data were fit to a third-order cumulants expression to yield a z-average hydrodynamic diameter dsph. Further information on size distribution was obtained by re-analysis of autocorrelation data using CONTIN. Average scattered intensity at 90° scattering angle, I(90°), was measured at the same time. After ~22–24 h, scattered intensity as a function of angle was measured and information on average particle molecular mass, shape, and dimensions was obtained as described in detail previously (Shen et al. 1993; Murphy and Pallitto 2000).
Atomic force microscopy
Aβ samples were prepared as described above and were used directly, or with fourfold or 10-fold dilution. Samples were adsorbed onto freshly cleaved mica attached to a stainless steel disk. Samples were immediately imaged in fluid with a MultiMode Nanoscope IIIA Scanning Probe Microscope (Model MMAFM-2, Digital Instruments) without using an O-ring. Cantilevers with oxide-sharpened silicon nitride tips (Digital Instruments) having a nominal spring constant of 0.32 N/m were used in tapping mode with a type E scanner. The drive frequency was 8–9 kHz, and the free amplitude was between 0.25 and 0.4 V, corresponding to 5–8 nm.
Mathematical modeling
A model of Aβ aggregation kinetics was developed previously (Pallitto and Murphy 2001); a schematic is shown in Figure 6 ▶. Model equations are:
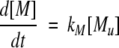 |
(1) |
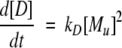 |
(2) |
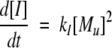 |
(3) |
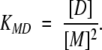 |
(4) |
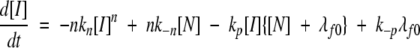 |
(5) |
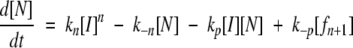 |
(6) |
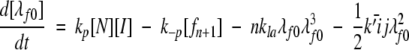 |
(7) |
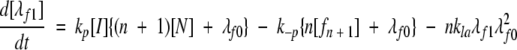 |
(8) |
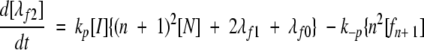 |
(9) |
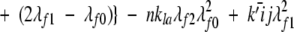 |
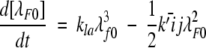 |
(10) |
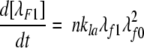 |
(11) |
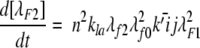 |
(12) |
where the parameters and species are defined in Figure 6 ▶, and λfi and λFi are the ith moments of filament and fibril distributions, respectively. In this work, a part of this model was modified (see Results). Model equations were solved numerically using DDASAC, and model parameters were fit to the data using the nonlinear regression package GREG, as described in detail (Pallitto and Murphy 2001). Unless otherwise specified (see Table 4), model parameters were assumed independent of urea concentration.
Acknowledgments
Acknowledgments
Funding for this research was provided by NIH grant AG 14079 from the National Institute of Aging. A.S.M. was supported by grants from the Alzheimer’s Association (IIRG-9901175) and the American Health Assistance Foundation (A1999057). The work was partially supported by the University of Chicago MRSEC Program of the NSF DMR0213745.
Abbreviations
Aβ, β-amyloid
AFM, atomic force microscopy
CD, circular dichroism, DLS, dynamic light scattering
PrP, prion protein
SEC, size exclusion chromatography
Article published online ahead of print. Article and publication date are at http://www.proteinscience.org/cgi/doi/10.1110/ps.04847404.
References
- Antzutkin, O.N., Leapman, R.D., Balbach, J.J., and Tycko, R. 2002. Supramolecular structural constraints on Alzheimer’s β-amyloid fibrils form electron microscopy and solid-state nuclear magnetic resonance. Biochemistry 41 15436–15450. [DOI] [PubMed] [Google Scholar]
- Ayed, A. and Duckworth, H.W. 1999. A stable intermediate in the equilibrium unfolding of Escherichia coli citrate synthase. Protein Sci. 8 1116–1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baskakov, I.V., Legname, G., Baldwin, M.A., Prusiner, S.B., and Cohen, F.E. 2002. Pathway complexity of prion protein assembly into amyloid. J. Biol. Chem. 277 21140–21148. [DOI] [PubMed] [Google Scholar]
- Baskakov, I.V., Legname, G., Gryczynski, Z., and Prusiner, S.B. 2004. The peculiar nature of unfolding of the human prion protein. Protein Sci. 13 586–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booth, D.R., Sunde, M., Bellotti, V., Robinson, C.V., Hutchinson, W.L., Fraser, P.E., Hawkins, P.N., Dobson, C.M, Radford, S.E., Blake, C.C., et al. 1997. Instability, unfolding and aggregation of human lysozyme variants underlying amyloid fibrillogenesis. Nature 385 787–793. [DOI] [PubMed] [Google Scholar]
- Creighton, T.E. 1993. Proteins: Structures and molecular properties, 2nd ed. W.H. Freeman, New York.
- Cymes, G.D., Grosman, C., Delfino, J.M., and Wolfenstein-Todel, C. 1996. Detection and characterization of an ovine placental lactogen stable intermediate in the urea-induced unfolding process. Protein Sci. 5 2074–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harper, J.D., Lieber, C.M., and Lansbury Jr., P.T. 1997. Atomic force microscopic imaging of seeded fibril formation and fibril branching by the Alzheimer’s disease amyloid-β protein. Chem. Biol. 4 351–358. [DOI] [PubMed] [Google Scholar]
- Kheterpal, I., Zhou, S., Cook, K.D., and Wetzel, R. 2000. Aβ amyloid fibrils possess a core structure highly resistant to hydrogen exchange. Proc. Natl. Acad. Sci. 97 13597–13601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, J.R. 2004. “Characterization of β-amyloid aggregation and its modulation.” Ph.D. thesis, University of Wisconsin, Madison, WI.
- Kim, Y.S., Cape, S.P, Chi, E., Raffen, R., Wilkins-Stevens, P., Stevens, F.J., Manning, M.C., Randolph, T.W., Solomon, A., and Carpenter, J.F. 2001. Counteracting effects of renal solutes on amyloid fibril formation by immunoglobulin light chains. J. Biol. Chem. 276 1626–1633. [DOI] [PubMed] [Google Scholar]
- Kirkitadze, M.D., Bitan, G., and Teplow, D.B. 2002. Paradigm shifts in Alzheimer’s disease and other neurodegenerative disorders: The emerging role of oligomeric assemblies. J. Neurosci. Res. 69 567–577. [DOI] [PubMed] [Google Scholar]
- Kowalewski, T. and Holtzman, D.M. 1999. In situ atomic force microscopy study of Alzheimer’s β-amyloid peptide on different substrates: New insights into mechanism of β-sheet formation. Proc. Natl. Acad. Sci. 96 3688–3693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakdawala, A.S., Morgan, D.M., Liotta, D.C., Lynn, D.G., and Snyder, J.P. 2002. Dynamics and fluidity of amyloid fibrils: A model of fibrous protein aggregates. J. Am. Chem. Soc. 124 15150–15151. [DOI] [PubMed] [Google Scholar]
- Lansbury Jr., P.T. 1999. Evolution of amyloid: What normal protein folding may tell us about fibrillogenesis and disease. Proc. Natl. Acad. Sci. 96 3342–3344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merz, P.A., Wisniewski, H.M., Somerville, R.A., Bobin, S.A., Masters, C.L., and Iqbal, K. 1983. An ultrastructural morphology of amyloid fibrils from neuritic and amyloid plaques. Acta. Neuropathol. (Berl.) 60 113–124. [DOI] [PubMed] [Google Scholar]
- Murphy, R.M. and Pallitto, M.M. 2000. Probing the kinetics of β-amyloid self-association. J. Struct. Biol. 130 109–122. [DOI] [PubMed] [Google Scholar]
- Nichols, M.R., Moss, M.A., Reed, D.K., Lin, W.L., Mukhopadhyay, R., Hoh, J.H., and Rosenberry, T.L. 2002. Growth of β-amyloid(1–40) protofibrils by monomer elongation and lateral association. Characterization of distinct products by light scattering and atomic force microscopy. Biochemistry 41 6115–6127. [DOI] [PubMed] [Google Scholar]
- Pallitto, M.M., and Murphy, R.M. 2001. A mathematical model of the kinetics of β-amyloid fibril growth from the denatured state. Biophys. J. 81 1805–1822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petkova, A.T., Ishii, Y., Balbach, J.J., Antzutkin, O.N., Leapman, R.D., Delaglio, F., and Tycko, R. 2002. A structural model for Alzheimer’s β-amyloid fibrils based on experimental constraints from solid state NMR. Proc. Natl. Acad. Sci. 99 16742–16747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pike, C.J., Burdick, D., Walencewicz, A.J., Glabe, C.G., and Cotman, C.W. 1993. Neurodegeneration induced by β-amyloid peptides in vitro: The role of peptide assembly state. J. Neurosci. 13 1676–1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Provencher, S.W. 1982. A constrained regularization method for inverting data represented by linear algebraic or integral equations. Comp. Phys. Commun. 27 213–227. [Google Scholar]
- Seilheimer, B., Bohrmann, B., Bondolfi, L., Muller, F., Stuber, D., and Dobeli, H. 1997. The toxicity of the Alzheimer’s β-amyloid peptide correlates with a distinct fiber morphology. J. Struct. Biol. 119 59–71. [DOI] [PubMed] [Google Scholar]
- Selkoe, D.J. 1997. Alzheimer’s disease: Genotypes, phenotype and treatments. Science 275 630–631. [DOI] [PubMed] [Google Scholar]
- Serpell, L.C. 2000. Alzheimer’s amyloid fibrils: Structure and assembly. Bio-chim. Biophys. Acta 1502 16–30. [DOI] [PubMed] [Google Scholar]
- Shen, C.-L., Scott, G.L., Merchant, F., and Murphy, R.M. 1993. Light scattering analysis of fibril growth from the amino-terminal fragment β(1–28) of β-amyloid peptide. Biophys. J. 65 2383–2395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stine Jr., W.B., Dahlgren, K.N., Krafft, G.A., and LaDu, M.J. 2003. In vitro characterization of conditions for amyloid-β peptide oligomerization and fibrillogenesis. J. Biol. Chem. 278 11612–11622. [DOI] [PubMed] [Google Scholar]
- Walsh, D.M., Lomakin, A., Benedek, G.B., Condron, M.M., and Teplow, D.B. 1997. Amyloid β-protein fibrillogenesis: Detection of a protofibrillar intermediate. J. Biol. Chem. 272 22364–22372. [DOI] [PubMed] [Google Scholar]
- Wang, A. and Bolen, D.W. 1997. A naturally occurring protective system in urea-rich cells: Mechanism of osmolyte protection of proteins against urea denaturation. Biochemistry 36 9101–9108. [DOI] [PubMed] [Google Scholar]
- Yong, W., Lomakin, A., Kirkitadze, M.D., Teplow, D.B., Chen, S.H., and Benedek, G.B. 2002. Stucture determination of micelle-like intermediates in amyloid β-protein fibril assembly by using small angle neutron scattering. Proc. Natl. Acad. Sci. 99 150–154. [DOI] [PMC free article] [PubMed] [Google Scholar]