

Abstract
The wealth of biological information provided by structural and genomic projects opens new prospects of understanding life and evolution at the molecular level. In this work, it is shown how computational approaches can be exploited to pinpoint protein structural features that remain invariant upon long evolutionary periods in the fold-type I, PLP-dependent enzymes. A nonredundant set of 23 superposed crystallographic structures belonging to this superfamily was built. Members of this family typically display high-structural conservation despite low-sequence identity. For each structure, a multiple-sequence alignment of orthologous sequences was obtained, and the 23 alignments were merged using the structural information to obtain a comprehensive multiple alignment of 921 sequences of fold-type I enzymes. The structurally conserved regions (SCRs), the evolutionarily conserved residues, and the conserved hydrophobic contacts (CHCs) were extracted from this data set, using both sequence and structural information. The results of this study identified a structural pattern of hydrophobic contacts shared by all of the superfamily members of fold-type I enzymes and involved in native interactions. This profile highlights the presence of a nucleus for this fold, in which residues participating in the most conserved native interactions exhibit preferential evolutionary conservation, that correlates significantly (r = 0.70) with the extent of mean hydrophobic contact value of their apolar fraction.
Keywords: PLP-dependent enzymes, remote homology, molecular evolution, conserved hydrophobic contacts, structural stability
It is broadly accepted that protein three-dimensional structural features are conserved among proteins sharing a common ancestor, despite low-sequence identity (Lesk and Chothia 1980; Chothia and Lesk 1986; Rodionov and Blundell 1998). A particularly interesting problem is how highly divergent sequences fold to similar structures (Michnick and Shakhnovich 1998). In many cases, no significant sequence similarity can be detected, except for regions of particular structural importance or, concerning enzymes, residues involved in the catalytic mechanism (Russell and Barton 1994). This observation implies that not all of the residues of a protein sequence are equally involved in the determination of its final three-dimensional structure. This raises several questions, such as,
Is it possible to detect sequence information necessary to maintain a particular fold and discriminate between this signal and the noise derived from variable regions?
To what extent can we relate sequence conservation at a superfamily level (throughout this work, the term “superfamily” is used according to the SCOP [Murzin et al. 1995] definition) with structural fold and function?
Can we expect to find in all of the members belonging to a superfamily a similar three-dimensional pattern of interacting residues that is reflected by a property conservation at those sites?
In an effort to address these questions, the interacting hydrophobic residues conserved at primary and tertiary structure levels have been investigated in the case of fold-type I, pyridoxal-5′-phosphate (PLP)-dependent enzymes. Although there are at least five evolutionarily unrelated superfamilies of PLP-dependent enzymes, each displaying a completely different fold, by far the largest and best-characterized is known as fold-type I, α family, or aspartate aminotransferase family (Jansonius 1998; Schneider et al. 2000). This large group of enzymes, which are found in all organisms and together cover the whole range of enzymatic activities cataloged by the Enzyme Commission (John 1995), bears several interesting characteristics; its members are highly divergent enzymes that display structural homology with almost undetectable sequence similarity; thanks to the recent massive sequencing of several genomes and advances in protein structure determination, a good wealth of experimentally well-characterized information is now available for this superfamily.
On the basis of such consideration, the present work was aimed at detecting the evolutionarily conserved structural patterns possibly responsible for the maintenance of the fold of this protein superfamily. The analysis was carried out in two steps; initially, a structural study extracted from a nonredundant set of 23 superposed crystallographic structures the features shared by this superfamily of enzymes, that is, the structurally conserved regions (SCRs) and the conserved hydrophobic contacts (CHCs); then, the initial multiple structural alignment was extended by adding sequence homologs to the enzymes whose structure is known, and an evolutionary analysis was undertaken on the final multiple alignment of 921 sequences to detect the most conserved sequence sites. Finally, the structure-based and the sequence-based analyses were compared.
The role played by conserved residues in the stabilization of the native structure and their possible involvement in the mechanism of protein folding was then discussed in light of the most recent studies on PLP-dependent enzymes.
Results
The data collection used in this work included 23 crystallographic structures and 921 sequences of fold-type I, PLP-dependent enzymes (Table 1) from different sources comprising the three domains of life, Eukarya, Bacteria, and Archaea. The level of sequence identity between the superimposed structures guaranteed the coverage of values inside the “twilight zone” (Rost 1999), ranging from 6% to 27% (mean 12%, SD ± 3%). Despite the low-sequence identity, this superfamily of enzymes displays a remarkable structural conservation, with a mean secondary structure agreement computed over six states (α-helix, 3–10 helix, β-bridge, extended strand, bend, hydrogen-bonded turn, and loop) of 64% ± 4% and with a maximum pairwise RMSD of 4.2 Å (Table 2).
Table 1.
Fold-type I enzymes data set
| PDB code | Enzyme description | Source | Resolution | R-value | Reference | Min. % identity | Number of homologs found |
| 1BJ4 | Serine hydroxymethyltransferase | H. sapiens | 2.65 | 0.210 | Renwick et al. 1998 | 35 | 101 |
| 1BS0 | 8-amino-7-oxononanoate synthase | E. coli | 1.65 | 0.178 | Alexeev et al. 1998 | 35 | 47 |
| 1FG3 | Histidinol phosphate aminotransferase | E. coli | 2.20 | 0.198 | Sivaraman et al. 2001 | 35 | 32 |
| 1JG8 | Threonine aldolase | T. maritima | 1.80 | 0.207 | Kielkopf and Burley 2002 | 40 | 21 |
| 1ECX | Nifs-like protein | T. maritima | 2.70 | 0.207 | Kaiser et al. 2000 | 40 | 101 |
| 1BJW | Aspartate aminotransferase | T. thermophilus | 1.80 | 0.215 | Nakai et al. 1999 | 40 | 43 |
| 1D2F | Maly protein | E. coli | 2.50 | 0.201 | Clausen et al. 2000b | 40 | 2 |
| 1C7N | Cystalysin | T. denticola | 1.90 | 0.208 | Krupka et al. 2000 | 30 | 35 |
| 1ELQ | L-cysteine/L-cystine C-S lyase | Synechocystis | 1.80 | 0.198 | Clausen et al. 2000a | 30 | 8 |
| 1DGD | Dialkylglycine decarboxylase | B. cepacia | 2.80 | 0.178 | Hohenester et al. 1994 | 35 | 98 |
| 1BJN | Phosphoserine aminotransferase | E. coli | 2.30 | 0.175 | Hester et al. 1999 | 40 | 38 |
| 1AY4 | Aromatic amino acid aminotransferase | P. denitrificans | 2.33 | 0.175 | Okamoto et al. 1998 | 40 | 21 |
| 1DTY | Adenosylmethionine aminotransferase | E. coli | 2.14 | 0.196 | Alexeev et al. 1998 | 40 | 49 |
| 2GSA | Glutamate semialdehyde aminomutase | C. biosynthesis | 2.40 | 0.183 | Hennig et al. 1997 | 40 | 58 |
| 1B9H | 3-amino-5-hydroxybenzoate synthase | A. mediterranei | 2.00 | 0.218 | Eads et al. 1997 | 35 | 9 |
| 1AX4 | Tryptophanase | P. vulgaris | 2.10 | 0.186 | Isupov et al. 1998 | 40 | 15 |
| 1CL1 | Cystathionine beta-lyase | E. coli | 1.83 | 0.151 | Clausen et al. 1996 | 40 | 23 |
| 1GTX | 4-aminobutyrate aminotransferase | Sus scrofa | 3.00 | 0.186 | Storici et al. 1999 | 40 | 9 |
| 1JS6 | Dopa decarboxylase | Sus scrofa | 2.60 | 0.206 | Burkhard et al. 2001 | 35 | 27 |
| 1ORD | Ornithine decarboxylase | Lactobacillus sp. | 3.00 | 0.219 | Momany et al. 1995 | 35 | 17 |
| 1QGN | Cystathionine gamma-synthase | N. tabacum | 2.90 | 0.201 | Steegborn et al. 1999 | 35 | 108 |
| 1LK9 | Alliin lyase | Allium sativum | 1.53 | 0.193 | Kuettner et al. 2002 | 40 | 2 |
| 1MDX | Arnb aminotransferase | S. typhimurium | 1.96 | 0.206 | Noland et al. 2002 | 35 | 57 |
Several criteria were adopted to reduce any possible redundancy in the data set being analyzed, only significant hits (E-value ≤0.001) were retained; hits displaying a sequence identity >80% with the query protein were rejected, as well as distant homologs (<30% sequence identity) for which we could not be sure of the accuracy of their alignments to the sequences of known structure. Moreover, hits with sequence length <80% of the query sequence were rejected in order to get in the final alignment only full sequences, avoiding fragments.
Table 2.
Pairwise sequence identity, RMSD, and the number of Cα atoms superposed between the structures used as data set
| PDB | 1BJ4 | 1BS0 | 1FG3 | 1JG8 | 1ECX | 1BJW | 1D2F | 1C7N | 1ELQ | 1DGD | 1BJN | 1AY4 | 1DTY | 2GSA | 1B9H | 1AX4 | 1CL1 | 1GTX | 1JS6 | 1ORD | 1QGN | 1LK9 | 1MDX |
| Lengtha | 470 | 384 | 358 | 347 | 384 | 382 | 390 | 399 | 390 | 432 | 360 | 394 | 429 | 432 | 388 | 467 | 395 | 472 | 486 | 730 | 445 | 448 | 393 |
| 1BJ4 | 0.13 | 0.16 | 0.13 | 0.13 | 0.15 | 0.12 | 0.12 | 0.13 | 0.13 | 0.08 | 0.14 | 0.10 | 0.12 | 0.11 | 0.14 | 0.11 | 0.10 | 0.14 | 0.11 | 0.15 | 0.07 | 0.13 | |
| 1BS0 | 2.9 | 0.14 | 0.17 | 0.11 | 0.12 | 0.14 | 0.10 | 0.16 | 0.17 | 0.10 | 0.13 | 0.17 | 0.18 | 0.16 | 0.11 | 0.14 | 0.10 | 0.16 | 0.14 | 0.11 | 0.12 | 0.20 | |
| 324 | |||||||||||||||||||||||
| 1FG3 | 2.9 | 3.0 | 0.15 | 0.17 | 0.16 | 0.14 | 0.12 | 0.15 | 0.11 | 0.09 | 0.14 | 0.11 | 0.12 | 0.13 | 0.11 | 0.13 | 0.11 | 0.10 | 0.12 | 0.10 | 0.14 | 0.14 | |
| 308 | 303 | ||||||||||||||||||||||
| 1JG8 | 2.9 | 2.9 | 2.7 | 0.12 | 0.14 | 0.12 | 0.12 | 0.15 | 0.14 | 0.08 | 0.12 | 0.12 | 0.13 | 0.15 | 0.20 | 0.16 | 0.13 | 0.12 | 0.12 | 0.15 | 0.10 | 0.17 | |
| 316 | 314 | 303 | |||||||||||||||||||||
| 1ECX | 3.3 | 2.5 | 3.5 | 2.7 | 0.13 | 0.09 | 0.11 | 0.18 | 0.12 | 0.12 | 0.09 | 0.08 | 0.14 | 0.11 | 0.12 | 0.09 | 0.15 | 0.11 | 0.12 | 0.14 | 0.12 | 0.13 | |
| 335 | 316 | 312 | 305 | ||||||||||||||||||||
| 1BJW | 3.6 | 3.3 | 2.6 | 3.2 | 3.9 | 0.16 | 0.17 | 0.12 | 0.11 | 0.12 | 0.18 | 0.11 | 0.16 | 0.16 | 0.17 | 0.14 | 0.11 | 0.08 | 0.13 | 0.14 | 0.17 | 0.14 | |
| 322 | 313 | 319 | 304 | 320 | |||||||||||||||||||
| 1D2F | 3.9 | 3.5 | 2.6 | 3.7 | 3.9 | 2.1 | 0.27 | 0.13 | 0.11 | 0.08 | 0.14 | 0.11 | 0.14 | 0.10 | 0.13 | 0.13 | 0.08 | 0.10 | 0.06 | 0.14 | 0.16 | 0.16 | |
| 332 | 315 | 321 | 313 | 322 | 343 | ||||||||||||||||||
| 1C7N | 3.9 | 3.5 | 2.7 | 3.6 | 3.7 | 3.9 | 1.7 | 0.10 | 0.09 | 0.07 | 0.11 | 0.11 | 0.13 | 0.09 | 0.11 | 0.09 | 0.09 | 0.10 | 0.10 | 0.08 | 0.13 | 0.10 | |
| 329 | 313 | 320 | 311 | 320 | 314 | 387 | |||||||||||||||||
| 1ELQ | 3.1 | 2.9 | 3.5 | 2.9 | 2.3 | 4.0 | 3.9 | 3.9 | 0.14 | 0.09 | 0.15 | 0.14 | 0.12 | 0.13 | 0.10 | 0.12 | 0.10 | 0.11 | 0.11 | 0.15 | |||
| 326 | 313 | 316 | 296 | 338 | 301 | 315 | 314 | ||||||||||||||||
| 1DGD | 3.2 | 2.6 | 3.6 | 3.5 | 3.3 | 3.7 | 3.9 | 3.2 | 3.1 | 0.12 | 0.23 | 0.19 | 0.12 | 0.12 | 0.12 | 0.24 | 0.12 | 0.08 | 0.13 | 0.09 | 0.13 | ||
| 334 | 334 | 322 | 326 | 323 | 315 | 322 | 332 | 314 | |||||||||||||||
| 1BJN | 3.8 | 3.9 | 3.9 | 3.6 | 3.4 | 3.8 | 3.9 | 3.9 | 3.6 | 3.9 | 0.09 | 0.07 | 0.08 | 0.10 | 0.08 | 0.09 | 0.07 | 0.10 | 0.07 | 0.09 | 0.10 | 0.10 | |
| 315 | 315 | 300 | 294 | 326 | 288 | 293 | 292 | 337 | 318 | ||||||||||||||
| 1AY4 | 3.9 | 3.4 | 3.0 | 3.7 | 3.9 | 3.1 | 3.0 | 3.2 | 3.0 | 3.5 | 3.8 | 0.11 | 0.14 | 0.14 | 0.12 | 0.13 | 0.10 | 0.11 | 0.08 | 0.12 | 0.11 | 0.14 | |
| 321 | 304 | 300 | 297 | 304 | 361 | 334 | 332 | 280 | 323 | 278 | |||||||||||||
| 1DTY | 3.5 | 3.0 | 3.6 | 3.8 | 3.5 | 3.6 | 3.8 | 3.9 | 3.3 | 2.1 | 4.0 | 3.4 | 0.20 | 0.11 | 0.12 | 0.09 | 0.16 | 0.11 | 0.12 | 0.14 | 0.08 | 0.13 | |
| 337 | 344 | 297 | 321 | 315 | 304 | 313 | 312 | 325 | 398 | 315 | 293 | ||||||||||||
| 2GSA | 3.5 | 2.5 | 3.4 | 3.6 | 3.3 | 3.7 | 3.3 | 3.9 | 3.3 | 2.2 | 3.8 | 3.7 | 2.4 | 0.14 | 0.11 | 0.10 | 0.17 | 0.12 | 0.08 | 0.15 | 0.08 | 0.12 | |
| 329 | 331 | 293 | 315 | 322 | 296 | 309 | 308 | 315 | 390 | 307 | 289 | 385 | |||||||||||
| 1B9H | 2.9 | 2.8 | 3.0 | 3.3 | 3.6 | 3.6 | 3.6 | 3.6 | 3.7 | 3.3 | 3.7 | 3.7 | 3.4 | 3.2 | 0.13 | 0.12 | 0.14 | 0.10 | 0.08 | 0.13 | 0.12 | 0.27 | |
| 309 | 294 | 296 | 295 | 325 | 309 | 316 | 314 | 325 | 307 | 296 | 303 | 292 | 296 | ||||||||||
| 1AX4 | 3.2 | 3.0 | 3.3 | 2.6 | 3.1 | 3.0 | 3.3 | 3.3 | 3.6 | 3.6 | 3.8 | 3.6 | 3.8 | 3.5 | 3.3 | 0.11 | 0.10 | 0.12 | 0.10 | 0.11 | 0.10 | 0.13 | |
| 335 | 333 | 322 | 329 | 332 | 326 | 339 | 336 | 333 | 334 | 279 | 316 | 349 | 325 | 278 | |||||||||
| 1CL1 | 2.8 | 2.4 | 2.8 | 2.9 | 3.3 | 3.2 | 3.8 | 3.9 | 3.0 | 3.0 | 3.6 | 3.6 | 3.1 | 3.1 | 3.0 | 2.9 | 0.10 | 0.12 | 0.09 | 0.25 | 0.11 | 0.14 | |
| 285 | 281 | 285 | 284 | 316 | 276 | 300 | 298 | 288 | 289 | 279 | 279 | 281 | 280 | 299 | 291 | ||||||||
| 1GTX | 3.5 | 3.1 | 3.7 | 3.6 | 3.5 | 3.6 | 4.0 | 3.5 | 3.6 | 2.0 | 3.5 | 3.9 | 2.6 | 2.2 | 3.6 | 3.4 | 3.0 | 0.09 | 0.10 | 0.11 | 0.09 | 0.10 | |
| 334 | 345 | 311 | 318 | 321 | 314 | 332 | 265 | 328 | 394 | 304 | 309 | 401 | 380 | 299 | 315 | 278 | |||||||
| 1JS6 | 2.9 | 3.0 | 2.8 | 3.0 | 3.6 | 3.1 | 3.4 | 3.2 | 3.5 | 3.3 | 3.6 | 3.2 | 3.6 | 3.1 | 3.6 | 3.2 | 3.3 | 3.7 | 0.10 | 0.13 | 0.08 | 0.11 | |
| 349 | 332 | 211 | 325 | 338 | 318 | 321 | 316 | 327 | 330 | 317 | 311 | 324 | 314 | 312 | 339 | 325 | 341 | ||||||
| 1ORD | 3.8 | 3.1 | 3.6 | 3.3 | 3.0 | 3.9 | 3.8 | 4.0 | 3.3 | 3.7 | 3.6 | 3.3 | 3.2 | 3.8 | 4.2 | 3.8 | 3.3 | 3.8 | 3.6 | 0.10 | 0.08 | 0.11 | |
| 352 | 299 | 291 | 308 | 304 | 310 | 278 | 311 | 310 | 315 | 285 | 212 | 231 | 298 | 301 | 316 | 294 | 306 | 309 | |||||
| 1QGN | 3.0 | 2.5 | 2.8 | 2.9 | 2.8 | 3.3 | 3.5 | 3.3 | 2.9 | 3.0 | 3.7 | 3.3 | 2.9 | 3.1 | 2.9 | 3.1 | 1.6 | 2.9 | 3.0 | 3.1 | 0.14 | 0.14 | |
| 290 | 287 | 280 | 286 | 294 | 383 | 278 | 276 | 283 | 285 | 287 | 263 | 274 | 282 | 297 | 298 | 365 | 276 | 287 | 289 | ||||
| 1LK9 | 3.3 | 3.7 | 3.7 | 3.9 | 3.9 | 2.7 | 3.7 | 2.6 | 3.8 | 3.7 | 3.8 | 3.8 | 4.0 | 3.6 | 3.8 | 3.4 | 3.8 | 3.7 | 3.6 | 3.9 | 3.1 | 0.12 | |
| 361 | 357 | 243 | 264 | 261 | 277 | 302 | 277 | 258 | 255 | 269 | 321 | 419 | 231 | 266 | 360 | 255 | 241 | 280 | 238 | 264 | |||
| 1MDX | 3.4 | 2.7 | 3.1 | 3.7 | 3.7 | 3.3 | 3.6 | 3.3 | 3.3 | 3.5 | 4.0 | 3.6 | 3.1 | 2.8 | 1.9 | 3.2 | 2.9 | 2.9 | 2.8 | 3.5 | 2.8 | 3.0 | |
| 353 | 237 | 284 | 250 | 250 | 272 | 320 | 272 | 252 | 221 | 213 | 308 | 251 | 242 | 331 | 244 | 261 | 245 | 276 | 265 | 269 | 296 |
Pairwise sequence identity is reported on the upper half-matrix, RMSD (Å) and the number of Cα atoms superposed on the lower half-matrix.
a Sequence length of each monomeric unit is also reported.
To identify the common core regions and the residues of these proteins involved in structural and functional roles, the study was focused on protein segments that conserve a similar main-chain conformation in all of the three-dimensional structures analyzed (SCRs; see Materials and Methods section), excluding the intervening regions whose structure differs markedly among different proteins. The SCRs were subjected to similar constraints during the divergent evolution of these enzymes from a common ancestor; therefore, they possibly contain most of the determinants necessary to maintain the fold. Seventeen regions with a mean positional RMSD ≤3.0 Å, lacking insertions and deletions were detected (Figs. 1 ▶, 2A ▶). Positional RMSD values ranged from 0.8 Å in position 65 (residue numbering refers to sites in Fig. 1 ▶), to 3.1 Å in position 126 (Table 3). Figure 2A ▶ shows that an extensive and evident common structural organization of the main chain around PLP is responsible for the appropriate positioning of key residues previously identified as structural determinants for binding the cofactor (Grishin et al. 1995). Five SCRs are mainly implied in the constitution of this common core as follows: one α-helix (α3, which displays a mean positional RMSD of 1.59 Å) and four β-strands, forming a β-sheet (β6, β9, β10, and β11, with a mean positional RMSD of 1.76 Å, 1.54 Å, 1.41 Å, and 1.52 Å, respectively).
Figure 1.

Alignment of the SCRs in the fold-type I enzymes. Structurally conserved regions (SCRs) are represented as blocks separated by dashes. The top line represents absolute position of the alignment. Alignment columns are colored according to the color scheme of Figure 2B ▶. Each sequence is labeled according to the PDB code of its corresponding structure (see Table 1). Boxes at the bottom represent secondary structure elements, and are labeled as follows: (α), α-helix; (β), β-strand; (L) loop; (T) turn.
Figure 2.
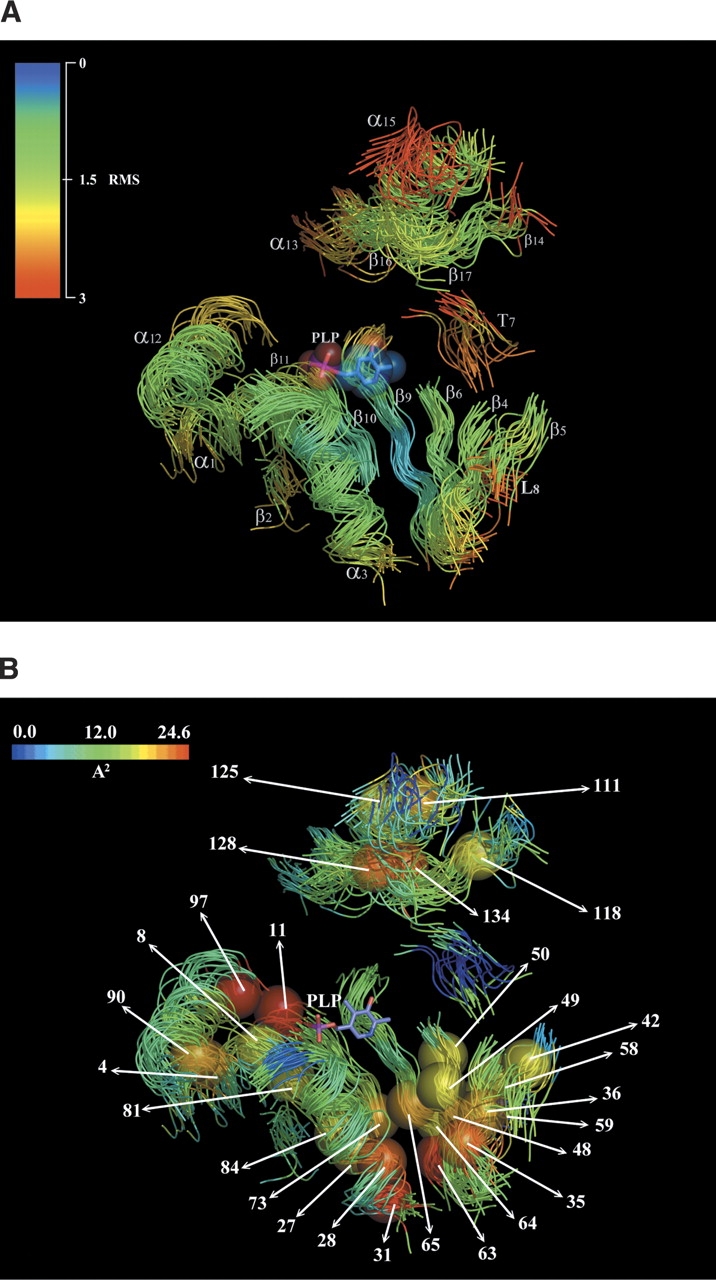
(A) Superimposition of the SCRs found in fold-type I enzymes. The backbones of the 23 superposed structures are shown as solid oval ribbon. Seventeen regions with a mean positional RMSD ≤3.0 Å, lacking insertions and deletions were detected, and the corresponding coordinates colored according to the RMSD value. PLP is displayed as slate CPKs, with oxygen atoms colored red; nitrogen atoms, blue; and phosphorus, purple. Each SCR is labeled as follows: (α), α-helix; (β), β-strand; (L) loop; (T) turn. (B) Representation of sites involved in making conserved hydrophobic contacts (CHCs). Positions involved in the strongest conserved CHCs (see also Table 4) are represented as colored space-filled spheres, and labeled according to absolute position of the alignment shown in Figure 1 ▶. The backbones of the 23 superposed structures are shown as a solid oval ribbon and colored according to the mean value of hydrophobic contact. PLP is displayed as slate sticks, with oxygen atoms colored red; nitrogen atoms, blue; and phosphorus, purple.
Table 3.
Structural and sequence attributes of the SCRs
| SCR | Mean RMS value | Site | RMS | Residues found in each position of the structural alignmenta | Max apolar contact surface (Å2) | Conservation score | Interaction with SCR, site |
| 1 | 2.2 | EHPIMLNTIVAEAEVYHVEATSN | 15.9 | 0.49 | 12, 90 | ||
| 2 | 2.2 | TQKNERDEAVEPINNYFETVVFQ | 6.1 | 0.46 | 1, 5 | ||
| 3 | 2.1 | LAAEKEEEQDEEEVSDSKVAVEE | 11.0 | 0.58 | 12, 94 | ||
| 4 | 1.8 | CLVLAAFYLLAFLLFLLLMALLL | 19.0 | 0.87 | 12, 90 | ||
| 5 | 1.8 | QEIERLLKRAEQCAEKQRMEEEE | 8.2 | 0.44 | 2, 15 | ||
| α1 | 2.0 | 6 | 2.1 | KEEREAAKQTKKRERDQEDKEKA | 6.7 | 0.83 | 1, 9 |
| 7 | 1.9 | RENLKEATARDAKMEKASWHKTA | 15.3 | 0.38 | 12, 97 | ||
| 8 | 1.7 | ALYAVKIVLLFMLVFAMLLAIIF | 17.6 | 1.15 | 12, 97 | ||
| 9 | 1.8 | LAAAAFAKAARGVNAKCLGASKC | 7.1 | 0.91 | 11, 83 | ||
| 10 | 2.0 | QEQEKRHKENDEADAEESKRAER | 5.7 | 0.92 | 1, 7 | ||
| 11 | 1.9 | AWYTVRWWTILLMAHLLVMVLLL | 24.5 | 0.90 | 12, 97 | ||
| 12 | 2.0 | YLAFLEFMFTLITVHFEALYEHT | 14.7 | 0.43 | 11, 83 | ||
| β2 | 2.1 | 14 | 2.4 | GRQESETWTDKTEEHDGSGKEYY | 6.2 | −0.46 | 11, 84 |
| 15 | 2.3 | VAVAEEVIIRVACMAYCQGTSIA | 9.7 | 0.21 | 11, 83 | ||
| 16 | 1.7 | NLLAITVITALTVVLIVLVYTVV | 7.4 | 0.31 | 11, 83 | ||
| 18 | 1.5 | YIRPSVPADSGVDNTHPAGGAVS | 9.5 | 0.57 | 3, 22 | ||
| 19 | 1.5 | SSGSCGSGNTGGSSNQCCSGSGS | 2.9 | 1.60 | 3, 22 | ||
| 20 | 1.5 | GGAGAGVVVGGGGGGGGGASGVA | 7.1 | 1.91 | 9, 69 | ||
| 21 | 1.4 | SFDTTKIVTARTSTTRASSSMTT | 9.0 | 0.97 | 3, 24 | ||
| 22 | 1.2 | PAEMEQYPTEGGVEHGACENCQA | 9.5 | 0.83 | 3, 18 | ||
| 23 | 1.3 | AAGGSAMAGSQAAAAAASAAALG | 12.6 | 1.92 | 10, 75 | ||
| 24 | 1.2 | NNINILVVCNFLVCLEVNTNSIM | 12.7 | 0.68 | 6, 49 | ||
| α3 | 1.5 | 25 | 1.3 | FQEQNFSFDEAREMENAELNTHH | 9.3 | 0.50 | 3, 27 |
| 26 | 1.4 | AALVWNENIAAQVALINNVTVGI | 11.4 | 0.75 | 11, 82 | ||
| 27 | 1.4 | VVLSILLAVAVAAVALSAAVMLA | 19.4 | 1.38 | 9, 65 | ||
| 28 | 1.4 | YIIILFIVLIPLMLLFIFLTLVL | 21.3 | 1.35 | 4, 35 | ||
| 29 | 1.8 | TARMKQRRWRLEKRQPLKLSLIM | 8.3 | 0.62 | 3, 26 | ||
| 30 | 2.1 | AAAATAQEGMNLMLVVATAAASA | 5.4 | 0.54 | 3, 27 | ||
| 31 | 1.9 | LMFHVIWFLAIAAMMLFIALLLL | 24.5 | 0.96 | 9, 63 | ||
| 32 | 2.3 | VMCTALSTDKLRLRGLIFRVVSG | 12.0 | 0.46 | 4, 35 | ||
| 34 | 2.2 | RRAETEGGEEARRKEPHSVLHKE | 10.3 | 0.52 | 5, 40 | ||
| 35 | 1.9 | IIIVIVVVIIDVFIVVVIAVIVV | 21.3 | 1.90 | 3, 28 | ||
| β4 | 1.9 | 36 | 1.7 | MALIIIVILVYFLIIFLLYLVVI | 18.5 | 1.82 | 6, 48 |
| 37 | 1.7 | GAYLTVIILGVVTKVIMSAFTAT | 15.9 | 0.61 | 6, 49 | ||
| 38 | 1.7 | LDCETLHITFDSFFPSTFSDTHP | 10.3 | 0.40 | 5, 42 | ||
| 40 | 2.5 | MRRHKVVIRFVQLLVASPKVTGV | 14.3 | 0.45 | 4, 36 | ||
| β5 | 2.0 | 41 | 1.8 | PRTPYVMIFAFTFTPIWIAYVNM | 11.8 | 0.42 | 4, 37 |
| 42 | 1.9 | YFVVVEPEFIDYATVNFAILIAV | 17.3 | 0.89 | 4, 36 | ||
| 43 | 1.7 | KAPPPVVCPPARPPDIDPPQDAD | 3.3 | 0.54 | 4, 38 | ||
| 45 | 2.7 | PQVTFTCNRLAGIITITVPRVQT | 13.0 | 0.56 | 4, 36 | ||
| 46 | 2.1 | KQKSLKKKLAYDAAKVKAFLNYK | 12.0 | 0.70 | 3, 32 | ||
| 47 | 1.7 | LMVLVAIAVAMMAGVAIGFALIA | 12.8 | 1.13 | 9, 65 | ||
| β6 | 1.7 | 48 | 1.3 | IVVISLMLIFHVVVIIVIVVFEI | 18.5 | 1.50 | 4, 36 |
| 49 | 1.3 | IVYAIVLLLIYLIIMVFIVIFMI | 16.9 | 1.55 | 9, 65 | ||
| 50 | 1.6 | ATVIMVLFSACLILPSLVAQTVP | 17.0 | 0.98 | 8, 58 | ||
| 51 | 1.3 | GECEANCCHEPHEEVTEETLETV | 7.8 | 0.75 | 9, 67 | ||
| 53 | 2.8 | SGGGGGGGGIIGMIGGMHCGLGG | 8.9 | 1.00 | 15, 122 | ||
| T7 | 2.7 | 54 | 2.5 | RDQRTAKRQEAAYVLQEASTRLA | 0.0 | 0.40 | — |
| 55 | 2.7 | NSLVIVVVVLINHPMPVSFICLP | 1.0 | 0.61 | 6, 51 | ||
| 56 | 2.7 | LAIVQYWWLPDLPDAVHDDYVRA | 12.0 | 0.86 | 6, 50 | ||
| 58 | 1.8 | LIFIVLMIILFIILLVILVVVAI | 17.0 | 1.78 | 6, 50 | ||
| L8 | 2.4 | 59 | 2.5 | RQRCTAAKMKGARRAYVRGVSVY | 17.9 | 0.65 | 9, 64 |
| 60 | 3.0 | KQTTRRDDARASKEKEADPKKIA | 0.0 | 0.46 | — | ||
| 62 | 2.0 | AGAITFVLVMVAIAVIACIDACI | 13.2 | 1.09 | 6, 48 | ||
| 63 | 1.5 | YWINLYRMRLVLLLPFIAWYLKP | 24.5 | 1.43 | 3, 31 | ||
| 64 | 1.0 | LLVVVLVLVLVPLLLVIFLIVSV | 17.9 | 1.63 | 8, 59 | ||
| 65 | 0.8 | MMVHHVIWLIALIVLVMLHECII | 19.1 | 1.40 | 3, 27 | ||
| β9 | 1.5 | 66 | 0.9 | AVAIVSSSVLAIAFQMIVVFIYE | 13.0 | 0.93 | 10, 74 |
| 67 | 1.1 | DDDDDDDDDDDDDDDDDDDDDDD | 7.9 | 3.27 | 6, 51 | ||
| 68 | 1.4 | MDEGAEEEGEFLEEASNEASGMA | 13.4 | 0.85 | 10, 74 | ||
| 69 | 1.7 | AAAAVIIIAASAIVAATVAATVA | 10.4 | 1.43 | 10, 77 | ||
| 70 | 2.2 | HHYRQYHHQQSYAMHRWQYWFYH | 10.9 | 1.20 | 15, 122 | ||
| 71 | 2.3 | IGIITEMFSTTQTTAFATAVAYA | 11.3 | 0.80 | 9, 68 | ||
| 73 | 1.0 | VLASYLAIYIVLILIAVVSILIT | 18.3 | 1.41 | 3, 27 | ||
| 74 | 1.1 | VLIVATLTYLIILTALSMFVVLA | 15.0 | 1.37 | 11, 83 | ||
| β10 | 1.4 | 75 | 1.1 | TVLMSVLFATYACTTTITNVLLI | 12.8 | 0.93 | 11, 82 |
| 76 | 1.5 | TVRFFNTTFLAALLFMQFFQHFF | 13.3 | 0.56 | 1, 12 | ||
| 77 | 2.1 | TTTCSGSATSGSGGSSASNSSTS | 10.5 | 1.55 | 9, 69 | ||
| 79 | 1.5 | AGCGVITGVLLTLVGGITCTAFG | 14.2 | 0.70 | 12, 93 | ||
| 80 | 1.7 | GAGSGGGMGATGSGGGGGSSGGG | 6.7 | 1.90 | 10, 77 | ||
| β11 | 1.5 | 81 | 1.4 | MAFVIYASGAICAAALTGAQCWI | 17.5 | 1.95 | 1, 8 |
| 82 | 1.4 | IVTVTAYNLIVLTYVVAFMIIAV | 12.8 | 1.37 | 10, 75 | ||
| 83 | 1.5 | FLLVYCGIYVILLGVAVFWHSLV | 15.0 | 0.75 | 10, 74 | ||
| 84 | 1.5 | YCAGIGIIITVATGFICHVKGIT | 18.2 | 0.60 | 10, 73 | ||
| 86 | 1.9 | HPSANDSTADPPNNNADDRFNPP | 7.2 | 0.61 | 12, 89 | ||
| 87 | 1.6 | NPTGVTVTYPTFPPEGAPRYPRD | 8.0 | 0.40 | 12, 90 | ||
| 88 | 1.7 | HAPVPILLPLFHLLFRDSFPNEL | 9.2 | 0.59 | 12, 91 | ||
| 89 | 1.5 | AQVLGAAGQPAGAASDTKRMATN | 12.2 | 0.65 | 11, 79 | ||
| 90 | 1.5 | IAAAIQLYYAWACMAMANSYAQA | 19.0 | 0.93 | 1, 4 | ||
| α12 | 1.9 | 91 | 1.7 | AQDAVWTKAAYKATSAYLLAYLA | 9.6 | 0.92 | 12, 94 |
| 92 | 1.8 | GAIAGAAAGVLIAAVAILKALRI | 8.0 | 1.02 | 12, 89 | ||
| 93 | 1.7 | VLAGATHCLGSVAGLMTLMLISA | 14.2 | 0.67 | 1, 4 | ||
| 94 | 2.0 | ARAIALIELLGSNIRVSAWDILL | 14.7 | 0.52 | 1, 3 | ||
| 95 | 2.4 | VAQIREAIARLTAKAQREFVRKA | 7.6 | 0.74 | 12, 92 | ||
| 96 | 2.4 | ASAAAAACAVVVSTQGGVVNGVQ | 8.6 | 1.03 | 12, 93 | ||
| 97 | 2.5 | LLLLMLYYLLFLLLLLLIFAMLL | 24.5 | 1.70 | 1, 11 | ||
| 99 | 3.2 | VAEDEMAGEAEAQGEEVSAKQFA | 8.4 | 0.81 | 13, 102 | ||
| 100 | 2.6 | YRRRAALCRRMVQTQYRNYLQGL | 7.0 | 0.52 | 13, 103 | ||
| 101 | 2.2 | QRVLARRIYADRVYILLAIWQFN | 10.5 | 0.50 | 13, 104 | ||
| 102 | 2.6 | HEAKKEIKQNKSAEAHRARHNKA | 8.4 | 0.83 | 13, 99 | ||
| 103 | 2.5 | QKQEHAYVAVIGDYVYQHKDSKR | 7.0 | 0.72 | 13, 100 | ||
| 104 | 2.1 | VLIDMYLIIMNMILRRHAHLTLR | 10.5 | 0.60 | 13, 101 | ||
| 105 | 1.9 | VAIHERKDCGQLEDDIHGVLARA | 7.6 | 0.50 | 13, 108 | ||
| α13 | 1.2 | 106 | 2.3 | AAAEKRDKQDQRVQEGEKQILEA | 6.4 | 0.74 | 13, 109 |
| 107 | 2.2 | NLENLRNNRRKLQIRQSVLTRRI | 10.8 | 0.57 | 17, 136 | ||
| 108 | 1.7 | CIRARRLQSLARLTWVSLSTMWA | 14.7 | 0.70 | 17, 136 | ||
| 109 | 1.9 | RTERSDTREREERKTKLLHIAVA | 8.3 | 0.67 | 13, 112 | ||
| 110 | 2.2 | ARYFKLYIFRLQERLYKTEEENQ | 7.2 | 0.53 | 13, 107 | ||
| 111 | 1.9 | LFLLLLIVLGLLQLLLVGFAIIY | 18.4 | 1.04 | 17, 134 | ||
| 112 | 1.7 | SRIAVLAKWLYALSSGALERLTH | 15.2 | 0.63 | 14, 116 | ||
| 113 | 2.0 | EAALSEDDRLGGADRDELAKEAQ | 6.0 | 0.76 | 13, 110 | ||
| 115 | 1.8 | LIYMTYAQLLNFIMAALFLKAYL | 13.4 | 1.07 | 17, 133 | ||
| 116 | 1.8 | IQIVLVWWVLVSGFMVFCVFVAF | 15.2 | 1.15 | 13, 112 | ||
| β14 | 2.0 | 117 | 1.7 | LPLISLLISGPRVGFFSSCMSWI | 13.3 | 0.41 | 17, 133 |
| 118 | 1.8 | VLALVMDDFVFLVFRVFFFLFVI | 16.5 | 1.20 | 17, 134 | ||
| 119 | 2.0 | DIRRSDLFTEQGEFIDVDRTEKR | 6.1 | 0.52 | 15, 122 | ||
| 120 | 2.9 | LVFTFTRRVILATFPCLTLTVCV | 13.8 | 1.16 | 17, 132 | ||
| 122 | 2.9 | EAFIQAKEVTDKMHVILILATFM | 14.2 | 0.56 | 17, 132 | ||
| 123 | 2.8 | KEKENEAFQRKEARDNASENAQA | 6.6 | 0.60 | 15, 126 | ||
| α15 | 2.9 | 124 | 2.8 | VKSALRLMKELEAGRANIRYKNS | 0.0 | 0.57 | — |
| 125 | 2.9 | LLLLLLIILCFFLMLLYAILFGL | 17.1 | 1.55 | 16, 128 | ||
| 126 | 3.1 | ERWRSLEHEMLGQLVYLRNRVRK | 6.6 | 0.00 | 15, 123 | ||
| β16 | 2.0 | 128 | 2.0 | CVLAVVIFLMLYRLAAILLPDTT | 20.8 | 0.84 | 17, 134 |
| 129 | 2.0 | NTRNSVMDRNKMPAFVLGVEQQG | 9.0 | 0.41 | 17, 133 | ||
| 130 | 2.1 | KADATPPETIGVFPAEAGPKPNL | 11.9 | 0.62 | 17, 132 | ||
| 132 | 2.0 | LLLIIVVEIFMSLFVMIILIVVC | 14.9 | 1.75 | 16, 128 | ||
| 133 | 1.7 | RRRRRRRRRRRRIEWRRRRLRRS | 13.4 | 2.00 | 14, 115 | ||
| β17 | 1.9 | 134 | 1.8 | LLILILLIAIAIYALLLFFFFLL | 20.8 | 1.43 | 16, 128 |
| 135 | 1.8 | GTTVSSNNCASNLGHTHRALSSP | 11.4 | 0.76 | 14, 115 | ||
| 136 | 1.8 | TLVTLYALCPIIMFHIIPIMFLL | 14.7 | 0.95 | 13, 108 | ||
| 137 | 2.4 | PTGHCAGAHPYAPTRAGTCTGIF | 9.5 | 0.67 | 13, 104 |
a Each SCR is labeled according to the following scheme: (α) α-helix; (β) β-strand; (L) Loop; (T) turn.
Given the structural conservation of the residues involved in the SCRs and their possible relevance to the stability of type I superfamily of enzymes, an analysis was carried out to infer to what extent their properties, and consequently their functional role, was preserved during evolution. To get more general information on the conservation of physico-chemical properties of each position in the multiple-structure alignment, sequence homologs for each structure collected were retrieved and aligned. Several criteria were adopted to reduce the presence of any possible redundancy in the data set being analyzed (see Materials and Methods section); hits displaying a sequence identity >80% with any other protein were rejected, as well as distant homologs (<30% sequence identity) for which accuracies of the alignments to sequences of known structure cannot be assured.
The number of sequences retrieved for each structure is shown in Table 1. The multiple-structure alignment obtained from the superposition of the crystallographic structures was then used as a guide to merge the 23 multiple-sequence alignments comprising 921 nonredundant sequences. A total of 376,573 of the 422,740 pairwise sequence comparisons displayed a sequence identity in the interval 0%–20% (mean 16%, SD ±6%), which suggests that the data set can sample very distant evolutionary events. After obtaining the multiple-sequence alignment, a method for the identification of evolutionarily conserved residues was applied. Because in extensive tests of sequence alignments (Vogt et al. 1995) the BLOsum62, on average, gave superior results compared with most other matrices, it seemed appropriate to adopt this mutational matrix to assign a score for the amino acid exchanges. A weighting scheme based upon sequence similarity was also adopted, to incorporate in the algorithm corrections for sequence evolutionary distance and residue frequency (see Materials and Methods section). The results obtained for the SCRs, expressed in units of SD from the mean conservation value (R), are shown in Table 3. The structural role played by SCRs in maintaining the fold of this superfamily of enzymes is reflected by the high sequence conservation of the corresponding positions of the multiple alignment. Scores displayed by the SCRs are, in fact, all above the mean conservation value, with the only exception being site 14, which obtained a negative score. In particular, residues interacting with the PLP moiety are the most conserved; Asp 67, which is known to interact with the pyridinium nitrogen of PLP (Mehta and Christen 1998), was found in 919 of 921 sequences aligned (the only exceptions are 8-amino-7-oxononanoate synthase from Mesorhizobium loti and Cystathionine β-lyase from Bifidobacterium longum, GI 13475018 and GI 23336039, respectively [Holm and Sander 1998], in which Asp was replaced by Gly and Asn, respectively), scoring at a significance of 3.3 SDs from the mean conservation value; a comparable value (R = 3.2) was seen only by the Schiff base-forming lysine, which is placed in a variable loop between SCRs β10 and β11 (Christen and Mehta 2001). Taken together, these two residues represent the major signature of this superfamily of enzymes. Other sites involved in interactions with the cofactor or the substrates are strongly conserved, that is, position 70, interacting with the phenol oxygen of PLP (R = 1.2), the ring moiety stacking on the re side of PLP (data not shown; R = 1.6), the residue stacking on the si side (site 69, R = 1.4), the so-called glycine-rich region (positions 19, 20, and 21; R = 1.6, 1.9, 1.0, respectively), the 5′-phosphate-binding residue in position 77 (R = 1.5), and the Arg residue ion-paired with the α carboxyl group of many substrates bound to the fold-type I enzymes (site 133, R = 2.0).
In addition to these positions, other sites not directly involved in any interaction with the cofactor or the substrates show a high degree of sequence conservation, comparable to the conservation measured for functionally important residues (R ≥ 1.0; Table 3). These sites might be grouped in two distinct categories as follows: (1) Gly/Ala-rich sites; (2) positions mainly occupied by residues with a hydrophobic character (position 97, for example, scoring at a significance of 1.7 SDs from the mean conservation value, is almost invariantly occupied by a Leu or an aromatic residue in all of the 921 sequences considered, although it seems not to be implied in any functional role).
The positions mainly occupied by Gly or Ala residues (23, 80, and 92), that show a high degree of sequence conservation (1.9, 1.9, and 1.0, respectively), might play important functions other than binding the PLP moiety or being involved in hydrophobic contacts. For example, two Ala rich sites (23 and 92) are found in the middle of an α-helix spine; it was observed that Ala show the strongest preference over any other residue for a middle-helix location (Richardson and Richardson 1989). This, in turn, is due to the structurally unique features shown by Ala, which direct and stabilize the α-helix fold (Blaber et al. 1993). The other Gly-rich site (80) was found in β11, where it could be helpful in modulating the curvature of the sheet (Richardson and Richardson 1989).
To test whether the conservation of the physicochemical properties of the second group of positions was driven by selective pressure to maintain the stability of fold-type I, PLP-dependent enzymes through the involvement of the corresponding residues in hydrophobic interactions, an analysis of the conserved hydrophobic contacts (CHCs) was performed on the SCRs previously identified. Previous comparative studies that have been focused on the relationship between sequence conservation of a protein family and the hydrophobic contacts of the corresponding structures available (see, for example, Ptitsyn 1998; Hill et al. 2002; Gromiha et al. 2004; Gunasekaran et al. 2004) have considered two residues to be in contact if the distance between their Cα atoms or between one atom and any other atom was below an arbitrary threshold. In this work, a different criterion was adopted, which is based on the comparative analysis of the pairwise residue apolar contact areas for every possible pair of residues belonging to the SCRs. CHCs are, therefore, defined as residue hydrophobic contacts involving only apolar atoms (Drabløs 1999), observed in at least two of the structures analyzed. This approach permitted us to quantify the strength of a hydrophobic contact and to assess the correlation between this quantity and the evolutionary conservation of the corresponding sites. The strongest CHCs for each site belonging to the SCRs and the corresponding site involved in the hydrophobic interaction are shown in Table 3.
Figure 3 ▶ shows the mean conservation values between pairs of sites involved in CHCs in comparison with their mean hydrophobic contact values. Residues interacting with the cofactor PLP as well as the Ala/Gly-rich sites described above were not plotted, as their high evolutionary conservation reflects functions other than the stabilization of this superfamily fold through the involvement in hydrophobic contacts. A significant linear coefficient (r = 0.70) resulted between the two variables. The statistical significance of r was assessed with the t-test, assuming r = 0 as the null hypothesis. This gave a P-value ≅1.7e-53, indicating that there is a statistically significant relationship between the strength of a CHC and the extent of conservation of the involved residues during evolution. At values >16 Å2, the mean conservation grade becomes comparable to the values measured for catalytically important residues (R ≥ 1.0). CHCs with the highest values of mean apolar contact area (Table 4) may be grouped in three main clusters (Fig. 2B ▶); a first cluster of CHCs is located at the buried bottom region of the PLP-binding, conserved common core of the major domain constituted by the six SCRs (α3 β6, L8, β9, β10, and β11); a second small cluster of interacting residues is centered around position 133 of the minor domain (α13, β14, α15, β16, and β17); a third cluster of CHCs forms a hinge between SCRs α1 and α12, which are positioned at the beginning and at the end of the major domain, respectively (Fig. 2B ▶). Amino acids belonging to the first cluster of CHCs occur at positions 27, 28, and 31 in α3; 35 and 36 in β4; 42 in β5; 48, 49, and 50 in β6; 58 and 59 in L8; 63, 64, and 65 in β9; 73 in β10; 81 and 84 in β11 (Fig. 2B ▶; Table 4). The five residues participating in the formation of the second cluster (111 in α13, 118 in β14, 125 in α15, 128 in β16, and 134 in β17), are located in proximity of position 133 of β17, which is occupied mainly by an Arg residue (18 of 23 structures analyzed); the α carboxyl group of many substrates bound to the fold-type I enzymes is often ion-paired to this arginine (Jansonius 1998). Residues forming the third cluster of CHCs are involved in interhelical contacts in 22 of the 23 structures considered (the only exception is represented by 1BJW, in which only two CHCs involving site 8 are conserved; Table 4). These residues (positions 4, 8, and 11 of SCR α1; 90 and 97 of SCR α12) form a vertical strip down each side of the helices that delimit the major domain, lying at sites i, i + 4, and i + 7. Site 97, which was described above (R = 1.7), is engaged in the constitution of the two most extensive CHCs measured for the α1–α12 hinge (11–97 and 8–97 [Fig. 2B ▶]; the mean apolar contact areas are, according to Table 3, 24.6 Å2 and 17.6 Å2, respectively), and in an additional conserved contact with position 7 (15.3 Å2).
Figure 3.

Mean conservation values between pairs of sites involved in CHCs in comparison with their mean hydrophobic contact values. The correlation coefficient between the mean conservation grade and the CHC value, expressed in square angstroms, is 0.70. The statistically significant relationship between the strength of a CHC and the extent of conservation of the involved residues is supported by a P-value <0.0001.
Table 4.
Sites involved in the strongest conserved hydrophobic contacts (CHCs with a mean hydrophobic contact > 16.0 Å2)
| Hydrophobic contacts (Å2) | ||||||||||||||||||
| I SCR, site interacts with II SCR, site | 1, 4 | 1, 8 | 1, 8 | 1, 11 | 3, 27 | 3, 27 | 3, 28 | 3, 31 | 4, 36 | 4, 36 | 6, 49 | 6, 50 | 8, 59 | 10, 73 | 13, 111 | 15, 125 | 16, 128 | |
| 12, 90 | 11, 81 | 12, 97 | 12, 97 | 9, 65 | 10, 73 | 4, 35 | 9, 63 | 6, 48 | 5, 42 | 9, 65 | 8, 58 | 9, 64 | 11, 84 | 17, 134 | 16, 128 | 17, 132 | ||
| Molecule (PDB code) | 1BJ4 | 18.5 | 17.7 | 17.5 | 16.3 | 17.9 | 19.7 | 26.0 | 32.1 | 17.2 | 20.2 | 20.2 | 23.0 | 29.9 | 22.4 | 25.9 | 20.0 | 23.6 |
| 1BS0 | 18.2 | 22.4 | 18.5 | 32.7 | 22.5 | 24.5 | 27.1 | 29.1 | 15.7 | 9.9 | 24.2 | 17.8 | 16.7 | 18.2 | 21.0 | 27.6 | 27.6 | |
| 1FG3 | 17.4 | 43.0 | 26.0 | 15.7 | 22.2 | 15.3 | 29.7 | 34.4 | 21.5 | 23.9 | 18.2 | 27.0 | — | 19.7 | 24.6 | 30.8 | 39.1 | |
| 1JG8 | 19.0 | 18.7 | 11.8 | 16.0 | 12.9 | 2.8 | 21.7 | 5.7 | 31.1 | 22.6 | 5.5 | 37.4 | 21.0 | 12.4 | 32.1 | 11.9 | 20.3 | |
| 1ECX | 17.3 | 28.8 | 22.8 | 23.4 | 16.7 | 34.5 | 10.6 | 16.5 | — | 23.6 | — | — | 20.9 | 11.9 | 29.4 | 26.6 | 22.2 | |
| 1BJW | — | 12.2 | 20.0 | — | 24.3 | 41.5 | 18.5 | 27.3 | 22.7 | — | 17.2 | 25.8 | 24.2 | 15.9 | 27.4 | 27.5 | 25.2 | |
| 1D2F | 38.5 | — | 17.8 | 40.0 | 23.0 | 20.0 | 29.3 | 27.4 | 19.6 | — | 20.7 | 15.7 | 7.4 | — | 25.7 | — | 32.8 | |
| 1C7N | 32.6 | — | 18.1 | 44.5 | 20.3 | 13.2 | 16.7 | 32.6 | 20.5 | — | 28.0 | 26.6 | — | — | 23.4 | — | 29.5 | |
| 1ELQ | 28.7 | 3.2 | 21.9 | 8.5 | 23.3 | 39.5 | 17.9 | 3.6 | — | 20.9 | — | — | 31.0 | 34.6 | 19.1 | 28.8 | 25.0 | |
| 1DGD | 18.2 | 19.4 | 16.4 | 29.3 | 17.7 | 19.3 | 32.5 | 26.7 | 22.8 | 19.5 | 27.2 | 12.2 | 37.9 | 10.6 | — | 30.0 | 27.7 | |
| 1BJN | 10.8 | 31.5 | 31.0 | 27.8 | 13.8 | 17.4 | — | 17.3 | 19.4 | — | — | — | — | 24.9 | 15.6 | 23.3 | 21.5 | |
| 1AY4 | 21.7 | 22.5 | 8.2 | 31.3 | 22.4 | 12.0 | 19.0 | 15.4 | 22.6 | 18.7 | 23.1 | 23.6 | — | 18.4 | 19.2 | — | — | |
| 1DTY | 26.8 | 20.8 | 16.7 | 33.4 | 19.0 | 13.7 | 42.0 | 17.8 | 22.4 | 13.4 | 29.3 | 23.8 | 6.9 | 10.1 | — | — | — | |
| 2GSA | 29.7 | 2.8 | 14.7 | 10.6 | 19.7 | 26.7 | 23.9 | 20.7 | 24.0 | 20.0 | 20.3 | 24.3 | 18.5 | 10.5 | — | 31.4 | — | |
| 1B9H | 14.3 | 17.3 | 20.3 | 13.2 | 19.1 | 13.0 | 28.1 | 20.8 | 34.0 | 29.3 | 17.5 | 24.0 | 14.8 | 33.2 | 20.4 | 18.4 | 21.9 | |
| 1AX4 | 26.6 | 12.9 | 2.2 | 23.0 | 18.1 | 12.4 | — | 37.3 | — | — | 15.3 | — | 32.5 | 21.4 | 26.2 | 9.5 | 19.2 | |
| 1CL1 | 19.4 | 17.5 | 17.4 | 33.7 | 12.2 | 6.6 | 20.9 | 37.5 | 21.5 | 24.2 | 19.0 | 31.5 | 32.8 | 27.7 | 14.7 | 11.4 | 25.0 | |
| 1GTX | 1.8 | — | 3.3 | 30.9 | 19.9 | 14.2 | 32.5 | 20.9 | 28.1 | 17.7 | 25.2 | 15.0 | 31.0 | 22.8 | — | 26.2 | 31.3 | |
| 1JS6 | 11.8 | 14.6 | 22.8 | 39.9 | 15.6 | 2.8 | 13.7 | 31.4 | 21.7 | 31.8 | 18.5 | 17.9 | 21.8 | 20.0 | 52.0 | 28.2 | 27.1 | |
| 1ORD | 12.4 | 0.8 | 13.8 | 12.1 | 14.9 | 25.2 | 0.4 | 37.0 | — | 34.4 | — | — | 22.1 | 18.6 | — | 23.7 | 22.0 | |
| 1QGN | 18.6 | 27.4 | 23.8 | 25.4 | 20.0 | 12.2 | 25.0 | 22.0 | 19.9 | 22.1 | 18.0 | 0.2 | 14.2 | 14.0 | — | — | — | |
| 1LK9 | 16.2 | 40.1 | 15.3 | 28.8 | 23.0 | 29.9 | 33.0 | 21.9 | 12.2 | 16.0 | 28.6 | 15.4 | — | 23.5 | 22.9 | — | 21.4 | |
| 1MDX | 18.9 | 29.1 | 24.5 | 28.3 | 21.0 | 5.2 | 20.6 | 27.6 | 27.7 | 29.2 | 12.2 | 29.0 | 28.6 | 12.9 | 24.3 | 18.7 | 16.0 | |
| Mean | 19.0 | 17.5 | 17.6 | 24.6 | 19.1 | 18.3 | 21.2 | 24.5 | 18.4 | 17.3 | 16.9 | 17.0 | 17.9 | 18.2 | 18.4 | 17.1 | 20.8 | |
The full list of CHCs values is available upon request to the authors.
Discussion
The present work was aimed at the detection of structural features remaining invariant over long evolutionary periods in the fold-type I PLP-dependent enzymes. This protein superfamily is particularly suited for such an analysis, because its members are related by a long divergent evolution. It was proposed that these enzymes were already present in the universal ancestor cell some 1500 millions years ago (Mehta and Christen 1998). Whereas the structural homology among the different members is still recognizable, the extent of sequence similarity is not sufficient to establish a common ancestry. For these reasons, this superfamily can be considered, per se, a model of protein evolutionary structural plasticity. The structural similarity among members of this superfamily of enzymes is distributed accordingly to the locally different functions accomplished by the motifs of secondary structure. Approximately 30% of the residues form a well-conserved, structural common core of secondary elements. This observed structural conservation is probably due to the spatial restraints imposed by the similar binding mode of the cofactor PLP, which is accommodated inside of a hydrophobic cleft at the interface between two subunits. Apart from this common core, secondary structure lengths and loops in these distantly related structures vary substantially, which results are also evident in the large multiple alignment of 921 sequences, where indels-free conserved blocks are sparse. Large structural adaptations have probably taken place during the divergent evolution of this superfamily from a common ancestor for the adjustment of a catalytic apparatus required to change reaction and substrate specificity. Likely, the loops surrounding the active site entrance were mainly affected by these structural changes (Contestabile et al. 2001).
Our work focused on the conservation of hydrophobic contacts between the structurally conserved regions resulting from the comparison of 23 distantly related type I PLP-dependent enzymes. The conservation of hydrophobic contacts is the result of the selective pressure exerted during the molecular evolution to maintain a functionally competent fold. We identified three clusters of conserved hydrophobic contacts; the first and the second clusters of CHCs (Fig. 2B ▶) are located in proximity of key residues responsible for the proper positioning of the cofactor and the substrate in the active site. Regarding the first cluster, the separation of residues involved in a functional role (interaction with the PLP moiety and modulation of its activity), all located at the top of the conserved core region (constituted by SCRs α3, β6, β9, β10, and β11) (Fig. 2A,B ▶) and residues involved in a structural role (maintenance of structural stability throughout CHCs), positioned, instead, at the inner bottom core of the same functional unit, is remarkable. This functional and spatial arrangement, comprising a stable scaffold folded around a mutable functional core of residues, can be found in many other evolutionarily successful (Nagano et al. 2002; Selvaraj and Gromiha 2003) structural units exploited by nature during the course of evolution (i.e., TIM-barrel and Ig-like domains), and it seems to provide a suitable way to solve the compromise between three-dimensional stability and plasticity of function, broadening substrate, and reaction specificity, without affecting protein fold and conformation (Todd et al. 2001; Wierenga 2001; Nagano et al. 2002).
In apparent contrast to the two previously described clusters, the helix α1–helix α12 cluster of CHCs (Fig. 2B ▶) seems not to be involved in the proper positioning or stability of any active site residue. Examination of the contact network showed that the CHCs lie along one side of each helix, forming a buried spine at positions i, i + 4, and i + 7. This particular pattern of almost absolutely conserved residue–residue contacts was previously identified by Hill et al. (2002) and Ptitsyn (1998) in the case of the cytokines and c-type cytochromes superfamily of proteins, respectively. In both studies, it was concluded that these residues were of critical importance for protein folding. In the case of PLP-dependent enzymes, previous experimental studies have suggested the presence of three structural nuclei responsible for the proper fold-type I enzymes folding pattern and stability. Herold et al. (1991) demonstrated that the excised PLP-binding domain of aspartate aminotransferase from Escherichia coli, corresponding to the first and third domain in which CHCs are located, is able to fold autonomously both in vivo and in vitro and bind PLP. More recently, Fu et al. (2003) proposed that the folding mechanism of serine hydroxymethyltransferase from E. coli can be divided into two phases, a first fast phase in which two domains, corresponding to the first and second domains in which CHCs are located, have folded into their native state, and a slow final phase in which an interdomain segment, comprising the helix α12, folds into its native conformation, interacting with the N-terminal α1 helix of the major domain. This last step is thought to be involved in PLP binding. The present analysis supports this hypothesis and suggests a possible mechanistic explanation for these experimental studies, serving as a basis for further experiments to establish sequence-structure correlation, and to investigate the role of individual residues and pairwise interactions in the folding and stability of this superfamily of proteins.
A main goal of this work was to determine whether the common structural constraints found for the packing of interacting residues within the protein core of the type-I PLP enzymes was reflected by a sequence conservation pattern observed for the hydrophobic positions in the multiple alignment of the fold-type I superfamily. A plot of the mean conservation grade of two interacting sites of the SCRs against the extent of mean hydrophobic contact value of their apolar fraction can be fit by a linear relationship (r = 0.70). In the present analysis, the mean amino acid pairwise conservation was considered in addition to single-site, positional conservation. A significant advantage of considering pairwise conservation is that it allows one to take into account compensating mutations that may occur in the amino acid sequence during evolution. It should be noted that conserved positions are not invariant; on the contrary, correlated mutations can be detected by comparing different structures. Therefore, it seems that what is really conserved is the three-dimensional location of the hydrophobic interaction and its hydrophobic effect, rather than the specific identity of the side chains participating in a CHC. Although the 23 PLP enzymes taken into consideration are very distantly related, they contain a structural pattern of conserved hydrophobic contacts, whose potential importance in stabilizing the native fold is supported by a preferential conservation throughout the homologous sequences.
Finally, we suggest that the significant correlation between sequence conservation and CHC values and the strategy and the algorithms described to determine it, could be extended to other superfamilies for which suitable sequence and structural information is known, to properly train statistical predictors of protein contact maps (Fariselli and Casadio 2000; Pollastri et al. 2001) and to help in planning protein folding and design experiments.
Materials and methods
Structural alignments
An initial search for nonredundant, representative members of each fold-type I family whose three-dimensional structure had been previously solved was carried out. Using the classification of these families in several structural databases (SCOP [Murzin et al. 1995], CATH [Orengo et al. 2003], and NCBI’s MMDB [Chen et al. 2003]), we retrieved an exhaustive set of crystallographic structures, from which we selected 27 representative members, on the basis of a hierarchical set of criteria; initially, engineered enzymes bearing residue mutations were discharged; then, in the presence of orthologous enzymes, the one with the highest resolution was chosen; finally, at comparable resolution values, the highest R-factor was also taken into consideration. All structures were retrieved by the Protein Data Bank (PDB; Berman et al. 2000).
An initial multiple alignment was obtained automatically by using the combinatorial extension algorithm, implemented in the program CE (Shindyalov and Bourne 1998). The resulting alignment was utilized as a starting point to build a manually refined structural alignment. Every possible pair of structures was visually inspected and, where necessary, modified to optimize the matching of several structural features, including observed secondary elements, functionally conserved residues known to interact with the PLP moiety and hydrophobic regions, in order to give the most accurate structural alignment. In a few cases of ambiguity, that is, some insertions or deletions in which visual inspection could not discern the optimal matching between two regions, the residue similarity measured by the BLOsum62 (Henikoff and Henikoff 1992) mutational matrix was adopted as a guide criterion. At the end of the manual refinement, structures displaying >30% sequence identity were discharged, leading to a nonredundant ensemble of 23 representatives of fold-type I enzymes with a maximum pairwise sequence identity of 27% and a maximum pairwise RMSD of 4.2 Å.
Identification of the structurally conserved regions
The structural alignment obtained as described above was utilized to identify the common core and the structurally conserved regions between members of this superfamily (SCRs). SCRs were defined as regions displaying similar local conformation, with a mean positional RMSD of the equivalent α-carbon positions of every structure superposed ≤ 3.0 Å (Hill et al. 2002), lacking indels (insertions and deletions) in all of the structures considered and composed of at least three consecutive residues. A C-language routine was developed to extract from the three-dimensional coordinates of the superimposed structures and their associated multiple alignment the candidate SCRs. For every structurally equivalent position of the multiple structural alignment, the RMSD from the center of mass of the structurally equivalent Cα atoms was computed. To avoid the presence of SCRs with indels, positions with gaps were not considered. A window of size w = 3 positions was then scrolled through the alignment and used to define seed positions with a mean RMSD ≤ 3.0 Å. Each time a seed position was found, w was increased iteratively by one position until the mean score did not raise above 3.0 Å, or until the window reached the end of the alignment.
Identification of the conserved hydrophobic contacts
Computation of conserved hydrophobic contacts (CHCs) performed on the crystallographic structures retrieved is based on the program pdb_np_cont (Drabløs 1999), which computes pairwise atom contact areas between nonpolar atoms from structural protein data in a standard PDB coordinate file. The output of this program was utilized to calculate the pairwise residue contact areas for every possible pair of residues belonging to the SCRs of the structures analyzed. If two positions of the joint multiple structural alignment, x and y, have residues in hydrophobic contact in at least two of the structures, then a candidate CHC was detected. CHCs were then classified on the basis of their strength sxy, defined as:
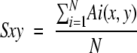 |
(1) |
where Ai is the apolar contact area of the i-th structure between residues at absolute positions x and y of the structural alignment, and N is the number of superposed structures.
Collection and alignment of sequence homologs
Sequence search was performed against the nonredundant database NRDB (Holm and Sander 1998) with the program BLAST (Altschul et al. 1997), using each of the sequences of the 23 superposed structures as probes. When applicable, the following criteria were adopted to collect or discharge each sequence: (1) Hits were considered to be significant if the E-value was ≤0.0001—if less than 10 sequences were collected, this value was increased to ≤0.001; (2) hits were filtered to assure that no sequence with identity >80% or <30% with any other sequence of the multiple alignment was present in the final alignment; (3) hits with sequence length <80% of the query sequence were rejected to avoid the presence of fragmented sequences in the final alignment.
Sequences filtered were aligned to each corresponding query sequence using the program CLUSTALW (Thompson et al. 1994). The 23 multiple alignments were then merged using as a guide the structural alignment of the 23 PLP enzyme sequences (Pascarella and Argos 1992). The final alignment, comprising 973 sequences, was further checked for redundancy. At the end of this final step, a total number of 921 sequences was obtained.
Identification of the evolutionarily conserved positions
To measure the sequence conservation, each position of the final multiple sequence alignment was assigned a score according to:
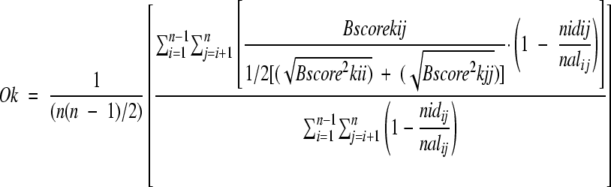 |
(2) |
where Ok is the score assigned for every position k of the multiple sequence alignment, n is the number of sequences included in the alignment, i and j refers to the i-th and the j-th sequence, respectively, Bscorekij, Bscorekii, and Bscorekjj are the scores assigned to the residue exchange in position k between the i-th and the j-th sequence according to the BLOsum62 mutational matrix, nidij is the number of identical residues, and nalij is the number of aligned residues between the i-th and the j-th sequence, respectively. Therefore, for every possible exchange at a particular position of the multiple alignment, a normalized conservation index is computed, based on the BLOsum62 mutational matrix. Because the BLOsum62 matrix scores for matching the same amino acids vary for different residues, conservation indices for invariant positions of the multiple-sequence alignment would depend on residue type; normalization is used to avoid different conservation scores for invariant positions. The mean Ō and the standard deviation (SD) σ for the distribution of Ok values were determined; the significance R of every conservation index of the alignment was then calculated by dividing the difference between Ok and Ō by σ.
Acknowledgments
Acknowledgments
This work was supported in part by the Italian “Ministero dell’ Università e della Ricerca” (MIUR). This work will be submitted by A.P. in partial fulfillment of the requirements of the degree of Dottorato di Ricerca at the Università di Roma “La Sapienza.” Structural and sequence alignments and the source code of the software developed for the analysis are available on request from the authors.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.04938104.
References
- Alexeev, D., Alexeeva, M., Baxter, R.L., Campopiano, D.J., Webster, S.P., and Sawyer, L. 1998. The crystal structure of 8-amino-7-oxononanoate synthase: A bacterial PLP-dependent, acyl-CoA-condensing enzyme. J. Mol. Biol. 284 401–419. [DOI] [PubMed] [Google Scholar]
- Altschul, S.F., Madden, T.L., Schaffer, A.A., Zhang, J., Zhang, Z., Miller, W., and Lipman, D.J. 1997. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 25 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman, H.M., Westbrook, J., Feng, Z., Gilliland, G., Bhat, T.N., Weissig, H., Shindyalov, I.N., and Bourne P.E. 2000. The Protein Data Bank. Nucleic Acids Res. 28 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaber, M., Zhang, X.J., and Matthews, B.W. 1993. Structural basis of amino acid α helix propensity. Science 260 1637–1640. [DOI] [PubMed] [Google Scholar]
- Burkhard, P., Dominici, P., Borri-Voltattorni, C., Jansonius, J.N., and Malashkevich, V.N. 2001. Structural insight into Parkinson’s disease treatment gained from drug-inhibited dopa decarboxylase. Nat. Struct. Biol. 8 963–967. [DOI] [PubMed] [Google Scholar]
- Chen, J., Anderson, J.B., DeWeese-Scott, C., Fedorova, N.D., Geer, L.Y., He, S., Hurwitz, D.I., Jackson, J.D., Jacobs, A.R., Lanczycki, C.J., et al. 2003. MMDB: Entrez’s 3D-structure database. Nucleic Acids Res. 31 474–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chothia, C. and Lesk, A. 1986. The relation between the divergence of sequence and structure in proteins. EMBO J. 5 823–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christen, P. and Mehta, P.K. 2001. From cofactor to enzymes. The molecular evolution of pyridoxal-5′-phosphate-dependent enzymes. Chem. Rec. 1 436–447. [DOI] [PubMed] [Google Scholar]
- Clausen, T., Huber, R., Laber, B., Pohlenz, H.D., and Messerschmidt, A. 1996. Crystal structure of the pyridoxal-5′-phosphate dependent cystathionine β-lyase from Escherichia coli at 1.83 Å. J. Mol. Biol. 262 202–224. [DOI] [PubMed] [Google Scholar]
- Clausen, T., Schlegel, A., Peist, R., Schneider, E., Steegborn, C., Chang, Y.S., Haase, A., Bourenkov, G.P., Bartunik, H.D., and Boos, W. 2000a. X-ray structure of MalY from Escherichia coli: A pyridoxal 5′-phosphate-dependent enzyme acting as a modulator in mal gene expression. EMBO J. 19 831–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clausen, T., Kaiser, J.T., Steegborn, C., Huber, R., and Kessler, D. 2000b. Crystal structure of the cystine C-S lyase from Synechocystis: Stabilization of cysteine persulfide for FeS cluster biosynthesis. Proc. Natl. Acad. Sci. 97 3856–3861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Contestabile, R., Paiardini, A., Pascarella, S., di Salvo, M.L., D’Aguanno, S., and Bossa, F. 2001. 1-Threonine aldolase, serine hydroxymethyltransferase and fungal alanine racemase. A subgroup of strictly related enzymes specialized for different functions. Eur. J. Biochem. 268 6508–6525. [DOI] [PubMed] [Google Scholar]
- Drabløs, F. 1999. Clustering of non-polar contacts in proteins. Bioinformatics 15 501–509. [DOI] [PubMed] [Google Scholar]
- Eads, J.C., Beeby, M., Scapin, G., Yu, T.W., and Floss, H.G. 1997. The crystal structure of 3-amino-5-hydroxybenzoic acid Ahba synthase. Biochemistry 38 9840–9849. [DOI] [PubMed] [Google Scholar]
- Fariselli, P. and Casadio, R. 2000. Prediction of the number of residue contacts in proteins. Proc. Int. Conf. Intell. Syst. Mol. Biol. 8 146–151. [PubMed] [Google Scholar]
- Fu, T.F., Boja, E.S., Safo, M.K., and Schirch, V. 2003. Role of proline residues in the folding of serine hydroxymethyltransferase. J. Biol. Chem. 278 31088–31094. [DOI] [PubMed] [Google Scholar]
- Grishin, N.V., Phillips, M.A., and Goldsmith, E.J. 1995. Modeling of the spatial structure of eukaryotic ornithine decarboxylases. Protein Sci. 4 1291–1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gromiha, M.M., Pujadas, G., Magyar, C., Selvaraj, S., and Simon, I. 2004. Locating the stabilizing residues in (α/β)8 barrel proteins based on hydrophobicity, long-range interactions, and sequence conservation. Proteins 55 316–329. [DOI] [PubMed] [Google Scholar]
- Gunasekaran, K., Hagler, A.T., and Gierasch, L.M. 2004. Sequence and structural analysis of cellular retinoic acid-binding proteins reveals a network of conserved hydrophobic interactions. Proteins 54 179–194. [DOI] [PubMed] [Google Scholar]
- Henikoff, S. and Henikoff, J.G. 1992. Amino acid substitution matrices from protein blocks. Proc. Natl. Acad. Sci. 89 10915–10919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hennig, M., Grimm, B., Contestabile, R., John, R.A., and Jansonius, J.N. 1997. Crystal structure of glutamate-1-semialdehyde aminomutase: An α2-dimeric vitamin B6-dependent enzyme with asymmetry in structure and active site reactivity. Proc. Natl. Acad. Sci. 94 4866–4871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herold, M., Leistler, B., Hage, A., Luger, K., and Kirschner, K. 1991. Autonomous folding and coenzyme binding of the excised pyridoxal 5′-phosphate binding domain of aspartate aminotransferase from Escherichia coli. Biochemistry 30 3612–3620. [DOI] [PubMed] [Google Scholar]
- Hester, G., Stark, W., Moser, M., Kallen, J., Markovic-Housley, Z., and Jansonius, J.N. 1999. Crystal structure of phosphoserine aminotransferase from Escherichia coli at 2.3 Å resolution: Comparison of the unligated enzyme and a complex with α-methyl-l-glutamate. J. Mol. Biol. 286 829–850. [DOI] [PubMed] [Google Scholar]
- Hill, E.E., Morea, V., and Chothia, C. 2002. Sequence conservation in families whose members have little or no sequence similarity: The four-helical cytokines and cytochromes. J. Mol. Biol. 322 205–233. [DOI] [PubMed] [Google Scholar]
- Hohenester, E., Keller, J.W., and Jansonius, J.N. 1994. An alkali metal ion size-dependent switch in the active site structure of dialkylglycine decarboxylase. Biochemistry 33 13561–13570. [DOI] [PubMed] [Google Scholar]
- Holm, L. and Sander, C. 1998. Removing near-neighbour redundancy from large protein sequence collections. Bioinformatics 14 423–429. [DOI] [PubMed] [Google Scholar]
- Isupov, M.N., Antson, A.A., Dodson, E.J., Dodson, G.G., Dementieva, I.S., Zakomirdina, L.N., Wilson, K.S., Dauter, Z., Lebedev, A.A., and Harutyunyan, E.H. 1998. Crystal structure of tryptophanase. J. Mol. Biol. 276 603–623. [DOI] [PubMed] [Google Scholar]
- Jansonius, J. 1998. Structure, evolution and action of vitamin B6-dependent enzymes. Curr. Opin. Struct. Biol. 8 759–769. [DOI] [PubMed] [Google Scholar]
- John, R.A. 1995. Pyridoxal phosphate-dependent enzymes. Biochim. Biophys. Acta. 1248 81–96. [DOI] [PubMed] [Google Scholar]
- Kaiser, J.T., Clausen, T., Bourenkow, G.P., Bartunik, H.D., Steinbacher, S., and Huber, R. 2000. Crystal structure of a NifS-like protein from Thermotoga maritima: Implications for iron sulphur cluster assembly. J. Mol. Biol. 297 451–464. [DOI] [PubMed] [Google Scholar]
- Kielkopf, C.L. and Burley, S.K. 2002. X-ray structures of threonine aldolase complexes: Structural basis of substrate recognition. Biochemistry 41 11711–11720. [DOI] [PubMed] [Google Scholar]
- Krupka, H.I., Huber, R., Holt, S.C., and Clausen, T. 2000. Crystal structure of cystalysin from Treponema denticola: A pyridoxal 5′-phosphate-dependent protein acting as a haemolytic enzyme. EMBO J. 19 3168–3178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuettner, E.B., Hilgenfeld, R., and Weiss, M.S. 2002. The active principle of garlic at atomic resolution. J. Biol. Chem. 277 46402–46407. [DOI] [PubMed] [Google Scholar]
- Lesk, A. and Chothia, C. 1980. How different amino acid sequences determine similar protein structures: The structure and evolutionary dynamics of the globins. J. Mol. Biol. 136 225–270. [DOI] [PubMed] [Google Scholar]
- Mehta, P.K. and Christen, P. 1998. The molecular evolution of Pyridoxal-5′-phosphate-dependent enzymes. In Advances in enzymology and related areas of molecular biology: Mechanism of enzyme action, Part B (ed. D.L. Purich), pp. 129–184. John Wiley & Sons, Inc., New York. [DOI] [PubMed]
- Michnick, S.W. and Shakhnovich, E. 1998. A strategy for detecting the conservation of folding-nucleus residues in protein superfamilies. Fold. Des. 3 239–251. [DOI] [PubMed] [Google Scholar]
- Momany, C., Ernst, S., Ghosh, R., Chang, N.L., and Hackert, M.L. 1995. Crystallographic structure of a PLP-dependent ornithine decarboxylase from Lactobacillus 30a to 3.0 Å resolution. J. Mol. Biol. 252 643–655. [DOI] [PubMed] [Google Scholar]
- Murzin, A.G., Brenner, S.E., Hubbard, T., and Chothia, C. 1995. SCOP: A structural classification of proteins database for the investigation of sequences and structures. J. Mol. Biol. 247 536–540. [DOI] [PubMed] [Google Scholar]
- Nagano, N., Orengo, C.A., and Thornton, J.M. 2002. One fold with many functions: The evolutionary relationships between TIM barrel families based on their sequences, structures and functions. J. Mol. Biol. 321 741–765. [DOI] [PubMed] [Google Scholar]
- Nakai, T., Okada, K., Akutsu, S., Miyahara, I., Kawaguchi, S., Kato, R., Kuramitsu, S., and Hirotsu, K. 1999. Structure of Thermus thermophilus HB8 aspartate aminotransferase and its complex with maleate. Biochemistry 38 2413–2424. [DOI] [PubMed] [Google Scholar]
- Noland, B.W., Newman, J.M., Hendle, J., Badger, J., Christopher, J.A., Tresser, J., Buchanan, M.D., Wright, T.A., Rutter, M.E., Sanderson, W.E., et al. 2002. Structural studies of Salmonella typhimurium Arnb Pmrh aminotransferase: A 4-amino-4-deoxy-L-arabinose liposaccharide modifying enzyme. Structure 10 1569–1580. [DOI] [PubMed] [Google Scholar]
- Okamoto, A., Nakai, Y., Hayashi, H., Hirotsu, K., and Kagamiyama, H. 1998. Crystal structures of Paracoccus denitrificans aromatic amino acid amino-transferase: A substrate recognition site constructed by rearrangement of hydrogen bond network. J. Mol. Biol. 280 443–461. [DOI] [PubMed] [Google Scholar]
- Orengo, C.A., Pearl, F.M., and Thornton, J.M. 2003. The CATH domain structure database. Meth. Biochem. Anal. 44 249–271. [DOI] [PubMed] [Google Scholar]
- Pascarella, S. and Argos, P. 1992. A data bank merging related protein structures and sequences. Protein Eng. 5 121–137. [DOI] [PubMed] [Google Scholar]
- Pollastri, G., Baldi, P., Fariselli, P., and Casadio, R. 2001. Improved prediction of the number of residue contacts in proteins by recurrent neural networks. Bioinformatics Suppl. 1 234–242. [DOI] [PubMed]
- Ptitsyn, O.B. 1998. Protein folding and protein evolution: Common folding nucleus in different subfamilies of c-type cytochromes? J. Mol. Biol. 278 655–666. [DOI] [PubMed] [Google Scholar]
- Renwick, S.B., Snell, K., and Baumann, U. 1998. The crystal structure of human cytosolic serine hydroxymethyltransferase: A target for cancer chemotherapy. Structure 6 1105–1116. [DOI] [PubMed] [Google Scholar]
- Richardson, J.S. and Richardson, D.C. 1989. Principles and patterns of protein conformation. In Prediction of protein structure and the principles of protein conformation (ed. G.D. Fasman), pp. 1–99. Plenum Press, New York.
- Rodionov, M.A. and Blundell, T.L. 1998. Sequence and structure conservation in a protein core. Proteins 33 358–366. [PubMed] [Google Scholar]
- Rost, B. 1999. Twilight zone of protein sequence alignments. Protein Eng. 12 85–94. [DOI] [PubMed] [Google Scholar]
- Russell, R.B. and Barton, G.J. 1994. Structural features can be unconserved in proteins with similar folds. An analysis of side-chain to side-chain contacts secondary structure and accessibility. J. Mol. Biol. 244 332–350. [DOI] [PubMed] [Google Scholar]
- Schneider, G., Käck, H., and Lindqvist, Y. 2000. The manifold of vitamin B6 dependent enzymes. Structure 8 1–6. [DOI] [PubMed] [Google Scholar]
- Selvaraj, S. and Gromiha, M.M. 2003. Role of hydrophobic clusters and long-range contact networks in the folding of α/β8 barrel proteins. Biophys. J. 84 1919–1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shindyalov, I.N. and Bourne, P.E. 1998. Protein structure alignment by incremental combinatorial extension CE of the optimal path. Protein Eng. 11 739–747. [DOI] [PubMed] [Google Scholar]
- Sivaraman, J., Li, Y., Larocque, R., Schrag, J.D., Cygler, M., and Matte, A. 2001. Crystal structure of histidinol phosphate aminotransferase HisC from Escherichia coli, and its covalent complex with pyridoxal-5′-phosphate and l-histidinol phosphate. J. Mol. Biol. 311 761–776. [DOI] [PubMed] [Google Scholar]
- Steegborn, C., Messerschmidt, A., Laber, B., Streber, W., Huber, R., and Clausen, T. 1999. The crystal structure of cystathionine γ-synthase from Nicotiana tabacum reveals its substrate and reaction specificity. J. Mol. Biol. 290 983–996. [DOI] [PubMed] [Google Scholar]
- Storici, P., Capitani, G., De Biase, D., Moser, M., John, R.A., Jansonius, J.N., and Schirmer, T. 1999. Crystal structure of gaba-aminotransferase, a target for antiepileptic drug therapy. Biochemistry 38 8628–8634. [DOI] [PubMed] [Google Scholar]
- Thompson, J.D., Higgins, D.G., and Gibson, T.J. 1994. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22 4673–4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todd, A.E, Orengo, C.A., and Thornton, J.M. 2001. Evolution of function in protein superfamilies, from a structural perspective. J. Mol. Biol. 307 1113–1143. [DOI] [PubMed] [Google Scholar]
- Vogt, G., Etzold, T., and Argos, P. 1995. An assessment of amino acid exchange matrices in aligning protein sequences: The twilight zone revisited. J. Mol. Biol. 249 816–831. [DOI] [PubMed] [Google Scholar]
- Wierenga, R.K. 2001. The TIM-barrel fold: A versatile framework for efficient enzymes. FEBS Lett. 492 193–198. [DOI] [PubMed] [Google Scholar]