

Abstract
The self-association equilibrium of a tracer protein, apomyoglobin (apoMb), in highly concentrated crowded solutions of ribonuclease A (RNase A) and human serum albumin (HSA), has been studied as a model system of protein interactions that occur in crowded macromolecular environments. The rotational diffusion of the tracer protein labeled with two different fluorescent dyes, 8-anilinonaphthalene-1-sulfonate and fluorescein isothiocyanate, was successfully recorded as a function of the two crowder concentrations in the 50–200 mg/mL range, using picosecond-resolved fluorescence anisotropy methods. It was found that apoMb molecules self-associate at high RNase A concentration to yield a flexible dimer. The apparent dimerization constant, which increases with RNase A concentration, could also be estimated from the fractional contribution of monomeric and dimeric species to the total fluorescence anisotropy of the samples. In contrast, an equivalent mass concentration of HSA does not result in tracer dimerization. This different effect of RNase A and HSA is much larger than that predicted from simple models based only on the free volume available to apoMb, indicating that additional, nonspecific interactions between tracer and crowder should come into play. The time-resolved fluorescence polarization methods described here are expected to be of general applicability to the detection and quantification of crowding effects in a variety of macromolecules of biological relevance.
Keywords: time-resolved fluorescence anisotropy, macromolecular crowding, self-association, segmental flexibility, apomyoglobin, ribonuclease A, human serum albumin
A large variety of physical and chemical processes in living organisms take place in highly concentrated solutions (50–400 mg/mL) of different macromolecules, which occupy a large fraction of the solution volume (Minton 2001). As a result of that, there is a significative increase of nonspecific interactions, such as steric repulsion or electrostatic interactions, which have been proven to have large effects on the equilibrium and kinetic parameters of biomolecular reactions (Ellis 2001). Previous experimental and theoretical work demonstrated substantial effects of crowding on a broad range of biochemical and physiological processes (Zimmerman and Minton 1993; Minton 2001; Rivas et al. 2004), like macromolecular association (Minton 2000) and protein folding (Sasahara et al. 2003; Tokuriki et al. 2004). Thus, as would be expected, macromolecular crowding enhances the formation of those reaction products that exclude less volume to other species in the solution such as compact structures or polymers. The quantification of species populations and the determination of apparent equilibrium constants in crowded media will be crucial for the correct understanding of biological processes in physiological environments.
A crowded environment can be mimicked, as a first approximation, by adding to a given solution a high concentration of the crowder, in the form of unrelated synthetic or natural macromolecules (Ellis 2001). However, even in these simplified model systems, monitoring changes in the macromolecule of interest (the tracer) to isolate crowding effects is a difficult experimental challenge. Physical methods based on fluorescence spectroscopy appear very convenient for this purpose, due to the possibility of labeling with extrinsic fluorescent dyes only the tracer protein, which in this way can be easily distinguished from the crowding macromolecules. In fact, this approach has been applied before to the study of horse apomyoglobin (apoMb) dimerization in crowded solutions, by means of steady-state fluorescence anisotropy techniques (Wilf and Minton 1981).
Steady-state fluorescence anisotropy values of a labeled tracer protein depend on the rotational correlation time (φ) of the macromolecule and on the dye fluorescence lifetime, as expressed by Perrin’s equation (Lakowicz 1999). When the tracer protein forms oligomers the hydrodynamic volume of the rotating unit increases, and this results in an increase of φ and, consequently, in the fluorescence anisotropy value. However, the oligomerization process may also affect the label fluorescence lifetime and, in this case, the correct interpretation of the recorded fluorescence anisotropy changes is far from simple. On the other hand, time-resolved anisotropy methods often allow the direct determination of the tracer rotational correlation time, independently of the changes in the fluorescence lifetime, facilitating the correct identification of the molecular species present in the solution. In the present work, we have further extended the previous steady-state anisotropy study of apoMb in highly concentrated protein solutions by means of time-resolved fluorescence anisotropy techniques with picosecond resolution. Two proteins were selected as crowders: ribonuclease A (RNase A; 13,700 Da), as in the work of Wilf and Minton (1981), and human serum albumin (HSA; 66,500 Da), as one of the major plasma proteins. In the case of fluorescence studies in concentrated RNase A solutions, the tracer apoMb was labeled with 8-anilinonaphthalene-1-sulfonate (ANS). This dye was selected because of its convenient long fluorescence lifetime (~15 nsec) and the lack of interfering local ANS motions when bound to apoMb (Zorrilla et al. 2004a). However, the noncovalent ANS binding to apoMb prevents its use in the presence of HSA, which also contains ANS binding sites (Slavik 1982; Matulis and Lovrien 1998). Therefore, the experiments in crowded HSA solutions were carried out using a fluorescein dye covalently bound to apoMb.
The fluorescence polarization methods developed in this work served to characterize apoMb self-association in concentrated protein solutions and should be of general application to the study of other protein interactions. Moreover, these methods may also facilitate the extension of current high throughput analytical techniques based on fluorescence spectroscopy from diluted to highly concentrated, crowded solutions.
Results
Characterization of labeled apoMb
ApoMb labeled with fluorescein (apoMb-Fl) and apoMb labeled with ANS (apoMb-ANS) were first characterized independently in diluted solution, to determine the effect of fluorescent labeling on apoMb hydrodynamics and association. The steady-state anisotropy of apoMb-Fl, in a diluted buffer solution containing 2 μM of the labeled protein and 100 μM of unlabeled apoMb, was r = 0.120 ± 0.005 (λexc = 460 nm, λem =520 nm), and the decay of the fluorescence intensity was fitted to a triexponential function (Table 1), with an average lifetime (τm) of 3.4 ± 0.1 nsec. This complex decay is indicative of heterogeneity in the dye microenvironment, probably due to the lack of specificity in the labeling reaction. The decay of the fluorescence anisotropy in the same conditions was fitted to a biexponential function with two correlation times (Table 2). The faster correlation time of 0.2 nsec can be assigned to local, independent depolarizing motions of the fluorescein dye, while the slower one (10 ± 1 nsec) agreed with previous data for the global rotation of monomeric apoMb and Mb in diluted solutions (Wang et al. 1997; Tcherkasskaya et al. 2000). The experimental time-zero anisotropy, r(0) = 0.25 ± 0.01, was considerably lower than the fundamental anisotropy of fluorescein (~0.4; Chen and Bowman 1965). This decrease is due to ultrafast picosecond components in the anisotropy decay that could not be resolved. The fluorescence intensity decay of apoMb-ANS in similar diluted solutions is best described by a two-exponential function, with τm =14.7 ± 0.1 nsec (Zorrilla et al. 2004a). In these conditions, the fluorescence anisotropy of apoMb-ANS is monoexponential, with a single correlation time of 9.0 ± 0.2 nsec, which is again that expected for the global rotation of monomeric apoMb (Zorrilla et al. 2004a). Additional analytical ultracentrifugation experiments were run with the labeled samples of apoMb (100 μM total protein concentration, as in the crowding experiments). These data confirmed that both apoMb-Fl and apoMb-ANS are present in monomeric form in the above diluted solution conditions, with a molecular mass of 17,500 ± 2000 Da (data not shown).
Table 1.
Fluorescence intensity decay parametersa of apoMb-Fl as a function of increasing concentration of HSA
| [HSA] (mg/mL) | a1 ±0.02 | τ1 (ns) ±0.05 | a2 ±0.02 | τ2 (ns) ±0.3 | α3 ±0.02 | τ3(ns) ±0.1 | τm(ns) ±0.1 |
| 0 | 0.22 | 0.28 | 0.26 | 1.5 | 0.52 | 3.9 | 3.4 |
| 5 | 0.23 | 0.28 | 0.26 | 1.5 | 0.51 | 3.9 | 3.4 |
| 25 | 0.25 | 0.28 | 0.30 | 1.5 | 0.45 | 3.9 | 3.3 |
| 50 | 0.30 | 0.28 | 0.28 | 1.5 | 0.42 | 3.9 | 3.3 |
| 100 | 0.30 | 0.28 | 0.31 | 1.5 | 0.39 | 3.9 | 3.2 |
| 200 | 0.35 | 0.28 | 0.31 | 1.5 | 0.34 | 3.9 | 3.1 |
[apoMb-F1] = 2 μM; [apoMb]T = 100 μM; λexc = 460 nm and λem = 520 nm; 20 mM phosphate buffer, 150 mM NaCl, 0.1 mM EDTA, pH 7.4, 20°C.
a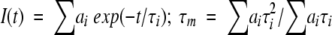
Table 2.
Time-resolved fluorescence anisotropy parameters of labeled apoMb as a function of increasing concentration of RNase A and HSA
| Tracer + crowder | [crowder] (mg/mL) | β1 ±0.06 | φ1 (ns) | β2 ±0.06 | φ2 (ns) |
| apoMb-ANSa | 0 | — | 9.0 (8.8–9.2) | — | — |
| 80 μM | 50 | 0.85 | 9.4 (8.8–10.1) | 0.15 | 24 (23–25) |
| +RNase A | 100 | 0.80 | 9.9 (9.0–10.8) | 0.20 | 28 (25–30) |
| 150 | 0.61 | 10.8 (9.9–11.9) | 0.39 | 36 (30–46) | |
| 225 | 0.51 | 12.2 (10.7–14.0) | 0.49 | 52 (41–69) | |
| 250 | 0.40 | 12.4 (10.7–14.6) | 0.60 | 54 (45–69) | |
| ApoMb-Flb,c | |||||
| 2 μM | |||||
| + RNase A | 200 | 0.30 | 0.28 ± 0.08 | 0.40 | 8.0 (5–14) |
| ApoMb-Flb | 0 | 0.40 | 0.2 ± 0.05 | 0.60 | 9.9 (8.8–11.4) |
| 2 μM | 5 | 0.40 | 0.2 ± 0.05 | 0.60 | 10.2 (8.7–11.8) |
| +HSA | 25 | 0.40 | 0.2 ± 0.05 | 0.60 | 10.6 (9.4–12.0) |
| 50 | 0.40 | 0.2 ± 0.05 | 0.60 | 10.7 (9.3–12.6) | |
| 100 | 0.40 | 0.2 ± 0.05 | 0.60 | 11.4 (10.3–12.9) | |
| 200 | 0.40 | 0.2 ± 0.05 | 0.60 | 13.6 (11.8–15.8) | |
The total concentration (labeled + unlabeled) of apoMb was 100 μM in all cases; 20 mM phosphate buffer, 150 mM NaCl, 0.1 mM EDTA, pH 7.4, 20°C.
aλexc = 393 nm, λem = 465 nm. The resolution was 12.2 ps/channel. r(0) = 0.35 ± 0.01.
bλexc = 460 nm, λem = 520 nm. The resolution was 13.1 ps/channel. r(0) = 0.25 ± 0.01.
cβ∞ = 0.3 ± 0.06.
Numbers in parenthesis represent the upper and lower limits of the recovered values (at the 67% confidence level) using rigorous error analysis, as described in Beechem et al. (1991).
A simple way to determine possible changes in apoMb structure due to covalent dye labeling is by recording the Förster resonance energy transfer (FRET) between ANS and fluorescein in the labeled protein. The binding of ANS to apoMb-Fl is taken as a test of protein integrity (Bismuto et al. 1996). If the dye is effectively bound to the protein the ANS-fluorescein separation should be less than the diameter of apoMb (~3.5 nm) (Papadopoulos et al. 2000). Because the Förster critical distance, R0, for the ANS-fluorescein pair is 5 nm (Sassaroli et al. 1984), the ANS → fluorescein energy transfer efficiency should be higher than 90%, and virtually all the ANS fluorescence would be quenched. The FRET assay was carried out by adding an excess (40 μM) of ANS to a 5 μM apoMb-Fl solution. In Figure 1 ▶ the emission spectra of the mixed solution is presented, together with those of two separated 5 μM apoMb-ANS and 5 μM apoMb-Fl solutions for comparison. It is shown that the fluorescence band at ~465 nm, corresponding to bound ANS, has been quenched, while that at ~520 nm (apoMb-Fl) increased significantly, indicating the efficient ANS binding to apoMb-Fl. Moreover, a similar assay can be used to estimate the affinity of ANS for the labeled protein. With this purpose, a 25 μM solution of unlabeled apoMb was added to a solution containing 25 μM apoMb-Fl and 25 μM ANS. The ANS emission intensity of the resulting mixture was ≈50% of a 25 μM apoMb-ANS control sample. This simple experiment indicates that both apoMb and apoMb-Fl are competent for ANS binding, with a dye dissociation constant to apoMb-Fl in the same range as that determined for unlabeled apoMb (3.4•10−6 M) (Stryer 1965).
Figure 1.

ANS binding to apoMb-Fl: Fluorescence emission spectra of 5 μM apoMb with 40 μM ANS (• •), 5 μM apoMb-Fl (——), and 5 μM apoMb-Fl with 40 μM ANS (- - -), in 20 mM phosphate, 150 mM NaCl, 0.1 mM EDTA (pH 7.4), 20°C, λexc =393 nm.
ApoMb self-association in RNase A concentrated solutions
Fluorescence anisotropy measurements were carried out to determine the possibility of apoMb association in crowded RNase A solutions. The steady-state fluorescence anisotropy of apoMb-ANS in the presence of RNase A (50–250 mg/mL) increased with RNase A concentration (Fig. 2 ▶). The polarization values were coincident, within the experimental error, with those published by Wilf and Minton (1981) at the same excitation and emission wavelength (λexc = 375 nm, λem = 465 nm) (data not shown).
Figure 2.

Steady-state fluorescence anisotropy (r) of labeled apoMb as a function of crowder concentration. (A) ApoMb-ANS in RNase A solutions; [apoMb-ANS] = 80 μM; λexc = 393 nm and λem = 465 nm. (B) ApoMb-Fl in HSA solutions; [apoMb-Fl] = 2 μM; λexc = 460 nm and λem = 520 nm. [apoMb]T = 100 μM. T = 20°C.
The fluorescence intensity decay of apoMb-ANS in the presence of RNase A (50–250 mg/mL) was biexponential, with an intensity averaged lifetime (τm) of 14.7 ± 0.2 nsec (Zorrilla et al. 2004a). Because this value remains practically unaltered upon addition of increasing concentrations of RNase A, the interpretation of the corresponding anisotropy changes was considerably simplified. When apoMb-ANS was placed in concentrated RNase A solutions (50–250 mg/mL), the anisotropy decay was biexponential and two well-separated rotational correlation times (Table 2) were determined at each RNase A concentration, which increased in parallel with the crowder concentration. The slowest correlation time (φ 2), which was not present in diluted apoMb solutions, increased from 24 ± 2 nsec to 54 ± 12 nsec for 50–250 mg/mL RNase A solutions. The value of φ 2 extrapolated to zero RNase A concentration was 22 ± 2 nsec, compatible with published values of the rotational time of apohemoglobin (apoHb), a dimeric protein with subunits homologous to monomeric apoMb (Sassaroli et al. 1986). On the other hand, the fastest correlation time (φ1) increased from 9.0 ± 0.2 nsec to 12.4 ± 2 nsec for 0–250 mg/mL RNase A solutions. This value, determined for zero RNase A concentration, is assigned to the global depolarizing rotational motion of the apoMb monomer (Zorrilla et al. 2004a). The experimental time-zero anisotropy, r(0) = 0.35 ± 0.01, was coincident with the ANS fundamental anisotropy (Anderson and Weber 1969) and was independent of RNase A concentration. This result, together with the absence of fast depolarization processes, indicates that the ANS chromophore is rigidly constrained within the hydrophobic apoMb heme pocket.
A parallel experiment was performed with apoMb-Fl in a 200 mg/mL RNase A solution, to test the possible influence of dye labeling on the hydrodynamic parameters of apoMb in RNase A solutions. In these conditions, the steady-state anisotropy of apoMb-Fl was r = 0.150 ± 0.005 (λexc = 460 nm, λem = 520 nm) and the decay of the fluorescence intensity was fitted to a triple exponential function, with lifetime values of 0.26 ± 0.05, 1.3 ± 0.3, and 3.7 ± 0.1 nsec; average lifetime τm = 3.2 ± 0.1 nsec. The anisotropy decay was best fitted to a function that included a residual anisotropy contribution, β∞, in addition to two rotational correlation times (Table 2).
Additional insight on apoMb association was obtained from steady-state anisotropy measurements of apoMb-ANS in samples where the RNase A concentration was fixed (100, 150, 200, and 250 mg/mL), while apoMb concentration was varied from 10 μM to 800 μM (Fig. 3 ▶). The background residual emission and the noncovalent ANS labeling prevented anisotropy measurements below 10 μM. Interestingly, the fluorescence anisotropy remained practically unaltered upon addition of apoMb to the solution in the studied concentration range. Furthermore, these values were not affected by the ANS/apoMb labeling ratio (0.2–0.8).
Figure 3.

Steady-state anisotropy (r) of apoMb-ANS as a function of apoMb concentration, in the presence of 250 mg/mL RNase A (○), 200 mg/mL RNase A (▾), 150 mg/mL RNase A (▵), and 100 mg/mL RNase A (▪). λexc = 393 nm; λem = 465 nm. Dashed/dotted lines correspond to theoretical curves computed from equation 11, for the lowest apparent equilibrium constant values compatible with experimental data.
ApoMb in HSA concentrated solutions
A parallel study of the association of apoMb was carried out in highly concentrated HSA solutions. The steady-state fluorescence anisotropy of apoMb-Fl in presence of HAS (5–200 mg/mL) increased with HSA concentration (Fig. 2 ▶), and the fluorescence intensity decay in these conditions was fitted to a triexponential function (Table 1). A small increase in the fractional contribution of the two shorter lifetimes was observed at high HSA concentration, without apparent changes in the lifetime values (Table 1).
The time-resolved fluorescence anisotropy of apoMb-Fl in the presence of a high concentration of HSA was best described by two correlation times (Table 2). The fastest correlation time (φ1) was practically independent of HAS concentration and, as indicated above, it is likely due to fast local flapping motions of the fluorescein dye bound to apoMb. The slowest correlation time (φ2) increased from 10 ± 1 nsec to 14 ± 2 nsec for 0–200 mg/mL HSA solutions, and its value determined for zero HSA concentration corresponds to the global rotation of monomeric apoMb. The r(0) value was independent on HSA concentration. Additional numerical analyses were run by including a residual anisotropy term in the fitting function. However, only in the case of the anisotropy decay in 200 mg/mL HSA solution the experimental data were compatible with this term, which may be assigned to a 5%–10% of dimeric apoMb (see Discussion).
Discussion
ApoMb self-associates in highly concentrated RNase A solutions
The relationship between rotational correlation time and hydrodynamic volume (V) of a spherical particle can be expressed by the Stokes-Einstein-Debye equation as:
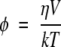 |
(1) |
where η is the solution viscosity, k is the Boltzmann constant, and T is the temperature. This expression has been shown to be of general applicability to globular proteins in diluted solutions (Ferrer et al. 2001; Garcia-Mayoral et al. 2004). Thus, in the case of apoMb solution at [RNase A] = 0, the rotational correlation time (φ1 = 9 ± 0.2 nsec) (Table 2) would correspond to the monomeric form of the protein, according to the volume estimated from equation 1 (Zorrilla et al. 2004a). On the other hand, when apoMb-ANS is present in solutions with increasing concentrations of RNase A, a second correlation time (φ2) was determined, that extrapolated to zero RNase A concentration takes a value (22 ± 2 nsec) compatible with the global motion of dimeric apoMb. In fact, this value largely exceeds that expected for the apoMb monomer in diluted solution (9–10 nsec). According to that, one possible way of interpreting the time-resolved anisotropy data of apoMb-ANS in RNase A crowded solutions would be by means of a rigid particle model, in which the correlation times φ1 and φ2 are associated to the global rotation of apoMb monomer and dimer, respectively, both considered as rigid particles in the nanosecond range. In that case, the fractional contributions (βi) from the anisotropy decay analysis (Table 2) could be directly associated to monomeric and dimeric apoMb molar fractions (xi). Thus, for example, in a 250 mg/mL RNase A solution in which β1 = 0.4 and β2 = 0.6, this would correspond to 40% of apoMb monomer and 60% of dimer, with rotational correlation times of about ~12 nsec and ~54 nsec, respectively. However, this model fails to explain the steady-state experiments reported in Figure 3 ▶, in which the viscosity of the crowded solution is kept constant and the monomer/dimer ratio is varied. The steady-state anisotropy values corresponding to apoMb monomer and rigid dimer, in the 250 mg/mL RNase A solution would be rm = 0.165 and rd = 0.275, respectively, as estimated from equation 12, and the spectroscopic parameters measured here. Then, the experimental steady-state fluorescence anisotropy (r), which is the weighted sum of the individual contribution from each species (r = xmrm + xdrd), would be very sensitive to changes in the molar fraction of apoMb monomer and dimer (see Materials and Methods). This expectation is not realized when the r value of apoMb is recorded at the fixed RNase A concentration of 250 mg/mL, neither at any other concentration values shown in Figure 3 ▶, despite the fact that the monomer/dimer ratio is expected to change appreciably. Furthermore, monomer and dimer fractional abundance estimations based on the rigid particle model did not agree with previous results (Wilf and Minton 1981), which indicate that apoMb in a 250 mg/mL RNase A solution is present essentially as a dimeric species. The lack of changes in r in Figure 3 ▶ could be understood if the apoMb dimer had a flexible structure. In fact, this possibility is strongly supported by the detection of flexible motions in the homologous dimeric protein apoHb (Sassaroli et al. 1986). In that case, the segmental motions of apoMb dimer would have the effect of decreasing the difference between the monomer (rm) and dimer (rd) anisotropy values. Because of that, the change in the summed anisotropy (r) as a function of the monomer/dimer ratio (given by the apoMb concentration) would be negligible in the range studied here (Fig. 3 ▶). Therefore, we interpret the results presented here in terms of a flexible particle model, in which the apoMb dimer is represented by two rigid subunits joined together with a flexible link. The depolarizing segmental motions of this dimer take place most likely in a time range close to that of the global rotation of apoMb monomer, because only two rotational times are experimentally observed (Table 2). This similarity was indeed reported in the case of the flexible apoHb dimer (Sassaroli et al. 1986). Accordingly, the correlation time φ1 is assigned to the combination of the segmental motion of apoMb dimer and the global motion of the monomer, while φ2 pertains to the dimer global rotation. A flexible apoMb dimer is also compatible with the increase in tracer anisotropy as a function of crowder concentration shown in Figure 2 ▶. In this case, the presence of increasing amounts of the crowder protein (RNase A or HSA) results in corresponding increases in the rotational viscosity affecting both monomeric and dimeric species (Zorrilla et al. 2004a), which is reflected in measurable changes in the anisotropy.
The anisotropy decay parameters of apoMb-Fl in a 200 mg/mL RNase A solution (Table 2) are fully consistent with the flexible particle model of apoMb dimerization proposed above. The φ2 value agrees with the fast correlation time determined for apoMb-ANS at the same RNase A concentration, which corresponds to the combined global motion of monomeric apoMb and segmental motion of dimeric apoMb. On the other hand, the correlation time of 0.2 nsec is assigned to independent motions of the fluorescein dye, as noted above. The short lifetime of the fluorescein chromophore (~3 nsec) and these fast depolarizing motions in apoMb-Fl (φ1 ~0.2 nsec) precluded the determination of the dimer correlation time, which appears instead as a residual term (β∞) in the anisotropy decay. This experiment also indicates that self-association of apoMb to form dimers is an intrinsic characteristic of the protein, independent of the bound ANS dye.
The data presented above also excluded specific interactions between tracer apoMb and crowder RNase A. These interactions, if present, would result in the heteroassociation of tracer and crowder. In that case, the absence of changes in anisotropy values shown in Figure 3 ▶ would indicate that all apoMb molecules were bound to RNase A forming heterodimers, for all RNase A and apoMb solutions studied here. Then, the two correlation times of apoMb determined in RNase A solutions (Table 2) would correspond to a heterodimer composed by two rigid subunits linked by a very flexible joint, in which β1 and β2 (Table 2) would be associated to the amplitudes of the flexible subunits and the overall heterodimer motion, respectively. Because β1 decreases from 0.85 to 0.40 in the 50–250 mg/mL RNase A concentration range, one needs to assume that the flexibility of the heterodimer would be strongly reduced at high RNase A concentrations. Previous results (Wilf and Minton 1981) show that the hydrodynamic volume of apoMb approaches that of dimeric apoHb as the RNase A concentration increases. Both are practically coincident in a 250 mg/mL RNase A solution, but markedly different at lower RNase A concentrations. Therefore, to explain the above results in terms of heteroassociation, a very flexible apoMb-RNase A complex would be necessary, with a variety of conformations different for each crowder concentration, which seems very unlikely. In conclusion, the simplest interpretation of all the fluorescence data presented above is that apoMb self-associates in highly concentrated RNase A solutions to yield a flexible dimer species. The monomer ↔ dimer equilibrium is almost completely shifted to the dimer side at RNase A concentrations of 250 mg/mL.
Fractional abundance of monomeric and dimeric apoMb in concentrated RNase A solutions
The apoMb-ANS monomer/dimer molar ratio in concentrated RNase A solutions can be estimated from time-resolved anisotropy data by assuming that the degree of flexibility of the apoMb dimer is independent on the RNase A concentration (see Materials and Methods). As it is shown in Figure 4 ▶, the molar fraction of dimeric apoMb increased almost linearly with RNase A concentration.
Figure 4.

The apoMb dimer molar fraction as a function of RNase A concentration, computed from the anisotropy parameters of Table 2 as described in the main text.
The apparent equilibrium dimerization constant (K′) could not be extracted from steady-state anisotropy experiments due to the limited apoMb concentration range that is accessible in these conditions, as noted above. Nevertheless, model calculations using equation 11 (see Materials and Methods), permit to estimate lower limits of K′ compatible with the steady-state anisotropy values and the fractional abundance of monomer and dimer apoMb species, as determined from the time-resolved parameters. In this way, the following values were obtained: K′ ≥2 × 104 M−1, K′ ≥2 × 105 M−1, and K′ ≥106 M−1, for RNase A concentration of 100 mg/mL, 150 mg/mL, and 200 to 250 mg/mL, respectively. The lower limit of the apparent dimerization constant shows a clear tendency to increase with the concentration of RNase A, which is consistent with the increasing apoMb dimer concentration in these crowded solutions detected by time-resolved measurements.
The method illustrated here is of general applicability to any homo- or heteroassociation reaction in a crowded environment, provided that the steady-state fluorescence anisotropy values of the monomeric and oligomeric species differ significantly. Because each experiment is run at a constant crowder concentration, the rotational friction is kept constant and the anisotropy changes would depend only on the hydrodynamic volume of each molecular species and on the dye fluorescence lifetime.
Differential effect of HSA and RNase A as crowder proteins
The data presented above show that apoMb self-associates in concentrated RNase A solutions; while in concentrated HSA solutions the tracer protein is in monomeric form, at least in the range studied here. This change in the apparent dimerization equilibrium constant can be analyzed first in terms of the different volume excluded to apoMb in HSA and RNase A solutions for the same mass concentration. In this regard, it is important to note that HSA itself is in monomeric state in the whole concentration range studied here (data not shown), while RNase A solutions are much more heterogeneous. Recent analytical ultracentrifugation experiments have shown that in the above concentration range RNase A molecules may be present as a mixture of monomers and trimers, monomers and tetramers, or monomers, dimers, and tetramers (Zorrilla et al. 2004b). Although two solutions of RNase A and HSA with the same mass concentration should have approximately the same total volume occupied by the macromolecule, the distribution of this occupied volume is markedly different, giving rise to a very different free volume available to apoMb, much lower in the case of an RNase A solution than in HSA. This is due to the higher number of macromolecules in RNase A solutions (Fig. 5 ▶). A crude estimate of the solution volume, from which apoMb is excluded in RNase A and HSA solutions, shows that the volume available to apoMb in 200 mg/mL HSA is comparable to that of an 80–100 mg/mL RNase A solution. For this last concentration of RNase A the apoMb dimer molar fraction detected here is ~30%, while this fraction is less than 10% in 200 mg/mL HSA. In conclusion, the differential effect of RNase A and HSA is parallel to the different free volume available to apoMb in these two crowded solutions.
Figure 5.

Idealized scaled representation of excluded and free volume in crowded solutions. (A) 200 mg/mL RNase A solution containing monomers and tetramers of the crowder. (B) 200 mg/mL RNase A solution containing monomers and trimers, and (C) 200 mg/mL HSA solution. All species are represented as spherical particles of equivalent volume, assuming an hydration of 0.3 g of water/g protein. The black circle represents an apoMb monomer molecule.
A semiquantitative estimate of the extent of excluded volume influence on the monomer/dimer association equilibrium of a tracer protein can be obtained from several existing theoretical models (Minton 1998). Thus, for example, the activity coefficient of a spherical tracer (apoMb in this case) due to a spherical crowder can be computed from the scaled particle theory of Boublik (Boublik 1986). Although in some of the experiments described above the real situation can be more complex, due to heterogeneity in crowder size distribution and a nonspherical dimer conformation, this computation may provide interesting worst-case expectation changes of the apparent association equilibrium constants. In fact, computations based on the above model (Minton 1998) predict that the K′ value should increase by a factor of ~2.2 for a change in RNase A crowding concentration from 100 to 200 mg/mL (data not shown), to be compared with the experimental change of about two orders of magnitude reported here. In the case of HSA the theoretically predicted change is ~1.5. One may conclude that this model correctly predicts, in a qualitative way, the observed difference between RNase A and HSA crowding effects, although fails short to account for each individual crowding effect. This large discrepancy is unlikely due to model limitations but, rather on the contrary, it is probably an indication of additional nonspecific crowder–tracer interactions in the solutions studied here, as, for example, those originated from electrostatic charges, which are different for the two crowder proteins.
Conclusions
The changes in steady-state and time-resolved fluorescence anisotropy of apoMb-ANS and apoMb-Fl, recorded in the presence of large concentrations of RNase A, indicate that the tracer protein in this crowding environment self-associates to yield flexible dimers. The spectroscopic techniques described here allowed the characterization of the hydrodynamic properties of the monomeric and dimeric species, as well as an estimate of the lower limit for the apparent dimerization equilibrium constant as a function of crowder concentration. On the other hand, it is also shown that self-association of apoMb does not take place in the presence of an equivalent mass concentration of HSA. The different effect of the two crowder proteins, RNase A and HSA, can only partially be interpreted by the large difference in free volume available to the tracer protein, which is much smaller in the first case. Additional, nonspecific crowder-dependent effects should also be operating.
Materials and methods
Chemicals
Proteins
Crystallized and lyophilized horse skeletal muscle myoglobin (Mb), crystallized and lyophilized HSA and five times crystallized pancreatic bovine RNase A were purchased from Sigma-Aldrich Chemie Gmbh. All the proteins were used without further purification, but extensively dialyzed against working buffer previously to any experiment.
Fluorescent labels: 8-anilinonaphthalene-1-sulfonic acid ammonium salt was purchased from Fluka Chemie AG, and fluorescein-5-isothiocyanate (FITC) was obtained from Molecular Probes, Inc.
All the experiments were carried out in 20 mM phosphate, 150 mM NaCl, 0.1 mM EDTA buffer (pH 7.4), at 20 ± 0.1°C.
Preparation and labeling of apomyoglobin
ApoMb was prepared from Mb by a modification of the acid/acetone method (Rossi-Fanelli et al. 1958; Wilf and Minton 1981). ApoMb yield was 60%–70%. The efficiency of heme group removal, determined from the absorbance ratio at 405 and 280 nm (Wilf and Minton 1981), was 98%. Protein concentration was determined by absorbance measurements performed on a Cary 3E spectrophotometer, using molar absorption coefficients of 15,700 M−1 cm−1 for apoMb at 280 nm (Wilf and Minton 1981); 9800 M−1 cm−1 for RNase A at 277.5 nm (Sela and Anfinsen 1957; Torrent et al. 1999); and 34,600 M−1 cm−1 for HSA at 280 nm (Mach et al. 1992; Peters Jr. 1996).
ApoMb was labeled with ANS, in the form of a stable 1:1 noncovalent complex (Stryer 1965). The concentration of added ANS was determined from its absorbance at 350 nm, using a molar absorption coefficient of 5000 M−1 cm−1 (Stryer 1965). Labeling ratios in the 0.2–0.8 range were estimated from the value of the binding constant (3.4 × 10−6 M) (Stryer 1965). The fluorescence emission of free ANS in buffer solution was negligible compared with that of the protein-bound form (Stryer 1965).
Nonspecific covalent amino terminal and lysine labeling of apoMb with FITC was performed by adding the fluorophore to an apoMb solution in a 5:1 molar ratio, in 0.2 M phosphate buffer at pH 8.0. The labeling reaction was carried out for 30 min at room temperature and the unreacted dye was removed with a HiTrap column (Pharmacia Biotech). The dye/protein ratio after labeling was 1.2, estimated from absorbance measurements at 490 and 280 nm, respectively. The molar absorption coefficient of bound fluorescein at 490 nm used here was 72,800 M−1 cm−1 (Diehl and Horchak-Morrils 1987).
Steady-state and time-resolved anisotropy measurements
Fluorescence spectra and anisotropy measurements were performed on a photon-counting SLM 8000D spectrofluorimeter fitted with Glan-Taylor polarizers in the excitation and emission channels, at 20 ± 0.1°C. The steady-state fluorescence anisotropy is defined as:
 |
(2) |
where III and I⊥ are the intensities observed in the parallel and perpendicular directions to the plane of polarization of the excitation beam, respectively. G is a scaling factor that accounts for differences in the detection efficiency for the two polarized intensities.
Picosecond-resolved fluorescence intensity and anisotropy measurements were carried out using two time-correlated single photon counting laser systems, one with a PicoQuant 393 nm diode laser beam as the excitation source (as described in Organero et al. 2002), and a second one with a Spectra Physics Ti:sa mode-locked laser, associated to a second harmonic generator tuned at 460 nm (as described in Lillo et al. 2002). The first one was used for apoMb-ANS samples and the second one in the apoMb-Fl experiments, with a time resolution of 12.2 and 13.1 psec/channel, respectively.
The fluorescence anisotropy decay r(t) was determined by simultaneous analysis of the parallel III (t) and perpendicular I⊥(t) emission intensity components:
 |
(3) |
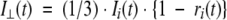 |
(4) |
 |
(5) |
where I(t), τi, and ai are the total fluorescence decay, the fluorescence lifetimes, and the preexponential factors, respectively (∑ai = 1). The intensity averaged fluorescence lifetime was calculated as 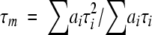 .
.
The fitting function was usually a sum of exponentials of the form (Lakowicz 1999; Valeur 2002):
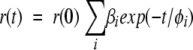 |
(6) |
where r(0) is the time-zero anisotropy, φi the rotational correlation times, and βi the fractional amplitudes. For anisotropy decays with a residual anisotropy term (r(0)•β∞), the fitting function was:
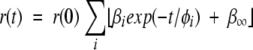 |
(7) |
Data analysis was performed using nonlinear least-squares global methods from the Globals Unlimited general-purpose program. The quality of the fit was determined from global χ2 values and visual inspection of the weighted residuals distribution.
Steady-state and time-resolved anisotropy measurements of apoMb-ANS in highly concentrated RNase A solutions were performed keeping the total apoMb concentration constant (100 μM; 1.7 mg/mL) while changing the RNase A concentration (50–250 mg/mL). In addition, some steady-state anisotropy measurements were performed for a constant RNase A concentration of 100, 150, 200, and 250 mg/mL and variable apoMb concentration (10–800 μM). In this case, the excitation and emission wavelengths were 393 nm and 465 nm for all the steady-state and time-resolved anisotropy determinations.
For highly concentrated HSA solutions, steady-state anisotropy measurements were carried out with samples containing 2 μM apoMb-Fl and additional unlabeled apoMb until a total apoMb concentration of 100 μM, with excitation and emission wavelengths at 460 nm and 520 nm, respectively. ApoMb-Fl in a 200 mg/mL RNase A solution was also studied in the same conditions.
The background fluorescence from unlabeled concentrated protein solutions was recorded and substracted for all the spectroscopic determinations. The fluorescence measurements were taken at least 1 h after solution preparation, to assure equilibrium conditions.
Determination of apoMb monomer and dimer molar fractions
The fluorescence anisotropy decay of apoMb-ANS in concentrated RNase A solutions was fitted to a double exponential function with rotational correlation times φ1 and φ2 (equation 6). For an apoMb solution containing labeled monomer and dimer species, the anisotropy decay is a weighted function of the individual monomer and dimer anisotropy decays, rm(t) and rd (t):
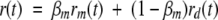 |
(8) |
where βm is the fractional intensity of monomeric apoMb. In the flexible particle model, φ1 was assigned to a combination of the global motion of monomeric apoMb and the segmental local motion of dimeric apoMb, while φ2 was assigned to the overall motion of dimeric apoMb (see above). Then equation 6 can be recast as follows:
 |
(9) |
where 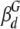 and
and  are the fractional amplitudes corresponding to the global and local motions of dimeric apoMb, respectively (
are the fractional amplitudes corresponding to the global and local motions of dimeric apoMb, respectively (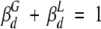 ). Assuming that the contribution of flexibility to the dimer depolarization was constant and equal to that found in a 250 mg/mL RNase A solution, where virtually all apoMb is in dimeric form (Wilf and Minton 1981), then
). Assuming that the contribution of flexibility to the dimer depolarization was constant and equal to that found in a 250 mg/mL RNase A solution, where virtually all apoMb is in dimeric form (Wilf and Minton 1981), then 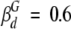 and
and . Finally, in the absence of spectroscopic changes in the fluorescent label upon dimer formation, the molar fraction of dimeric apoMb coincides with the fractional intensity of the dimer (xd = 1 -β m), and it can be estimated as:
. Finally, in the absence of spectroscopic changes in the fluorescent label upon dimer formation, the molar fraction of dimeric apoMb coincides with the fractional intensity of the dimer (xd = 1 -β m), and it can be estimated as:
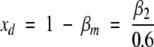 |
(10) |
Determination of the apparent equilibrium dimerization constant
For a solution containing only monomeric and dimeric fluorescent species of apoMb-ANS, detected at a given pair of excitation and emission wavelengths, the value of the steady-state anisotropy is given by: r = fdrd + fmrm. In this expression rd and rm are the dimer and monomer fluorescence anisotropies and fd and fm the corresponding fractional fluorescence intensities, which, in the absence of spectral changes in ANS upon dimer formation correspond to the respective molar fractions. On the other hand, the apparent equilibrium dimerization constant can be written as: K′ = [d]/[m]2, where [d] and [m] are the molar concentration of dimeric and monomeric apoMb at equilibrium, respectively. Combining this equation with that from above for the anisotropy of a monomer/dimer mixture, an expression that relates the steady-state anisotropy with the apparent dimerization equilibrium constant can be derived (Chauvin et al. 1994):
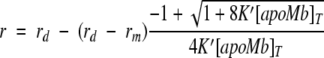 |
(11) |
where r is the steady-state anisotropy of apoMb measured for a total molar concentration [apoMb]T. Theoretical steady-state anisotropy curves were calculated from equation 11 for different values of the apparent equilibrium constant for each RNase A concentration. Because rd and rm values cannot be obtained directly in these solutions, these values were estimated from the time-resolved fluorescence parameters measured here for dimeric and monomeric apoMb at each RNase A concentration and the expression (Lakowicz 1999):
 |
(12) |
Acknowledgments
We thank Dr. A. Douhal for instrumentation facilities in the ANS-apoMb experiments. We also thank Dr. A.P. Minton for insightful discussion and advice, Dr. C. Alfonso for the performance of analytical ultracentrifugation experiments, and an unknown reviewer for very helpful criticisms. This work was supported by grants BQU/2000–1500, BIO99-0859-C03, BQU/2003-4430, and SAF/2003-04266 from the Spanish Dirección General de Enseñanza Superior e Investigación (DGESI), and grant 07B/0042/1999 from Comunidad de Madrid. S.Z. was a predoctoral fellow of the Comunidad de Madrid (CAM).
Article published online ahead of print. Article and publication date are at http://www.proteinscience.org/cgi/doi/10.1110/ps.04809404.
References
- Anderson, S.R. and Weber, G. 1969. Fluorescence polarization of the complexes of 1-anilino-8-naphthalenesulfonate with bovine serum albumin. Evidence for preferential orientation of the ligand. Biochemistry 8 371–377. [DOI] [PubMed] [Google Scholar]
- Beechem, J.M., Gratton, E., Ameloot, M., Knutson, J.R., and Brand, L. 1991. The global analysis of fluorescence intensity and anisotropy decay data: Second-generation theory and programs. In Topics in fluorescence spectroscopy, Vol. 2 (ed. J.R. Lakowicz), pp. 241–305. Plenum Press, New York.
- Bismuto, E., Sirangelo, I., Irace, G., and Gratton, E. 1996. Pressure-induced perturbation of apomyoglobin structure: Fluorescence studies on native and acidic compact forms. Biochemistry 35 1173–1178. [DOI] [PubMed] [Google Scholar]
- Boublik, T. 1986. Equations of state of hard body-fluids. Mol. Phys. 59 371–380. [Google Scholar]
- Chauvin, F., Brand, L., and Roseman, S. 1994. Sugar transport by the bacterial phosphotransferase system. Characterization of the Escherichia coli enzyme I monomer/dimer equilibrium by fluorescence anisotropy. J. Biol. Chem. 269 20263–20269. [PubMed] [Google Scholar]
- Chen, R.F. and Bowman, R.L. 1965. Fluorescence polarization: Measurement with ultraviolet-polarizing filters in a spectrophotometer. Science 147 729–732. [DOI] [PubMed] [Google Scholar]
- Diehl, H. and Horchak-Morris, N. 1987. Studies on fluorescein: The absorbance of fluorescein in the ultraviolet as a function of pH. Talanta 34 739–741. [DOI] [PubMed] [Google Scholar]
- Ellis, R.J. 2001. Macromolecular crowding: An important but neglected aspect of the intracellular environment. Curr. Opin. Struct. Biol. 11 114–119. [DOI] [PubMed] [Google Scholar]
- Ferrer, M.L., Duchowicz, R., Carrasco, B., García de la Torre, J., and Acuña, A.U. 2001. The conformation of serum albumin in solution: A combined phosphorescence depolarization-hydrodynamic modelling study. Biophys. J. 80 2422–2430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-Mayoral, M.F., García-Ortega, L., Lillo, M.P., Santero, J., Martínez del Pozo, A., Gavilanes, J.G., Rico, M., and Bruix, M. 2004. NMR structure of the non-cytotoxic α-sarcin mutant Δ(7–22): The importance of the native conformation of peripheral loops for activity. Protein Sci. 13 1000–1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakowicz, J.R. 1999. Principles of fluorescence spectroscopy, 2nd ed. (ed. J.R. Lakowicz). Kluwer Academic-Plenum Publishers, New York.
- Lillo, M.P., Cañadas, O., Dale, R.E., and Acuña, A.U. 2002. Location and properties of the taxol binding center in microtubules: A picosecond laser study with fluorescent taxoids. Biochemistry 41 12436–12449. [DOI] [PubMed] [Google Scholar]
- Mach, H., Middaugh, C.R., and Lewis, R.V. 1992. Statistical determination of the average values of the extinction coefficients of tryptophan and tyrosine in native proteins. Anal. Biochem. 200 74–80. [DOI] [PubMed] [Google Scholar]
- Matulis, D. and Lovrien, R. 1998. 1-Anilino-8-naphthalene sulfonate anion-protein binding depends primarily on ion pair formation. Biophys. J. 74 422–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minton, A.P. 1998. Molecular crowding: Analysis of effects of high concentrations of inert cosolutes on biochemical equilibria and rates in terms of volume exclusion. Methods Enzymol. 295 127–149. [DOI] [PubMed] [Google Scholar]
- ———. 2000. Implications of macromolecular crowding for protein assembly. Curr. Opin. Struct. Biol. 10 34–39. [DOI] [PubMed] [Google Scholar]
- ———. 2001. The influence of macromolecular crowding and macromolecular confinement on biochemical reactions in physiological media. J. Biol. Chem. 276 10577–10580. [DOI] [PubMed] [Google Scholar]
- Organero, J.A., Tormo, L., and Douhal, A. 2002. Caging ultrafast proton transfer and twisting motion of 1-hidroxi-2-acetonaphthona. Chem. Phys. Lett. 363 409–414. [Google Scholar]
- Papadopoulos, S., Jurgens, K.D., and Gros, G. 2000. Protein diffusion in living skeletal muscle fibers: Dependence on protein size, fiber type, and contraction. Biophys. J. 79 2084–2094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters Jr., T. 1996. All about albumin. Biochemistry, genetics and medical applications. Academic Press, San Diego, CA.
- Rivas, G., Ferrone, F., and Herzfeld, J. 2004. Life in a crowded world. EMBO Rep. 5 23–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi-Fanelli, A., Antonini, E., and Caputo, A. 1958. Structure of hemoglobin. I. Physicochemical properties of human globin. Biochim. Biophys. Acta 30 608–615. [DOI] [PubMed] [Google Scholar]
- Sasahara, K., McPhie, P., and Minton, A.P. 2003. Effect of dextran on protein stability and conformation attributed to macromolecular crowding. J. Mol. Biol. 326 1227–1237. [DOI] [PubMed] [Google Scholar]
- Sassaroli, M., Bucci, E., Liesegang, J., Fronticelli, C., and Steiner, R.F. 1984. Specialized functional domains in hemoglobin: Dimensions in solution of the apohemoglobin dimer labeled with fluorescein iodoacetamide. Biochemistry 23 2487–2491. [DOI] [PubMed] [Google Scholar]
- Sassaroli, M., Kowalczyk, J., and Bucci, E. 1986. Probe dependence of correlation times in heme-free extrinsically labeled human hemoglobin. Arch. Biochem. Biophys. 251 624–628. [DOI] [PubMed] [Google Scholar]
- Sela, M. and Anfinsen, C.B. 1957. Spectrophotometric and polarimetric experiments with ribonuclease. Biochim. Biophys. Acta 24 229–235. [DOI] [PubMed] [Google Scholar]
- Slavik, J. 1982. Anilinonaphthalene sulfonate as a probe of membrane composition and function. Biochim. Biophys. Acta 694 1–25. [DOI] [PubMed] [Google Scholar]
- Stryer, L. 1965. The interaction of a naphthalene dye with apomyoglobin and apohemoglobin. A fluorescent probe of non-polar binding sites. J. Mol. Biol. 13 482–495. [DOI] [PubMed] [Google Scholar]
- Tcherkasskaya, O., Ptitsyn, O.B., and Knutson, J.R. 2000. Nanosecond dynamics of tryptophans in different conformational states of apomyoglobin proteins. Biochemistry 39 1879–1889. [DOI] [PubMed] [Google Scholar]
- Tokuriki, N., Kinjo, M., Negi, S., Hocino, M., Goto, Y., Urabe, I., and Yomo, T. 2004. Protein folding by the effects of macromolecular crowding. Protein Sci. 13 125–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torrent, J., Connelly, J.P., Coll, M.G., Ribo, M., Lange, R., and Vilanova, M. 1999. Pressure versus heat-induced unfolding of ribonuclease A: The case of hydrophobic interactions within a chain-folding initiation site. Biochemistry 38 15952–15961. [DOI] [PubMed] [Google Scholar]
- Valeur, B. 2002. Molecular fluorescence principles and applications. Wiley-VCH, Weinheim, Germany.
- Wang, D., Kreutzer, U., Chung, Y., and Jue, T. 1997. Myoglobin and hemoglobin rotational diffusion in the cell. Biophys. J. 73 2764–2770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilf, J. and Minton, A.P. 1981. Evidence for protein self-association induced by excluded volume. Myoglobin in the presence of globular proteins. Biochim. Biophys. Acta 670 316–322. [DOI] [PubMed] [Google Scholar]
- Zimmerman, S.B. and Minton, A.P. 1993. Macromolecular crowding: Biochemical, biophysical, and physiological consequences. Annu. Rev. Biophys. Biomol. Struct. 22 27–65. [DOI] [PubMed] [Google Scholar]
- Zorrilla, S., Rivas, G., and Lillo, M.P. 2004a. Fluorescence anisotropy as a probe to study tracer proteins in crowded solutions. J. Mol. Recognit. 17 408–416. [DOI] [PubMed] [Google Scholar]
- Zorrilla, S., Jiménez, M., Lillo, M.P., Rivas, G., and Minton, A.P. 2004b. Sedimentation equilibrium in a solution containing an arbitrary number of solute species at arbitrary concentrations: Theory and application to concentrated solutions of ribonuclease. Biophys. Chem. 108 89–100. [DOI] [PubMed] [Google Scholar]