

Abstract
Human coactosin-like protein (CLP) shares high homology with coactosin, a filamentous (F)-actin binding protein, and interacts with 5LO and F-actin. As a tumor antigen, CLP is overexpressed in tumor tissue cells or cell lines, and the encoded epitopes can be recognized by cellular and humoral immune systems. To gain a better understanding of its various functions and interactions with related proteins, the crystal structure of CLP expressed in Escherichia coli has been determined to 1.9 Å resolution. The structure features a central β-sheet surrounded by helices, with two very tight hydrophobic cores on each side of the sheet. CLP belongs to the actin depolymerizing protein superfamily, and is similar to yeast cofilin and actophilin. Based on our structural analysis, we observed that CLP forms a polymer along the crystallographic b axis with the exact same repeat distance as F-actin. A model for the CLP polymer and F-actin binding has therefore been proposed.
Keywords: coactosin-like protein, crystal structure, F-actin binding protein, tumor antigen, actin depolymerizing protein, polymer
Human coactosin-like protein (CLP) was first identified from a yeast two-hybrid screen using 5-lipoxygenase (5LO) as bait from a human cDNA library (Provost et al. 1999). Human CLP shares significant homology with coactosin, an F-actin binding protein from Dictyostelium discoideum (de Hostos et al. 1993). CLP mRNA is widely distributed in tissue, and is expressed at high levels in placenta, lung, kidney, and peripheral blood leukocytes and low levels in brain, liver, and pancreas. To elucidate the molecular mechanism of all-trans-retinoic acid (ATRA)-induced differentiation of acute promyelocytic leukemia (APL) cells, the APL cell line NB4 was treated by ATRA. The results indicated that CLP expression could be modulated by ATRA (Liu et al. 2000). As a pancreatic cancer antigen, its mRNA is overexpressed in pancreatic cancer cell lines, compared to normal pancreatic tissues. Three epitopes of CLP have been recognized by HLA-A2 restricted and tumor-reactive CTL, and it has been suggested that peptides 15–24 and 104–113 of CLP could be appropriate candidates for humoral immunity (Nakatsura et al. 2001, 2002).
CLP interacts directly with 5LO both in vitro and in vivo. Immunoblot analysis in vitro using anti-5LO and anti-CLP antibodies ascertained that the high molecular mass band represents a 5LO–CLP complex; however, CLP has no direct effect on 5LO activity. 5LO plays a pivotal role in cellular leukotrene synthesis. Leukotrienes play central roles in immune response and tissue homeostasis. However, abnormal production of leukotrienes contributes to a variety of diseases. In resting cells, 5LO is localized in soluble compartments, in the cytosol and/or within the nucleus. Upon activation, 5LO becomes associated with the nuclear membrane. CLP might involve in this migration of 5LO, which is most probably of importance for regulation of the cellular 5LO activity (Kuebler et al. 2000). Site-directed mutagenesis studies have suggested an important role for Lys131 of CLP in mediating 5LO binding (Provost et al. 2001a). Further details of the interactions between CLP and 5LO, and the interaction and association of 5LO with the cell cytoskeleton, are still unknown.
CLP belongs to the actin-depolymerizing factor/cofilin family and is thought to interact with F-actin and 5LO via partially different binding sites. Under physiological conditions, monomeric globular actin (G-actin) is in equilibrium with filamentous actin (F-actin), which forms the actin cytoskeleton and is responsible for maintaining and modifying cell shape in motility, phagocytosis, and cytokinesis. The actin cytoskeleton is regulated by numerous actin binding proteins, which interact with actin and regulate the cytoskeleton in cells. Several actin binding proteins in the actin-depolymerizing factor/cofilin family have been demonstrated to associate with actin via electrostatic interactions involving basic and acidic amino acid residues (Carlier et al. 1997; Fedorov et al. 1997; McGough 1998). Among them, actophorin severs actin filaments and sequesters actin monomers (Leonard et al. 1997; Blanchoin and Pollard 1998). Cofilin plays a central role in regulating cytoskeletal dynamics, and can bind either monomers or filaments, while its conserved N terminus residues are critical for interactions with actin (Lappalainen et al. 1997; Paavilainen et al. 2002; Guan et al. 2002; Pope et al. 2004). The three-dimensional structure of destrin, together with the crystal structures of yeast cofilin and Acanthamoeba actophorin, is beneficial for a deeper understanding of the molecular mechanism through which these proteins stimulate F-turnover in vivo (Hatanaka et al. 1996). CLP associates with F-actin but does not form a stable complex with globular actin. However, CLP binds to actin filaments with a stoichiometry of 1:2 (CLP:actin subunits) but could be cross-linked to only one subunit of actin. In transfected mammalian cells, site-directed mutagenesis indicates that CLP can bind F-actin both in vitro and in vivo. CLP is also found to be colocalized with actin fibers and Lys75 has been shown to be essential for this interaction (Provost et al. 2001b).
In this study, we report the crystal structure of the CLP expressed in Escherichia coli and its comparison with yeast cofilin and actophilin. The potential regions of CLP that interact with 5LO and F-actin are surveyed in detail. From our structural analysis, we observe that CLP forms a polymer in the crystal lattice with a repeat distance corresponding to that of F-actin. A speculative model for CLP polymer and F-actin binding is proposed.
Materials and methods
Protein expression and purification
The coding sequence of the CLP from liver cDNA library was cloned into the BamH I and Xho I sites of the GST-Tag expression plasmid pGEX-6p-1 (Amersham Biosciences). The resulting pGEX-CLP plasmids were used to transform E. coli BL21 (DE3) cells. The soluble GST fusion protein, GST–CLP, was purified by GST–glutathione affinity chromatography and cleaved with GST–rhinovirus 3C protease (Zhu et al. 2003). The recombinant CLP was further purified by using Superdex 75 (Pharmacia). The purified and concentrated CLP (20 mg/mL) was stored in 20 mM Tris • HCl (pH 8.0).
Crystallization and data collection
The protein solution of CLP used for crystallization contained 20 mM Tris (pH 8.0) and 20 mg/mL protein. Crystals were obtained using the hanging drop vapor diffusion technique with reservoir solutions containing 30% PEG4000, 0.1 M Tris HCl (pH 8.5), 0.2 M Sodium acetate, in a drop formed by mixing 1 μL of protein solution and 1 μL reservoir solution at 291 K.
X-ray data of CLP were collected on a Rigaku RU2000 rotating Cu Kα anode source to 1.9 Å. Data sets were processed using DENZO and SCALEPACK (Otwinowski and Minor 1997), Data collection statistics are summarized in Table 1. The crystal belongs to the space group P21 and its Matthews coefficient suggests the presence of a monomer in the asymmetric unit, corresponding to a solvent content of 24% (Matthews 1968).
Table 1.
Data collection and refinement statistics
| Data collection and refinement | Refinement statistics | ||
| Data collection | Resolution (Å) | 50.0–1.9 | |
| Space group | P21 | Number of molecules/asymmetric unit | 1 |
| Unit cell | R-factor (%) | ||
| a/b/c (Å) | a = 25.6 | Working set | 16.6 |
| b = 55.2 | Test set | 21.8 | |
| c = 37.4 | RMSD | ||
| α/β/γ (°) | β = 96.0° | Bonds (Å) | 0.013 |
| Wavelength (Å) | 1.5418 | Angles (°) | 1.66 |
| Resolution (Å) | 50–1.9 | Ramachandran plot | |
| Completeness (%) | 96.0 (77.0) | Most favored | 93.4% |
| Reflections | Allowed | 6.6% | |
| Total | 25220 | Generously allowed | 0% |
| Unique | 7780 | Disallowed | 0% |
| Redundancy | 3.3 | ||
| Rmerge (%) | 6.9 (26.2) | ||
| I/σ(I) | 13.8 (3.0) | ||
Rmerge =ΣhΣl| Iih − <Ih> |/ΣhΣT <Ih>, where <Ih> is the mean of the observations Iih of reflection h.
Rwork = Σ (||Fobs|−Fcalc||)/Σ|Fobs|;
Rfree is the R factor for a subset (10%) of reflections that was selected prior refinement calculations and not included in the refinement.
Ramachandran plots were generated using PROCHECK (Laskowski et al. 1993).
Numbers in parentheses correspond to the highest resolution shell.
Structure determination and refinement
The CLP structure was determined by molecular replacement using a polyalanine model modified from a single monomer of actophorin (PDB code 1CNU; Blanchoin and Pollard 1998) as a search model. Actophorin shares only 18% sequence identity with CLP. A clear molecular replacement solution was found by cross-rotation function and translation function searches with the program CNS (Jones et al. 1991). Electron density maps were calculated to 2.5 Å from the MR model phases, and most of the hydrophobic residues were manually replaced by possible corresponding CLP residues using the program O (Brunger et al. 1998). According to maps calculated from the partially replaced model phases without refinement, further replacements were made for the remaining residues. The inserted residues were then added to the model according to a map calculated from the new postrefinement model phases. The resolution was extended to 1.9 Å data for further refinement and the model was subjected to simulated annealing, energy minimization, B-factor refinement, and water-picking using CNS. The refinement was alternated with manual model rebuilding using O-based on calculated difference Fourier maps. In the last stage of refinement, water molecules were allocated to peaks >3σ in the Fo − Fc map. The final model fits the electron density map very well with good geometry. The model includes 131 residues of CLP (of a total of 142 residues).
Results
CLP structure
The crystal structure of human CLP was determined to 1.9 Å resolution using the molecular replacement method with the actophorin crystal structure (PDB code 1CNU) as a search model (Blanchoin and Pollard 1998). Although the identity between CLP and actophorin is just 18%, only one molecule is located in the asymmetric unit. The R and free-R factors of the refined CLP model are 16.6% and 21.8%, respectively, with root-mean-square deviations (RMSD) of 0.013 Å for bond lengths and 1.74° for bond angles. All residues are well defined, with the exception of the C-terminal residues from 132 to 142. In addition, a Ser residue, which is induced by the clone, is found before the N-terminal methionine (Met 1). The final model has excellent stereochemistry with 93.4% of residues in the most favored region of the Ramachandran plot calculated by PROCHECK and no residues in disallowed regions.
The human CLP crystal belongs to the space group P21 and has unit cell dimensions of a = 25.6 Å, b = 55.2 Å, c = 37.5 Å, and β = 96.0°. Two molecules are packed tightly in a unit cell with only 24% of the volume occupied by solvent. Like other members of the actin depolymerizing protein superfamily, the basic secondary structure arrangement is a central β-sheet surrounded by helices, with very tight hydrophobic cores located on either side of the sheet (Fig. 1 ▶). The β-sheet consists of six strands: four antiparallel strands—β2 (25–33), β3 (34–43), β4 (55–67), β5 (72–83)—and two βstrands—β1 (3–6), β6 (109–115)—that are parallel with strands β3 and β5, respectively. A β-bulge is formed by Gly 39 and Glu 40 in the middle of the β3 strand. The central β-sheet is flanked on both sides by a total of six helices. Two α-helices, α1 (6–19) and α3 (87–98), and the distorted helix α4 (98–105) are located on the N-terminal side, which is a common structural feature of proteins belonging to this family. One 310 helix α5 (116–121) and two other α-helices, α2 (44–53) and α6 (121–131), are located on the C-terminal side. β2 and β3, α2 and β4, β5 and α3 are each linked by type I β-turns; α1 and β2, β4 and β5, α4 and β6, by coils; β3 and α2, β6 and α5 are linked directly. N-terminal residues (3–0) are extended straight from β1 while C-terminal residues after Lys 131 cannot be defined by the electron density map.
Figure 1.

The structure of CLP. A ribbon drawing of the structure of CLP prepared with MOLSCRIPT (Kraulis 1991). The secondary structure elements are labeled. Two IgG antibody binding loops, loop α1–β2 and loop α4–β6, are shown in yellow.
A notable feature of the molecular surface of human CLP (Fig. 3B ▶) is the distribution of positive and negative charges on opposite sides of the structure. The positively charged surface is defined by the N terminus, α3, α4, α5, α6 helices and the β6 strand; while the negatively charged surface is defined by α1, α2 helices and the β3 strand. Both sides are parallel to the BC plane of the cell. The positively charged surface residues can be divided into two groups: Lys 72, Arg 73, Lys 75, Arg 63, Lys 130, Lys 131, Lys 110, and Arg 18 are located in the top area; while the N terminus, Lys 4, Lys 7, Arg 11, Arg 57, Lys 93, Arg91, and Lys 30 are all located in the bottom area. Lys 118 is isolated on the surface. Located in the middle of the negatively charged side are the following negatively charged residues: Asp 6, Glu 8, Asp 19, Asp 32, Glu 40, Glu 44, Asp 54, Asp 55, Glu 84, Asp 116, Glu 121, Glu 122, and Asp 123. The residue Lys 131, which is important for binding 5LO, protrudes from the surface of the CLP molecule and is surrounded by a hydrophobic region and both positively charged and negatively charged areas.
Figure 3.
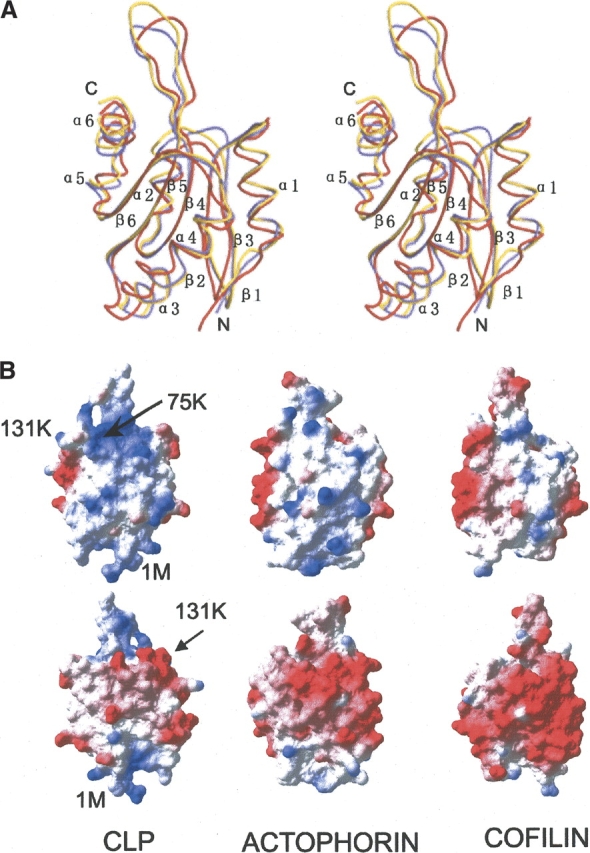
Comparison of CLP structure with actophorin and yeast cofilin. (A) Stereo view of CLP (red) superimposed with cofilin (yellow) and actophorin (purple). The figure was prepared with MOLSCRIPT. (B) Comparison of the CLP molecular surface charge distribution with those of cofilin and actophorin. The positive charges are shown in the first row; the negative, which is the opposite sides of the positive, are shown in the second row. The surface of CLP is more positively charged in the N-terminal and β4–β5 loop regions. This figure was prepared with SPDBV (Guex and Peitsch 1997).
According to the work by Nakatsura and colleagues, human CLP may cause humoral immune responses as a pancreatic cancer antigen (Nakatsura et al. 2002). The positive peptides, which bind to IgG antibodies, correspond to residues 15–24 and 104–113. The determination of the human CLP structure provides a structural basis for further investigation. Peptide 15–24, a negatively charged region, includes one turn of the α1 helix and the α1–β2 loop. Peptide 104–113 includes the α4–β6 loop and five residues in the β6 strand. α1–β2 and α4–β5 are adjacent loops.
Comparison of CLP structure with actophorin and yeast cofilin
Comparisons were made between the human CLP structure and homologous structures in the Protein Data Bank. Two structures, actophorin (1CNU) and yeast cofilin (1CFY) (Fedorov et al. 1997; Blanchoin and Pollard 1998), were found by sequence alignment from the Protein Data Bank using the program BLAST. Both structures can be superimposed with human CLP with an RMSD of 1.4 Å. Actin and cofilin belong to the actin depolymerizing protein superfamily and can bind G-actin and F-actin. Through the binding of G-actin, they can be prevented from forming polymers or from severing F-actin. Sequence identities are low with only 18% identity between CLP and actophorin, and 17% between CLP and yeast cofilin. In contrast, the sequence identity between actophorin and yeast cofilin is 43% (Fig. 2 ▶).
Figure 2.

Sequence alignment of human CLP with yeast cofilin and actophorin. The sequence alignment was made using the program CLUSTALW (Thompson et al. 1994) and modified according to structure alignment. The conserved residues are shaded.
The central β-sheets of CLP, actophorin, and yeast cofilin match each other very closely when superimposed together, and the secondary structures of the three proteins share the same arrangement (Fig. 3A ▶). Clear differences in length and shape are observed in several loop areas. The length of the α1–β2 loop in CLP is one residue greater than the equivalent loops of actophorin or yeast cofilin, causing it to stretch out further. The β2–β3 loop of CLP is two residues shorter and narrower than the equivalent loop in the other two structures. The β3–α2 loop of CLP is also two residues shorter than the corresponding loop in yeast cofilin and three residues shorter than in actophorin. In fact, β3 and α2 are linked directly in CLP, making the loop considerably less stretched. The β4–β5 loop of CLP is two residues shorter than the equivalent loop in yeast cofilin but equal in length to that in actophorin. The end of this loop is the widest of the three. Surrounded by loops α1–β2, β3–α2, α4–β6, and the helix α6, the β4–β5 loop stretches out of the β-sheet. All other corresponding loops in the three structures are equal in length.
The positional differences of the six helices are also notable when comparing the structures of CLP, actophorin, and yeast cofilin (Fig. 3A ▶). On the N-terminal side of the sheet, actophorin and yeast cofilin match each other very well. Relative to these two structures, the C-terminal end of α1 in CLP is closer to the β-sheet, and helix α3 is shifted by about 1 Å toward the strand β6, while α4 is closer to the β-sheet. On the C-terminal side, helix α2 is moved toward the β3 direction of the sheet. Because of a conserved hydrogen bond between Tyr 40 OH and Leu 120 O, the joint part of α5 and α6 moves together with the C-terminal end of α2.
The first visible N-terminal residues differ between CLP, actophorin, and yeast cofilin. There are two disordered N-terminal residues in the actophorin structure and three in yeast cofilin. However, in the case of CLP, an additional N-terminal residue induced by the clone is well defined in the electron density. The last visible C-terminal residues occur at the same position in all three structures, with varying lengths of undetectable disordered residues. The flexible C-terminal tail for CLP is 11 residues in length, while those for actophorin and yeast cofilin are 3 and 5, respectively.
Both CLP and yeast cofilin possess a KRSK motif located in the β4–β5 loop, which is believed to be related to F-actin binding (Provost et al. 2001b), while the last lysine residue (Lys 75) is known to be critically important. In actophorin, the equivalent motif is QRNK. In the proposed CLP–actin binding area, most residues have similar features with actophorin or yeast cofilin with the exception of Leu 89, Glu 103, Lys 110, Val 113, Ser 127, and Glu128. These differences may be at the cost of the ability of CLP to bind G-actin.
Surface charge distributions of the three proteins have a common feature: One side is positive and the opposite is negative (Fig. 3B ▶). Comparing molecular surfaces of the three structures, the CLP surface contains notably more positively charged residues than either actophorin and yeast cofilin in both the N-terminal and β4–β5-loop regions (Fig. 3B ▶). The surface of yeast cofilin is the least positively charged of the three structures presented here. The additional positive residues in CLP are Lys 4, Lys 7, Arg 11, Arg 57, and Lys 93 in the N-terminal region, and Arg 63, Lys 110, and Lys 126 in the β3–β4 loop area. In addition, CLP is the least negatively charged of the three structures, while the surface of yeast cofilin is the most negatively charged.
CLP polymerization
Human actin-depolymerizing factor (ADF) and cofilin are also actin-binding proteins belonging to the actin-depolymerizing factor (ADF) family. Human cofilin, a homolog of CLP, possesses the tendency for self-association, and can form dimers and oligomers that exhibit a biological activity distinct from the monomer. Adjacent cofilin monomers associate via two intermolecular disulfide bonds. These cofilin dimers and oligomers result in increased viscosity and light scattering of F-actin solutions, and possess actin bundling activity (Pfannstie et al. 2001). This property of human cofilin raises the possibility that human CLP might also form dimers and oligomers. Analysis of the CLP structure rules out the possibility of intermolecular disulfide bonds because the two cysteines in the CLP sequence are buried within the structure. However, from the crystal lattice, we observe the possibility of a CLP aggregated protein state.
Surveying molecular contacts within cell lattices, one type is much stronger than others. The two contact molecules are related by a 21 axis with a buried area of 2840 Å2 for one interface of this type. The CLP molecular surface is calculated to be only 7472 Å2 using a 1.4 Å probe radius (ViewerPro, Accelrys). Repeating this 21 operation, more molecules are involved in forming this type of contact, thus generating a polymer along the cell axis b (Fig. 4A ▶) with a repeat distance of 55.2 Å. Interestingly, this kind of polymer is not observed in the crystal lattices of either cofilin or actophorin.
Figure 4.
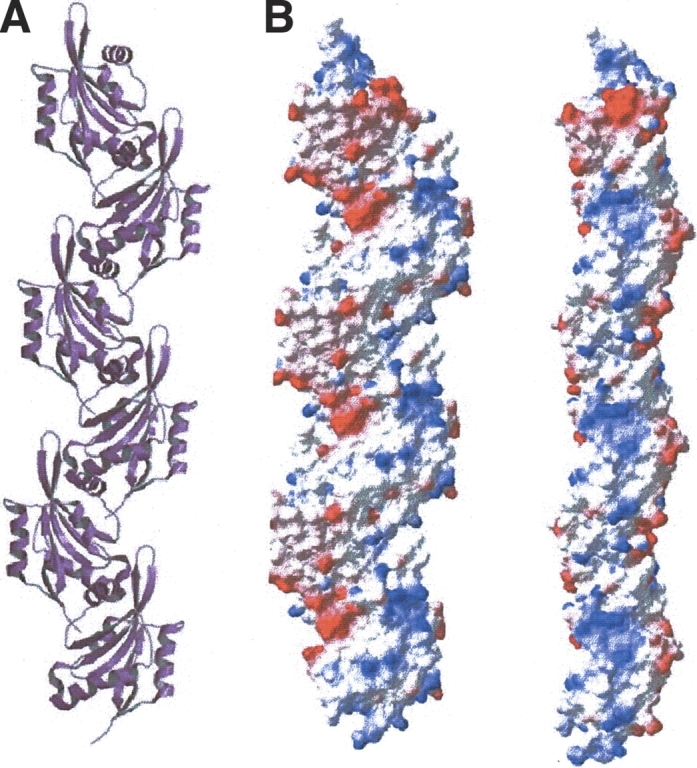
CLP Integration and polymerization. (A) A ribbon drawing of CLP polymer along cell axis b. This figure was prepared with MOLSCRIPT. (B) The distribution of molecular surface charges in the CLP polymer, shown in two views perpendicular to each other. This figure was prepared with SPDBV.
Eight structural elements are involved in intermolecular interactions. They are the N terminus, β2–β3, α2–β4, β5–α3 loops at the N-terminal end of the molecule, and α2, α5, α6 helices and loop β4–β5 at the β4–β5 end. Very few hydrophobic residues are located in the contact region. The hydrophobic interactions between the two contact surfaces are mediated mainly by the arms of long side chains. Residues forming hydrophobic interactions are Lys 118, Glu 121, Asp 123, Glu 122, Glu 46, Glu 44, Lys 126, Lys 130, Thr 65, and Arg 117 at the β4–β5 end; and Thr 53, Asp 55, Asn 85, Ser 87, Leu 89, Gln 90, Gly 33G, Val 56, and Met 1 at the N-terminal end. Eight hydrogen bonds, which directly link the two contact surfaces together, were found. The corresponding donor receptor pairs are 90OE1, 46NE2; 67O, 85ND2; 67O, 54OD1; 68O, 54OD1; 72NE, 84OE1; 72NE, 5OD1; 122OE2, 87N; 126NZ, 85O. Four additional hydrogen bonds between the two contact surfaces involve water molecules in the middle. The groups involved in these hydrogen bonds are 55O, S42, 126NE; 118O, S3, 33N; 87OG, S1, 46N; and 121OE1, S17, 57NH2, 93NZ. Through these contacts, the positive and the negative charges compensate each other very well. Figure 4B ▶ illustrates the two sides of the charged surface of the CLP polymer.
Discussion
Location of antibody recognition region
The antibody recognition loops α1–β2 and α4–β5 are adjacent in the N-terminal side of the molecule (Fig. 1 ▶), suggesting that these two loops are recognized by antibodies at the same time. The region containing the α1–β2 and α4–β5 loops is full of electrostatic charges. Lys 75, which is important for F-actin binding, and Lys 131, important for 5LO binding, are also located in this area. Furthermore, this area is not involved in the proposed interaction for CLP polymerization. Consequently, antibodies, F-actin, 5LO, and other proteins could interact with the CLP polymer if it exists other than in the crystal lattice. In fact, this area is the only loop area left unhindered after polymerization. In this case, it is possible that the region surrounding Lys 75 forms the binding area for most proteins that interact with CLP.
The function of CLP’s long disordered C terminus
It has previously been reported that the ability of F-actin to bind UNC-60B, an ADF/cofilin family protein, will be abolished when its flexible C-terminal tail is truncated or mutated (Ono et al. 2001). CLP also has a long flexible C terminus composed of 11 residues: AGGANYDAQTE. The first four residues form a hinge region, while the last seven make a good combination for molecular recognition. This flexible C terminus is sited not far from the region containing α1–β2, α4–β5 and β4–β5 loops. As a consequence, the flexible C terminus of CLP may be used to bind F-actin and/or other proteins (Fig. 5A ▶).
Figure 5.

A model for CLP polymer binding with F-actin and other proteins. (A) CLP polymer and F-actin have the same repeating distance. The morphologies of the two polymers match each other very well. Its long flexible C-terminal arm may help CLP binding other molecules. (B) A model for CLP polymer binding with F-actin. (C) CLP-F-actin copolymer binding with other proteins (red). The F-actin diagram is provided by Amy McGough (Baylor College of Medicine, Houston, TX). The models in B and C were prepared with Photoshop, and the charged surface model and the CPK (red) model were prepared with SPDBV and MOLSCRIPT, respectively.
A model for CLP-F-actin binding
As previously discussed, CLP has been shown to bind to F-actin. If CLP binds to F-actin in its polymerized form, then its repeat distance should correspond with that of F-actin. Comparing the repeat distance of our CLP polymer along the b axis in the crystal lattice with the repeat distance of F-actin subunits, we found that they are exactly equal, as we expected. The structure of F-actin resembles a twisted ladder with a 55 Å interval between consecutive rungs (Fig. 5A ▶). If the CLP polymer twists exactly like F-actin (Steinmetz et al. 1997), the protruding parts of the CLP polymer would be ideally placed to insert into the holes between frames and rungs, and thus bind into the F-actin helix grooves (Fig. 5B ▶). If the CLP polymer could not be twisted exactly like F-actin, however, it would still be possible for CLP oligomers to bind into the grooves. The proposed CLP contact surface is full of long, charged side chains, as illustrated in Figure 4B ▶. In the protruding part of the surface, both positive and negative charges are intensively distributed.
In the CLP–F-actin contact surface, besides the complementary morphology of the two polymers, their flexible C-terminal tails could assist the CLP polymer to bind to F-actin. The symmetry-related surface in the CLP polymer will face the surrounding environment. Again, the corresponding flexible C-terminal tails will help the CLP polymer to interact with other protein molecules (Fig. 5C ▶). CLP bound to F-actin could attach specific proteins, such as 5LO, to the cytoskeleton for protein localization or migration.
Conclusions
In summary, we have successfully determined the crystal structure of CLP to 1.9 Å resolution. Based on our structural analysis, we found that CLP can form a polymer along the b axis in the crystal lattice, unlike yeast cofilin and actophilin. The repeat distance of the CLP polymer is precisely the same as for F-actin, and thus a model for the CLP polymer and F-actin binding has been proposed. However, further work is required to confirm that CLP does indeed have a tendency for self-association and to test our model for CLP–F-actin binding. The determination of the human CLP structure should provide a structural basis for further investigation.
Acknowledgments
We thank Feng Xu and Feng Gao for assistance with data collection. This work was supported by the following grants: Project “863” 2002BA711A12; Project “973” no. G1999075602. Xuemei Li was supported by the NSFC.
Article published online ahead of print. Article and publication date are at http://www.proteinscience.org/cgi/doi/10.1110/ps.04937304.
References
- Blanchoin, L. and Pollard, T.D. 1998. Interaction of actin monomers with Acan-thamoeba actophorin (ADF/cofilin) and profilin. J. Biol. Chem. 273 25106–25111. [DOI] [PubMed] [Google Scholar]
- Brunger, A.T., Adams, P.D., Clore, G.M., DeLano, W.L., Gros, P., Grosse-Kunstleve, R.W., Jiang, J.S., Kuszewski, J., Nilges, M., Pannu, N.S., et al. 1998. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr. D Biol. Crystallogr. 54 905–921. [DOI] [PubMed] [Google Scholar]
- Carlier, M.F., Laurent, V., Santolini, J., Melki, R., Didry, D., Xia, G.X., Hong, Y., Chua, N.H., and Pantaloni, D. 1997. Actin depolymerizing factor (ADF/cofilin) enhances the rate of filament turnover: Implication in actin-based motility. J. Cell Biol. 136 1307–1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Hostos, E.L., Bradtke, B., Lottspeich, F., and Gerisch, G. 1993. Coactosin, a 17 kDa F-actin binding protein from Dictyostelium discoideum. Cell Motil. Cytoskel. 26 181–191. [DOI] [PubMed] [Google Scholar]
- Fedorov, A.A., Lappalainen, P., Fedorov, E.V., Drubin, D.G., and Almo, S.C. 1997. Structure determination of yeast cofilin. Nat. Struct. Biol. 4 366–369. [DOI] [PubMed] [Google Scholar]
- Guan, J.Q., Vorobiev, S., Almo, S.C., and Chance, M.R. 2002. Mapping the G-actin binding surface of cofilin using synchrotron protein footprinting. Biochemistry 41 5765–5775. [DOI] [PubMed] [Google Scholar]
- Guex, N. and Peitsch, M.C. 1997. SWISS-MODEL and the Swiss-PdbViewer: An environment for comparative protein modeling. Electrophoresis 18 2714–2723. [DOI] [PubMed] [Google Scholar]
- Hatanaka, H., Ogura, K., Moriyama, K., Ichikawa, S., Yahara, I., and Inagaki, F. 1996. Tertiary structure of destrin and structural similarity between two actin-regulating protein families. Cell 85 1047–1055. [DOI] [PubMed] [Google Scholar]
- Jones, T.A., Zou, J.Y., Cowan, S.W., and Kjeldgaard. 1991. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr. A 47 110–119. [DOI] [PubMed] [Google Scholar]
- Kraulis, P.J. 1991. MOLSCRIPT: A program to produce both detailed and schematic plots of protein structures. J. Appl. Crystallogr. 24 946–950. [Google Scholar]
- Kuebler, W.M., Borges, J., Sckell, A., Kuhnle, G.E., Bergh, K., Messmer, K., and Goetz, A.E. 2000. Role of L-selectin in leukocyte sequestration in lung capillaries in a rabbit model of endotoxemia. Am. J. Respir. Crit. Care Med. 161 36–43. [DOI] [PubMed] [Google Scholar]
- Lappalainen, P., Fedorov, E.V., Fedorov, A.A., Almo, S.C., and Drubin, D.G. 1997. Essential functions and actin-binding surfaces of yeast cofilin revealed by systematic mutagenesis. EMBO J. 16 5520–5530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskowski, R.A., MacArthur, M.W., Moss, D.S., and Thornton, J.M. 1993. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 26 283–291. [Google Scholar]
- Leonard, S.A., Gittis, A.G., Petrella, E.C., Pollard, T.D., and Lattman, E.E. 1997. Crystal structure of the actin-binding protein actophorin from Acanthamoeba. Nat. Struct. Biol. 4 369–373. [DOI] [PubMed] [Google Scholar]
- Liu, T.X., Zhang, J.W., Tao, J., Zhang, R.B., Zhang, Q.H., Zhao, C.J., Tong, J.H., Lanotte, M., Waxman, S., Chen, S.J., et al. 2000. Gene expression networks underlying retinoic acid-induced differentiation of acute promyelocytic leukemia cells. Blood 96 1496–1504. [PubMed] [Google Scholar]
- Matthews, B.W. 1968. Solvent content of protein crystals. J. Mol. Biol. 33 491–497. [DOI] [PubMed] [Google Scholar]
- McGough, A. 1998. F-actin-binding proteins. Curr. Opin. Struct. Biol. 8 166–176. [DOI] [PubMed] [Google Scholar]
- Nakatsura, T., Senju, S., Yamada, K., Jotsuka, T., Ogawa, M., and Nishimura, Y. 2001. Gene cloning of immunogenic antigens overexpressed in pancreatic cancer. Biochem. Biophys. Res. Commun. 281 936–944. [DOI] [PubMed] [Google Scholar]
- Nakatsura, T., Senju, S., Ito, M., Nishimura, Y., and Itoh, K. 2002. Cellular and humoral immune responses to a human pancreatic cancer antigen, coactosin-like protein, originally defined by the SEREX method. Eur. J. Immunol. 32 826–836. [DOI] [PubMed] [Google Scholar]
- Ono, S., McGough, A., Pope, B.J., Tolbert, V.T., Bui, A., Pohl, J., Benian, G.M., Gernert, K.M., and Weeds, A.G. 2001. The C-terminal tail of UNC-60B (actin depolymerizing factor/cofilin) is critical for maintaining its stable association with F-actin and is implicated in the second actin-binding site. J. Biol. Chem. 276 5952–5958. [DOI] [PubMed] [Google Scholar]
- Otwinowski, Z. and Minor, W. 1997. Processing of X-ray diffraction data collected in oscillation mode. In Macromolecular crystallography, part A, Vol. 276, pp. 307–326. Academic Press, New York. [DOI] [PubMed]
- Paavilainen, V.O., Merckel, M.C., Falck, S., Ojala, P.J., Pohl, E., Wilmanns, M., and Lappalainen, P. 2002. Structural conservation between the actin monomer-binding sites of twinfilin and actin-depolymerizing factor (ADF)/cofilin. J. Biol. Chem. 277 43089–43095. [DOI] [PubMed] [Google Scholar]
- Pfannstie, J., Cyrklaff, M., Habermann, A., Stoeva, S., Griffiths, G., Shoeman, R., and Faulstich, H. 2001. Human cofilin forms oligomers exhibiting actin bundling activity. J. Biol. Chem. 276 49476–49484. [DOI] [PubMed] [Google Scholar]
- Pope, B.J., Zierler-Gould, K.M., Kuhne, R., Weeds, A.G., and Ball, L.J. 2004. Solution structure of human cofilin: Actin binding, pH sensitivity, and relationship to actin-depolymerizing factor. J. Biol. Chem. 279 4840–4848. [DOI] [PubMed] [Google Scholar]
- Provost, P., Samuelsson, B., and Radmark, O. 1999. Interaction of 5-lipoxygenase with cellular proteins. Proc. Natl. Acad. Sci. 96 1881–1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Provost, P., Doucet, J., Hammarberg, T., Gerisch, G., Samuelsson, B., and Radmark, O. 2001a. 5-Lipoxygenase interacts with coactosin-like protein. J. Biol. Chem. 276 16520–16527. [DOI] [PubMed] [Google Scholar]
- Provost, P., Doucet, J., Stock, A., Gerisch, G., Samuelsson, B., and Radmark, O. 2001b. Coactosin-like protein, a human F-actin-binding protein: Critical role of lysine-75. Biochem. J. 359 255–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinmetz, M.O., Stoffler, D., Hoenger, A., Bremer, A., and Aebi, U. 1997. Actin: From cell biology to atomic detail. J. Struct. Biol. 119 295–320. [DOI] [PubMed] [Google Scholar]
- Thompson, J.D., Higgins, D.G., and Gibson, T.J. 1994. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22 4673–4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu, J., Li, P., Wu, T., Gao, F., Ding, Y., Zhang, C.W., Rao, Z., Gao, G.F., and Tien, P. 2003. Design and analysis of post-fusion 6-helix bundle of heptad repeat regions from Newcastle disease virus F protein. Protein Eng. 16 373–379. [DOI] [PubMed] [Google Scholar]