

Abstract
Pineal hormone melatonin (N-acetyl-5-methoxytryptamine) is thought to modulate the calcium/calmodulin signaling pathway either by changing intracellular Ca2+ concentration via activation of its G-protein–coupled membrane receptors, or through a direct interaction with calmodulin (CaM). The present work studies the direct interaction of melatonin with intact calcium-saturated CaM both experimentally, by fluorescence and nuclear magnetic resonance spectroscopies, and theoretically, by molecular dynamics simulations. The analysis of the experimental data shows that the interaction is calcium-dependent. The affinity, as obtained from monitoring 15N and 1H chemical shift changes for a melatonin titration, is weak (in the millimolar range) and comparable for the N- and C-terminal domains. Partial replacement of diamagnetic Ca2+ by paramagnetic Tb3+ allowed the measurement of interdomain NMR pseudocontact shifts and residual dipolar couplings, indicating that each domain movement in the complex is not correlated with the other one. Molecular dynamics simulations allow us to follow the dynamics of melatonin in the binding pocket of CaM. Overall, this study provides an example of how a combination of experimental and theoretical approaches can shed light on a weakly interacting system of biological and pharmacological significance.
Keywords: melatonin, calmodulin, molecular dynamics, NMR, fluorescence, weak interactions
Calmodulin (CaM) is one of the most abundant, ubiquitous, and conserved proteins in eukaryotic biology. Among vertebrates, the amino acid sequence of CaM appears to be completely invariant (Hoeflich and Ikura 2002). Higher eu-karyotes, including humans, possess three distinct bona fide CaM genes differentially regulated but which encode identical proteins (Hickie et al. 1983; Nojima 1989). The structure of CaM has a dumbbell shape, with two homologous Ca2+-binding domains linked together by a flexible tether (Barbato et al. 1992). CaM is known to interact with a large number of proteins important for Ca2+-dependent intracellular signaling, thus enabling the cell to control biological processes as diverse as muscle contraction, fertilization, cell proliferation, vesicular fusion, and apoptosis (Berridge et al. 1998). Despite their physiological diversity, two features appear to be common in the sequences of CaM binding partners: the presence of two hydrophobic anchors, one of which is often a tryptophan, normally separated by 14 residues, and the presence of a large number of basic residues (Crivici and Ikura 1995). Each of the hydrophobic anchors interacts mainly with one of the CaM domains.
Small, hydrophobic molecules bind to CaM and modify its function by inhibiting the interaction with other proteins (Prozialeck and Weiss 1982; Weiss et al. 1985; Cook et al. 1994; Vandonselaar et al. 1994; Craven et al. 1996; Osawa et al. 1998; Harmat et al. 2000). Some of these compounds used as pharmacological agents to block CaM-mediated enzyme activation may act as antipsychotics, muscle relaxants, antidepressants, minor tranquilizers, and local anesthetics (Weiss et al. 1982; Zhang et al. 1990). CaM antagonists (such as trifluoperazine [TFP] and many others) are also being evaluated for the treatment of cancers that are both sensitive and resistant to conventional anticancer drugs (Ford and Hait 1990; Liu et al. 2002), because diseases characterized by unregulated cell growth have been shown to present elevated levels of Ca2+-CaM (Hickie et al. 1983; Dai et al. 2002).
One of the endogenous CaM antagonists is thought to be the pineal hormone melatonin (N-acetyl-5-methoxytryptamine) (Fig. 1 ▶; Benitez-King et al. 1993, 1996; Cardinali et al. 1997). Melatonin is involved in several physiological functions, including the entrainment of both seasonal and circadian rhythms. It has been shown that melatonin prevents the Ca2+/CaM inhibition of microtubule polymerization both in situ and in vitro assays (Huerto-Delgadillo et al. 1994). More recently, it has been suggested that rat hypo-thalamic and striatal nitric oxide synthase (NOS) inhibition is regulated by the interaction of melatonin and some melatonin-related kynurenines with CaM (Pozo et al. 1997; León et al. 2000), although in the hamster retina NOS inhibition seems to be independent of CaM (Saenz et al. 2002).
Figure 1.

Structure of melatonin.
Melatonin has also been shown to exhibit cytotoxic activity in cancer cells, and is thus used as a chemotherapeutic drug. Both at physiological and pharmacological concentrations, melatonin acts as a differentiating agent in some cancer cells, lowering their invasive and metastatic status through alterations in adhesion molecules and maintenance of gap junctional intercellular communication (Blask et al. 2002). In other cancer cell types, melatonin, either alone or in combination with other agents, induces apoptotic cell death (Blask et al. 2002). Melatonin/CaM interactions may play an important role in mediating the growth-inhibitory effect of melatonin on human breast cancer cells (Dai et al. 2002). Despite its pharmacological and physiological significance, a structural characterization of the CaM/melatonin interaction is still lacking, and even the strength of the interaction remains controversial (Benitez-King et al. 1996; Ouyang and Vogel 1998).
In the present paper, conclusive atomistic evidence of the CaM/melatonin interaction is provided by fluorescence and nuclear magnetic resonance (NMR) studies, and by molecular dynamics (MD) simulations. NMR chemical shift mapping shows a significant and reversible binding of melatonin to both domains of CaM. Molecular docking and MD simulations provide a further atomistic characterization of the CaM–melatonin complex.
Results
Fluorescence spectroscopy characterization of CaM:melatonin interaction
Fluorescence intensity measurements were used to study the interaction of CaM with melatonin following the approach used previously by other researchers (Ouyang and Vogel 1998). Melatonin (0.5 mM) was titrated with Ca4-CaM up to a final concentration of 1.3 mM and fluorescence changes were monitored at 340 nm (Fig. 2 ▶). Because the interaction is weak (Kd > 2 mM) it was not possible to determine a precise value of the dissociation constant or to establish the stoichiometry of the interaction. A control titration with Ca2+-free CaM (apo-CaM) showed no change in fluorescence intensity, showing that the interaction is calcium-dependent.
Figure 2.

Titration of melatonin with Ca4-CaM monitored by fluorescence intensity changes at 340 nm.
NMR monitoring of melatonin binding to CaM
2D 1H-15N HSQC NMR spectra was used to map the interaction of melatonin onto each of the domains of CaM. The NMR spectral changes for the 15N-labeled CaM signals upon melatonin binding occur in fast exchange on the NMR chemical shift timescale leading to signals with averaged chemical shifts for the nuclei exchanging between bound and unbound species. Figure 3A ▶ shows the superposition of 1H-15N HSQC spectra taken at different [melatonin]/[CaM] molar ratios, showing the progressive shift of peaks corresponding to selected HN signals during the titration. Figure 3B ▶ shows changes in 1H and 15N chemical shifts observed upon complexation of CaM with melatonin for some representative residues.
Figure 3.

(A) Superposition of portions of 2D 15N-1H HSQC spectra at different melatonin additions showing the progressive shift of the HN signals with increasing melatonin concentration. (B) Melatonin titration plots at 35°C monitored by 15N chemical shifts for residues Glu 140 and Ala 88. (C) Melatonin titration plots at 35°C monitored by 1H chemical shifts for residues Leu 69 and Arg 74.
Independent fitting of the binding curves from representative residues of each of the two CaM domains suggests that the C-domain binds with comparable, although possibly marginally higher, affinity than the N-domain (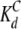 of 5 mM vs.
of 5 mM vs. 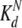 of 7 mM, Table 1). As expected for such weak binding contacts, no plateau could be reached in the titration even at high CaM–melatonin ratios, and no intermolecular NOE effects could be observed.
of 7 mM, Table 1). As expected for such weak binding contacts, no plateau could be reached in the titration even at high CaM–melatonin ratios, and no intermolecular NOE effects could be observed.
Table 1.
Estimation of CaM-melatonin Kd for N- and C-domains from 15N and 1H chemical shifts
| Domain | Residue | Kd (mM) | Errora | Initial (ppm) | Error | Final (ppm) | Error | χ2 |
| A. 15N chemical shifts | ||||||||
| N | Ser17 | 3.3 | 1.0 | 112.53 | 0.02 | 113.07 | 0.05 | 1.29 |
| Asp20 | 6.9 | 1.2 | 117.81 | 0.02 | 117.38 | 0.10 | 1.88 | |
| Ile27 | 10.4 | 2.0 | 127.08 | 0.02 | 127.28 | 0.13 | 1.34 | |
| Val55 | 8.6 | 1.2 | 108.46 | 0.02 | 110.56 | 0.16 | 1.58 | |
| Leu69 | 6.3 | 1.4 | 118.89 | 0.02 | 119.38 | 0.13 | 1.21 | |
| Met72 | 7.1 | 1.8 | 116.62 | 0.02 | 117.74 | 0.14 | 2.29 | |
| Arg74 | 7.8 | 1.7 | 116.75 | 0.02 | 115.48 | 0.14 | 1.89 | |
| Average | 7.2 | 2.2b | ||||||
| C | Ala88 | 6.3 | 0.8 | 122.15 | 0.02 | 120.28 | 0.11 | 1.40 |
| Met124 | 3.8 | 1.2 | 119.64 | 0.02 | 120.15 | 0.08 | 1.82 | |
| Glu127 | 4.5 | 0.9 | 116.02 | 0.02 | 116.92 | 0.08 | 1.52 | |
| Glu140 | 6.0 | 1.5 | 119.91 | 0.01 | 120.59 | 0.08 | 1.43 | |
| Thr143 | 8.5 | 3.4 | 116.71 | 0.02 | 117.87 | 0.23 | 1.43 | |
| Met145 | 2.5 | 0.5 | 115.06 | 0.01 | 115.64 | 0.03 | 1.55 | |
| Thr146 | 5.4 | 0.8 | 110.63 | 0.022 | 109.30 | 0.09 | 0.89 | |
| Average | 5.3 | 1.9b | ||||||
| B. 1H chemical shifts | ||||||||
| N | Ile27 | 6.0 | 1.1 | 9.76 | 0.01 | 9.87 | 0.09 | 1.99 |
| Met36 | 4.6 | 1.0 | 8.43 | 0.01 | 8.37 | 0.06 | 1.71 | |
| Asn53 | 6.1 | 1.8 | 8.53 | 0.01 | 8.64 | 0.04 | 1.22 | |
| Ala57 | 7.7 | 1.6 | 8.49 | 0.01 | 8.31 | 0.02 | 1.01 | |
| Leu69 | 6.9 | 1.2 | 8.32 | 0.01 | 8.60 | 0.03 | 0.93 | |
| Met71 | 7.3 | 1.4 | 7.78 | 0.01 | 7.44 | 0.04 | 0.89 | |
| Arg74 | 7.8 | 1.9 | 7.57 | 0.01 | 7.14 | 0.04 | 1.96 | |
| Average | 6.5 | 1.2b | ||||||
| C | Ala88 | 8.0 | 1.4 | 7.94 | 0.01 | 7.43 | 0.04 | 1.72 |
| Arg90 | 4.0 | 0.4 | 7.67 | 0.01 | 7.78 | 0.047 | 1.71 | |
| Phe92 | 3.4 | 0.3 | 7.56 | 0.01 | 7.50 | 0.053 | 1.63 | |
| Val121 | 3.1 | 0.4 | 8.07 | 0.01 | 7.98 | 0.041 | 1.29 | |
| Ile125 | 7.9 | 1.9 | 7.92 | 0.01 | 8.21 | 0.036 | 1.45 | |
| Asn129 | 6.2 | 1.6 | 7.88 | 0.01 | 8.01 | 0.017 | 1.73 | |
| Val142 | 5.5 | 0.5 | 8.49 | 0.01 | 8.71 | 0.009 | 1.75 | |
| Average | 5.4 | 2.0b | ||||||
a The errors are the errors reported by the fitting program for the particular fit and are not the errors in the Kd itself.
b The error assigned to the average is simply the standard deviation for the seven individual values and the true error in the Kd may be substantially higher.
Figure 4 ▶, A and B, show the 1H and 15N chemical shift differences between [melatonin]/[CaM] molar ratios of 0 and 9 for all residues in CaM. Figure 4C ▶ presents a weighted average of the chemical shift changes observed for each residue, and gives a rough indication of the extent to which a residue is affected by complexation with melatonin. Complex formation significantly affects ~22% of the CaM resonances. The most significant changes are localized mainly in the hydrophobic pockets of either domain (Fig. 4D ▶).
Figure 4.
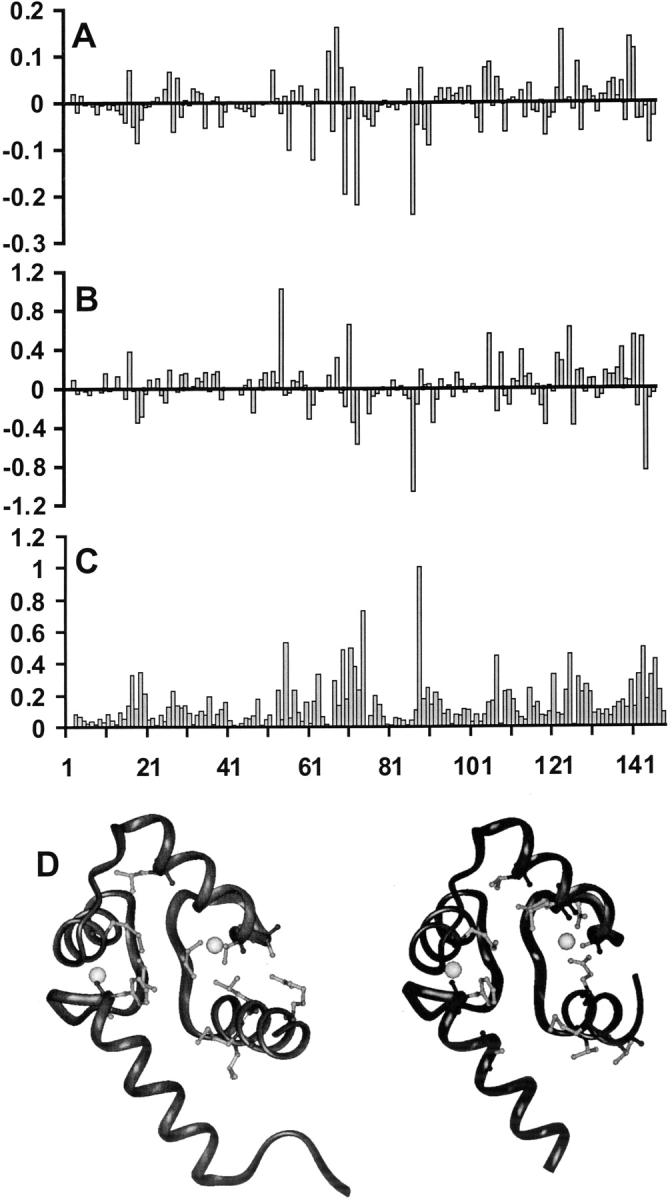
Plots of the change of (A) 1H and (B) 15N chemical shift for the HN signals occurring upon complexation of CaM with melatonin as a function of residue number. Graph (C) presents a normalized weighed average, [WA], over the chemical shift changes of both 1H and 15N, using the formula [WA] = Σ|δCaM-Mel – δCaM|/2|Δmax|, where δCaM-Mel and δCaM are the chemical shifts observed in the complex and in free CaM, and Δmax is the largest chemical shift change observed for each type of nucleus. The bottom figure (D) shows the mapping of the chemical shift changes onto the structure of CaM (PDB: 4CLN) for N-domain (left panel) and C-domain (right panel). The amino acids that have the larger chemical shift upon melatonin binding are shown with a light gray ball-and-stick model. The Ca2+ ions are represented as spheres.
Orientational freedom of the domains
To assess whether upon complexation the two CaM globular domains collapse, as observed in other CaM complexes, or remain independent from each other, we partially replaced diamagnetic Ca2+ by paramagnetic Tb3+. This allows the measurement of interdomain NMR pseudocontact shifts (PCS) and of residual dipolar couplings (RDC) induced by magnetic alignment (Biekofsky et al. 1999).
As shown in previous work, the Tb3+ added binds preferentially to the N-domain of CaM (Biekofsky et al. 1999). The binding of Tb3+ ions to the N-domain gives rise to three paramagnetic species, according to whether Tb3+ binds separately to sites I or II, or simultaneously to both these sites. For each amide NH group there will be up to four signals in an 1H-15N HSQC spectrum: the signal corresponding to the (Ca2+)4 species, and three signals corresponding to Tb3+(I)–(Ca2+)3, Tb3+(II)–(Ca2+)3, and Tb3+(I)–Tb3+(II)–(Ca2+)2–calmodulin (where I and II indicate the binding sites of the N-domain, III and IV those of the C-domain). Figure 5 ▶ shows the four peaks corresponding to C-domain residues Ile 100 and Val 136. Such patterns are seen throughout the spectrum for many residues. However, in crowded areas of the HSQC spectrum this leads to superposition of peaks, making the RDC and PCS measurement difficult. For residues very near to the Tb3+ ions, the NMR signals of Tb3+ species become broad and cannot be observed (mostly those in the N-domain). Such is the case, for example, of the N-domain residue Ile 27 shown in Figure 5 ▶.
Figure 5.

(A) A region of the 1H-15N HSQC spectrum for (Ca2+)4–CaM–melatonin showing the NH signals for Ile 27, Ile 100, and Asn 137. (B) The same region of the 1H-15N HSQC spectrum as in A for (Ca2+)4–CaM–melatonin with [Tb3+] = 0.66 eq added to the sample showing the NH signals for Ile 27, Ile 100, and Asn 137. The four signals for Ile 100 and Asn 137 correspond to, going in the upfield direction, the species (Ca2+)4, Tb3+(II)–(Ca2+)3, Tb3+(I)–(Ca2+)3, and Tb3+(I)–Tb3+(II)–(Ca2+)2–CaM (the assignments indicated on the spectrum). (C) The same region of the 1H-15N IPAP-HSQC spectrum as in A and B for (Ca2+)4–CaM–melatonin with [Tb3+] = 0.66 eq added to the sample showing both doublet components for the signals corresponding to the (Ca2+)4 and Tb3+/Ca2+ species found in B for Ile 27, Ile 100, and Asn 137.
The degree of alignment of the Tb3+-free C-domain depends on the degree of orientational freedom of this domain with respect to the N-domain containing the Tb3+ ions (Biekofsky et al. 1999). PCS and one-bond 1H-15N RDC values were measured for the Ca2+/Tb3+-CaM:melatonin complex (for a molar ratio of [Tb3+]/[CaM] = 0.66) and compared with those measured previously in the same conditions for CaM alone, where the domains have orientational freedom, and for the complex of CaM with a target peptide from skeletal muscle myosin light-chain kinase (MLCK), where the domains have fixed orientations with respect to each other (Biekofsky et al. 1999). Table 2 presents data measured for selected residues in these three systems to provide examples of the range of values obtained. The residues included in Table 2 were chosen from both domains, and show examples of both positive and negative pseudocontact shifts, and positive and negative dipolar coupling contributions to the NH splittings. For each residue shown in Table 2, the data measured for the four species in solution are given, except for the N-domain residues such as Gly 33 or Ala 57, for which the signals corresponding to some species could not be found.
Table 2.
Pseudocontact shifts and residual dipolar couplings for selected NH groups in (1) Ca2+/Tb3+–calmodulin–melatonin, (2) Ca2+/Tb3+–calmodulin, and (3) Ca2+/Tb3+–calmodulin–WFF peptide systems
| Ca2+/Tb3+–calmodulin–melatonin | Ca2+/Tb3+–calmodulin | Ca2+/Tb3+–calmodulin–WFF peptide | ||||
| Residue | δH (NH)a | 1H PCSb | δH (NH)a | 1H PCSb | δH (NH)a | 1H PCSb |
| A. Pseudocontact shifts (in ppm) | ||||||
| Gly33 | 8.62 | 0.00 | 8.66 | 0.00 | 8.74 | 0.00 |
| n/ae | — | n/ae | — | 5.57 | −3.17 | |
| Gly40 | 7.80 | 0.00 | 7.89 | 0.00 | 7.83 | 0.00 |
| 7.72 | −0.08 | 7.80 | −0.09 | 7.71 | −0.12 | |
| 7.51 | −0.29 | n/ae | — | 7.13 | −0.70 | |
| 7.08 | −0.72 | n/ae | — | 7.00 | −0.83 | |
| Ala57 | 8.42 | 0.00 | 8.58 | 0.00 | 8.03 | 0.00 |
| 8.35 | −0.07 | 7.34 | −1.24 | 6.56 | −1.47 | |
| Ile100 | 10.19 | 0.00 | 10.26 | 0.00 | 10.27 | 0.00 |
| 10.13 | −0.06 | 10.23 | −0.03 | 10.15 | −0.12 | |
| 10.09 | −0.10 | 10.17 | −0.09 | 10.02 | −0.25 | |
| 10.06 | −0.13 | 10.11 | −0.15 | 9.88 | −0.39 | |
| Leu116 | 7.98 | 0.00 | 7.98 | 0.00 | 8.11 | 0.00 |
| 7.96 | −0.02 | 8.01 | +0.03 | 8.18 | +0.07 | |
| 7.95 | −0.03 | 8.04 | +0.06 | 8.32 | +0.21 | |
| 7.93 | −0.05 | 8.06 | +0.08 | 8.38 | +0.27 | |
| Asn137 | 9.60 | 0.00 | 9.66 | 0.00 | 9.64 | 0.00 |
| 9.54 | −0.06 | 9.61 | −0.05 | 9.55 | −0.09 | |
| 9.50 | −0.10 | 9.56 | −0.10 | 9.41 | −0.23 | |
| 9.45 | −0.15 | 9.50 | −0.16 | 9.31 | −0.33 | |
| Ca2+/Tb3+–calmodulin–melatonin | Ca2+/Tb3+–calmodulin | Ca2+/Tb3+–calmodulin–WFF peptide | ||||
| Residue | Δ1 JNHc | RDCd | Δ1 JNHc | RDCd | Δ1 JNHc | RDCd |
| a1H chemical shift for NH groups in ppm referenced to DSS. For each residue, the value in the first row corresponds to the signal from the diamagnetic species and the values in the following row/rows correspond to signals from the paramagnetic species. | ||||||
| b1H pseudocontact shifts values for NH groups obtained as the difference between the1H chemical shift for the signal being measured minus the 1H chemical shift of the corresponding signal for the same residue in the diamagnetic (Ca2+)4 species. | ||||||
| c One-bond 1H-15N splittings given in Hz with a minimum uncertainty of ±0.5 Hz and measured at 600 MHz. For each residue, the value in the first row corresponds to the splitting for the (Ca2+)4 species, and the values in the following row/rows correspond to the splittings for the Tb3+-bound species. | ||||||
| d Residue dipolar coupling contributions to one-bond 1H-15N splittings given in Hz and obtained as the difference between the one-bond 1H-15N splitting for the signal being measured minus the corresponding one-bond 1H-15N splitting for the same residue in the (Ca2+)4 species. | ||||||
| e n/a = not assigned. | ||||||
| B. Residual dipolar couplings (in Hz) | ||||||
| Gly33 | −94.3 | 0.0 | −94.8 | 0.0 | −95.2 | 0.0 |
| n/ae | — | n/ae | — | −100.8 | −5.6 | |
| Gly40 | −94.0 | 0.0 | −95.1 | 0.0 | −93.4 | 0.0 |
| −91.4 | +0.5 | −94.6 | +0.5 | −107.6 | −14.2 | |
| −81.9 | — | n/ae | — | −96.0 | −2.6 | |
| −100.9 | — | n/ae | — | −110.7 | −17.3 | |
| Ala57 | −92.2 | 0.0 | −94.2 | 0.0 | −93.5 | 0.0 |
| −88.9 | +4.8 | −89.4 | +4.8 | −88.7 | +4.8 | |
| Ile100 | −92.1 | 0.00 | −92.7 | 0.00 | −92.6 | 0.0 |
| −91.5 | +0.2 | −92.5 | +0.2 | −95.2 | −2.6 | |
| −90.8 | +0.1 | −92.6 | +0.1 | −90.8 | +1.8 | |
| −92.8 | +0.2 | −92.5 | +0.2 | −92.5 | −0.1 | |
| Leu116 | −92.7 | 0.0 | −93.5 | 0.0 | −93.3 | 0.0 |
| −93.4 | +0.1 | −93.4 | +0.1 | −92.8 | +0.5 | |
| −93.4 | +0.1 | −93.4 | +0.1 | −86.8 | +6.5 | |
| −92.8 | −0.8 | −94.3 | −0.8 | −85.3 | +8.0 | |
| Asn137 | −92.9 | 0.0 | −93.8 | 0.0 | −94.0 | 0.0 |
| −92.7 | +0.2 | −93.4 | +0.4 | −88.0 | +6.0 | |
| −92.1 | +0.8 | −93.5 | +0.3 | −85.0 | +9.0 | |
| −91.8 | +0.9 | −92.8 | +1.0 | −78.3 | +15.7 | |
The comparison of the PCS and RDC data for the three systems indicates that the CaM:melatonin complex behaves like the uncomplexed CaM, that is, the domains in the complex present orientational freedom from one another. In particular, this can be seen from the RDC data corresponding to residues belonging to the Tb3+-free C-domain, which show negligible RDCs in the case of CaM and CaM–melatonin, compared to that of CaM–WFF peptide. The WFF peptide fixes both domains so that the paramagnetic Tb3+ ions bound to the N-domain can orient both domains with respect to the magnetic field, and hence, the residues corresponding to both domains have significant RDC values. As well, the smaller PCS values for the residues in the Tb3+-free C-domain in the case of CaM and CaM–melatonin are consistent with a longer distance for these residues, compared to the collapsed structure of the CaM–WFF peptide complex.
There is evidence that other small molecules binding to Ca2+/CaM induce interdomain compaction. For instance, the binding of two W-7 molecules to Ca2+/CaM in solution induces a globular shape for Ca2+/CaM, probably caused by the collapse of both domains (Osawa et al. 1999). As well, the crystal structures of Ca2+/CaM-(TFP)1 (Cook et al. 1994), Ca2+/CaM-(TFP)2 (Vertessy et al. 1988), and Ca2+/CaM-(TFP)4 (Vandonselaar et al. 1994) reveal juxtaposition of both CaM domains compared with the dumbbell structure of Ca2+/CaM (Babu et al. 1988). In these structures, a hydrophobic tunnel resulting from the juxtaposition of the hydrophobic patches of the N- and C-terminal domains of CaM is very similar to that seen in the peptide complexes. In the present work, it is important to make the case of autonomy of the domains in the CaM–melatonin complex, which allows us to use the strategy of characterizing computationally the binding of melatonin to individual domains.
MD of uncomplexed CaM
Based on the independent behavior of the two homologous domains in the CaM:melatonin complex, and to limit the computational load, we decided to restrict the MD simulations to the C-terminal domain. A MD trajectory (SC1 trajectory) was calculated for C-CaM. The root mean square displacement (RMSD) of the backbone α-carbon atoms of C-CaM and C-CaM:melatonin with respect to the initial structure is shown in Figure 6 ▶. Global translational and rotational motions of the protein were removed by least square superposition of the α-carbon atoms relative to the initial structure. The overall protein structure was found to be stable, because the RMSD values are of the order of 1 Å for C-CaM. Our results are consistent with simulations of full CaM solvated by a water cluster (Wriggers et al. 1998). In the C-CaM:melatonin complex the RMSD values are bigger (see below).
Figure 6.

Root mean square displacement (RMSD) of the α-backbone carbon atoms of C-CaM and C-CaM:melatonin with respect to the initial configurations.
Molecular docking
Docking of melatonin into C-CaM was performed with the goal of finding plausible structures of the melatonin:C-CaM complex. The docking procedure yields numerous structures all with very close free energy. The empirical binding free energy (Morris et al. 1998) for the most stable structure used for further simulations was 6.0 kcal/mole. This configuration was chosen as the starting point for further MD simulations, because it was the only one where melatonin was inside the hydrophobic pocket of C-CaM and the contacts were in excellent agreement with those observed experimentally by NMR.
MD simulations of the complex melatonin–CaM
In the course of a 2.8-nsec MD simulation (SMC1 trajectory) of the melatonin–C-CaM complex, the melatonin position with respect to C-CaM exhibited large fluctuations. We have selected four geometrical parameters to structurally characterize the interaction. The first two parameters are meant to represent the rotation of the melatonin heterocycle with respect to the initial orientation. We have defined two quasi-orthogonal axes: One is defined by the two centers of mass of each ring of the indole moiety, and the other one is the axis that goes along the bond that connects the two carbon atoms (atoms 10 and 12 of Fig. 1 ▶) linking the two rings. The angle of these axes with respect to their orientation in the first snapshot of the simulation is measured along the trajectory (Fig. 7B ▶). These angles give information about the rotation of the melatonin heterocycle. We also measured the distance of the indolic nitrogen, NA, and the methoxy oxygen, OE (atoms 9 and 16, respectively, in Fig 1 ▶), to the geometrical center determined by the α-carbon atoms of Ile 100 and Val 136 (Fig. 7A ▶). These residues were chosen because they are located at the bottom of the hydrophobic pocket and their positions fluctuate very little during the simulation. Global translational and rotational motions of the protein were removed by least-square superposition of the α-carbon atoms relative to the initial structure. The time evolution of these parameters is presented in Figure 7B ▶.
Figure 7.

(A) Definition of axis 1 and 2 (left panel), used to follow the dynamics of the indolic ring of melatonin during the MD trajectory. Definition (right panel) of the distances designed to monitor the position of the indolic moiety with respect to the hydrophobic pocket of CaM. (B) The top panel shows the angle formed between axis 1 at time t, and the same axis at time 0. The dashed curve refers to the same quantity for axis 2. The bottom panel shows the time evolution of the distances of methoxylic oxygen (OE) and indolic nitrogen (NA) to the center defined by CA of Ile 100 and CA of Val 136.
The complex is found in three different characteristic configurations during the simulation. The first configuration presents melatonin inside the hydrophobic pocket and is similar to the docking structure. In the second one, melatonin is still inside the pocket but is rotated with respect to the initial structure. In the third one, melatonin stays outside the pocket. Typical representative snapshots are shown in Figure 8 ▶, A–C. Due to the limited time of our simulation, it may be possible that we have not sampled other relevant configurations.
Figure 8.

Typical snapshots observed during the melatonin:CaM simulation. (A) Initial configuration, obtained using the docking scheme. (B) Configuration with melatonin exploring the surface of the protein. (C) Configuration with melatonin in the hydrophobic pocket.
During the first 0.2 nsec of simulation SMC1, melatonin stays inside the pocket, rotating about 60° around axis 1, and getting closer (from 12 Å to 8 Å) to the bottom of the pocket (Fig. 8A ▶). This configuration is very similar to that exhibited by the MLCK complex (Meador et al. 1992; see below). The amino acids whose average distance from the ligand, during the initial 0.2 nsec of simulation SMC1 is less than 6 Å, are shown in Table 3. It can be seen that they all belong to the hydrophobic pocket. To compare with other complexes of calmodulin the distances of the calmodulin amino acids to TRP(MLCK), TFP and W7 in their X-ray structures are also shown in Table 3. Interestingly, similar amino acids are found in the different complexes, indicating that the interaction with the C-terminal hydrophobic pocket seems to be an invariant of the CaM recognition: the two residue β-sheet side chains (Ile 100 and Val 136), the aromatic cluster (Phe 92, Phe 141), the aliphatic cluster (Ile 105, Ile 125, and Ala128), and the four methionine residues (Met 109, Met 124, Met 144, and Met 145). However, there is a difference in the W7 complex with respect to the other complexes: Amino acids 82 to 87 are found near the W7 ligand, at odds with other cases. Probably, this is due to the fact that W7 has a long positive chain interacting with residues Glu 82, Glu 83, Glu 84, and Glu 87. This tail is lacking in melatonin, while in the TRP(MLCK) case there are other amino acids with positive groups interacting with the negative glutamates, and there are other TFP molecules interacting with the negative groups of calmodulin in the 1:4 complex with TFP. This lack of a positive coil could be the reason for the weaker interaction of melatonin.
Table 3.
C-CaM amino acids within 6 Å distancea from bound ligand for different complexes: C-CaM:melatonin, C-CaM:TFP, C-CaM:W7, and C-CaM:TRP (MLCK)
| Residue number | Residue name | Distance from Melb (Å) | Distance from TFPc (Å) | Distance from W7d (Å) | Distance from TRP (MLCK)− (Å) |
| 81 | SER | 4.57 | — | — | — |
| 82 | GLU | — | — | 5.59 | — |
| 83 | GLU | — | — | 2.66 | — |
| 84 | GLU | — | — | 1.88 | — |
| 85 | ILE | — | — | 3.82 | — |
| 86 | ARG | — | — | 4.11 | — |
| 87 | GLU | — | — | 1.91 | — |
| 88 | ALA | 5.47 | — | 2.06 | — |
| 89 | PHE | — | — | 5.12 | — |
| 92 | PHE | 2.62 | 4.12 | 1.93 | 4.56 |
| 100 | ILE | 4.39 | 3.74 | 2.47 | 4.81 |
| 105 | LEU | 2.57 | 3.94 | 2.77 | 3.69 |
| 106 | ARG | — | — | — | 5.14 |
| 108 | VAL | 3.50 | — | 4.83 | — |
| 109 | MET | 2.42 | 5.05 | 3.46 | — |
| 112 | LEU | 2.69 | — | 5.99 | — |
| 114 | GLU | 4.16 | — | — | — |
| 116 | LEU | 5.47 | — | — | — |
| 121 | VAL | 5.22 | — | — | — |
| 123 | GLU | — | 5.23 | — | 5.48 |
| 124 | MET | 2.70 | 3.24 | 3.22 | 2.79 |
| 125 | ILE | 3.26 | 3.96 | 2.54 | 4.16 |
| 127 | GLU | — | 3.44 | 4.36 | 5.25 |
| 128 | ALA | 3.08 | 3.94 | 1.92 | 3.56 |
| 129 | ASP | — | — | 4.97 | — |
| 136 | VAL | 2.61 | 3.47 | 3.24 | 4.11 |
| 141 | PHE | 2.89 | 3.96 | 1.91 | 4.07 |
| 142 | VAL | — | — | — | — |
| 143 | GLN | — | 5.93 | — | — |
| 144 | MET | 2.55 | 3.32 | 2.91 | 2.84 |
| 145 | MET | 3.28 | — | 2.61 | 4.64 |
| 148 | LYS | 2.51 | — | — | — |
a The distance was calculated between the nearest atoms of melatonin and amino acid.
b Mean distance of the SMCl simulation for the first 200 psec.
c Distances calculated from the 1LIN PDB crystal structure of the complex.
d Distances calculated from the 1MUX PDB crystal structure of the complex.
e Distances calculated from the 1 CDL crystal structure of the complex.
Between 0.2 and 0.6 nsec melatonin gets out of the pocket and explores the protein surface in the vicinity of the pocket. This can be seen by monitoring the OE and NA distances (Fig. 8B ▶), that get as large as 12 Å and 15 Å, respectively. The indolic ring gets also parallel to helices 1 and 3, rotating both axes about 150° with respect to the initial structure. At about 0.6 nsec melatonin gets inside the hydrophobic pocket, but with a different orientation corresponding to a rotation of 100° of both axes, compared to the initial docking structure. The NA and OE distances are similar to those found in the first structure. This configuration, depicted in Figure 8C ▶, is found with little variations, until the end of the simulation. Analysis of the changes in energy contributions (internal, electrostatic, van der Waals) during the trajectory (not shown) suggests that a major role in determining melatonin position and conformation is played by electrostatic interaction with protein and with solvent.
Flexibility of C-CaM along the simulation
C-CaM complexed with melatonin exhibits large structural fluctuations along the simulation, as shown in Figure 6 ▶. To compare with the SC1 simulation we have again calculated the RMSD of the α-carbon backbone atoms with respect to the initial structure. Global translational and rotational motions of the protein were removed by least-square superposition of the α-carbon atoms relative to the initial structure. The average RMSD starts to oscillate around a value of about 2 Å and then increases to values around 4 Å. A more detailed analysis shows that large displacements are limited, apart from the terminations, to the coil that links helix 2 to helix 3, to helix 3 itself, and to the second loop (numbering of helices is made considering the C domain of CaM only). This is probably due to the fact that during the simulation melatonin changes its position with respect to calmodulin significantly, even leaving the hydrophobic pocket.
Notwithstanding the large RMSD, the secondary structure of C-CaM is well conserved along the Molecular Dynamics trajectory, preserving a well-defined hydrophobic pocket. Indication of major conformational change of CaM has been found in the case of binding to trifluoroperazine (Vandonselaar et al. 1994). In this case, the movement involves the bending of the central helix that links the two domains, but small changes have been observed in the C-and N-terminal domains in the molecular dynamics simulations of the 4TFP:CaM (Yamaotsu et al. 2001). This is in agreement with a stronger interaction between the drug and the protein resulting in small fluctuations of the TFP position. The occurrence of large concerted motions of C-CaM in the presence of melatonin may raise some doubts about the validity of the docking procedure, where a single protein configuration was considered. Actually, the importance of the hydrophobic pocket is confirmed both by direct experimental evidence (this work) and crystal structures of similar complexes (see Discussion).
Free energy of binding
In addition to the above MD simulations, a theoretical estimation of ΔG° associated to the binding process was also performed, following the double annihilation method (Jorgensen et al. 1988). The result ΔG° = −9 ± 0.4 kcal/mole, corresponding to Kd less than micromolar, is far from the experimental one (–3.2 ± 0.2 kcal/mole) and at odds with Molecular Dynamics results, where melatonin is seen to exit easily from the pocket. The reasons for such disagreement are (1) the poor sampling of relevant configurations of the complex, notwithstanding the considerable length of simulation SMC2 (15 nsec); (2) the force-field inaccuracies; and (3) the flaw intrinsic to the method (Gilson et al. 1997). Our computation confirms, in our opinion, the need for new, more reliable methods to estimate the absolute free energy of binding (Luo et al. 2002). This is especially true in weakly interacting complexes in which there are no specific interactions, such as the case for melatonin–calmodulin.
Discussion
Both experimental and MD results show that the calcium-dependent interaction of melatonin with CaM is weak. The acquisition of structural details for weakly interacting systems poses a challenge from the methodological point of view (Zuiderweg 2002). Often, these complexes do not crystallize either at all or in a biologically relevant conformation. Similarly, traditional NOE-based NMR methods for obtaining solution structures do not work, because they provide useful information only for a relatively strong complex (Kd < 10 μM). A static description of an average structure might be inappropriate anyway. We have therefore resorted to using a combined approach in which fluorescence and NMR experiments are complemented by MD simulations to provide a description of the dynamical properties of the CaM–melatonin complex.
Chemical shift perturbation allowed us to map the interaction in the hydrophobic cavities of each of the CaM globular domains. Based both on this evidence and on the experimental information that the two globular domains retain their orientational freedom in the complex, we chose to model the CaM–melatonin complex by following the dynamical behavior of one melatonin molecule in the pocket of the C-terminal domain. The most stable complex found by molecular docking, shows melatonin inside the hydrophobic pocket, with the O-methyl group pointing to the bottom of the pocket and the ethylamide side chain located in the entrance. The aromatic ring of melatonin is relatively immobilized, with the aliphatic tail extended into the solvent. Two main orientations were found for this arrangement, and within each orientation the ring retains some degree of translational freedom.
In the course of our MD simulations, melatonin explores different conformations, in some cases, even leaving the hydrophobic pocket. Melatonin binds to C-CaM mainly by hydrophobic interactions of the indolic moiety with the aliphatic and aromatic amino acids of the C-CaM hydrophobic pocket. However, from about 0.6 nsec to 2 nsec the indolic nitrogen also forms a hydrogen bond with the carbonyl of Ala 128. This is consistent with previous results which suggested an important role for Ala 128 in ligand binding to CaM (Osawa et al. 1998).
The main average structural features obtained for the melatonin–CaM complex are in excellent agreement with those of other CaM complexes formed both with peptides and with small molecules (Fig. 9 ▶). The interaction with the C-terminal pocket seems to be an invariant of the CaM recognition: In all available structures, a bulky hydrophobic residue binds inside the CaM pocket of the C-domain. In CaM–peptide complexes, this site is often occupied by a tryptophan. A second hydrophobic site anchors the molecule to the N-terminal domain, helped by the additional support of a basic group, which interacts with residues outside the pocket (Prozialeck and Weiss 1982; Weiss et al. 1985), thus resulting in a strong binding constant. Comparison of the CaM complexes with small molecules both obtained by crystallographic studies, such as those of TFP (PDB: 1CTR, 1A29, 1LIN), or DPD (PDB: 1QIV), or by NMR, such as those of W7 (PDB: 1MUX), also shows that the C-terminal hydrophobic pocket is always occupied. Additional binding sites may be present outside the pocket, but mostly sandwiched in between the two domains (as is the case of 1LIN).
Figure 9.

(A,B) The C-domain of CaM complexed with W7 (PDB: 1MUX) and TFP (PDB: 1LIN), respectively. (C) The structure of the C-CaM:melatonin complex obtained through the docking procedure. (D) The C-domain of the complex of CaM with MLCK (only the tryptophan of MLCK is shown; PDB: 1CBL).
Melatonin binds to calmodulin mainly by the indolic ring that fits very well inside the hydrophobic pocket of calmodulin, thus maximizing their interactions. Melatonin has an O-Methyl moiety in the indolic ring in contrast to tryptophan, but this does not affect the binding, as the methyl group interacts fairly well with the aliphatic aminoacids of the pocket. On the contrary, in the case of 5-hydroxy tryptophan and serotonin, the presence of an hydroxyl group instead of the O-Methyl makes the interaction weaker due the hydrophilicity of this group (Ouyang and Vogel 1998). However, melatonin does not have a basic group that may interact with the acidics groups that lay in the surroundings of the hydrophobic pocket, and this makes the interaction weaker compared with TFP or W7. In this sense, it has been shown that melatonin metabolites, kynuremines, which lack NH2 groups, do not bind to calmodulin (León et al. 2000). Moreover, tryptophan has an extra negative charge from its carboxilic group that is likely to be repelled by the acidics groups of CaM, explaining the lower interaction found with tryptophan in comparison with melatonin (Ouyang and Vogel 1998). Our results suggest that a melatonin analog with an NH2 group instead of the amido one would have a stronger binding constant, probably comparable to more potent calmodulin antagonists.
Biological implications
Both melatonin and CaM are ubiquitously found and their structures are phylogenetically well preserved. The fact that melatonin interacts weakly with CaM, does not detract the possibility that this interaction may occur in biological media. The weak melatonin–CaM interaction may allow the hormone to gently modulate many cellular functions in a rhythmical fashion. The cyclic rise in melatonin bloodstream levels stimulated by darkness would have the effect of producing a subtle inhibition of CaM’s interaction with other proteins. This inhibition has been shown in vitro for certain systems (Benitez-King et al. 1993; Cardinali et al. 1997; León et al. 2000), and remains, however, difficult to assess in vivo. A higher affinity for the CaM–melatonin interaction would probably render this mechanism unsuitable.
On the other hand, the formation of the complex in biological media will depend on the concentrations of both species in different physiological microenvironments within the different cells. In fact, while this interaction was observed in some tissues (Benitez-King et al. 1996; Pozo et al. 1997; León et al. 2000), it was not confirmed in other systems (Wolfler et al. 1998; Sáenz et al. 2002). Melatonin may accumulate in some tissues and cellular structures, such as cell membrane, cytosol, and nuclei, to about 103 times higher than circulating levels (Menendez-Pelaez et al. 1993). The affinity measured for this interaction, although low, is able to explain in vitro effects that have been observed in previous papers (Pozo et al. 1997; León et al. 2000).
Moreover, from the pharmacological point of view, melatonin has been found to be an important anticancer agent. The proposed mechanism of action of this drug is via its inhibition of Ca2+/calmodulin, and because pharmaceutical dosage of melatonin is high (but not toxic) the low Kd of the interaction is not a problem (MacNeil et al. 1988). The therapeutic use of melatonin as well as its safety, dosage, side effects, and contraindications have been extensively investigated (e.g., see Karasek et al. 2002). The drug has been shown to present a very low toxicity (Karasek et al. 2002), and therefore, it can be considered as a viable pharmacological alternative to other more toxic antagonists for inhibiting CaM activity.
Materials and methods
Protein expression and purification
Drosophila melanogaster wild-type CaM was expressed in Escherichia coli and purified as described previously (Biekofsky et al. 1998). The uniformly 15N-labeled proteins were made by incorporating 99% 15N-labeled (NH4)2SO4 as the sole nitrogen source into the growth media. The purities of the CaM samples were monitored by NMR, mass spectrometry, and UV measurements. The 15N-labeled protein samples used for the NMR studies were 1.08 mM solutions of protein in 90% H2O:10% D2O and 0.1 M KCl. The pH of the sample was adjusted to 6.8 (uncorrected for deuterium isotope effects).
Fluorescence spectroscopy
Uncorrected fluorescence emission spectra were recorded using a SPEX Fluoromax fluorimeter with λex = 295 nm (bandwidth 0–85 nm) and emission from 310 to 450 nm (bandwidth 5 nm). Spectra were recorded at 20°C in 25 mM Tris (pH 8), 100 mM KCl with 1 mM CaCl2 and 1 mM EGTA, as appropriate.
NMR spectroscopy
All NMR spectra were recorded at 500 and 600 MHz (1H frequency) and at 35°C on Varian Unity Plus 500 and 600 spectrometers. Two-dimensional 15N-1H HSQC (Bodenhausen and Ruben 1980; Mori et al. 1995) spectra, with WATERGATE sequence (Piotto et al. 1992; Sklenár et al. 1993), were acquired at 500 MHz with spectral widths of 7500 Hz (1H) and 2027 Hz (15N), respectively. A total of 110 t1 increments were acquired with an acquisition time of 0.15 sec, during which the 15N nuclei were decoupled using a GARP sequence (Shaka et al. 1985).
Assignment of 1H and 15N resonances for Ca2+-CaM was taken from Ikura and coworkers (Ikura et al. 1990), and confirmed by three-dimensional 15N-1H NOESY-HSQC and TOCSY-HSQC experiments (Marion et al. 1989; Zuiderweg and Fesik 1989). A 15N-1H NOESY-HSQC was acquired at 500 MHz with spectral widths as above, a mixing time of 100 msec, 125 increments in the indirect 1H dimension and 48 increments for the 15N dimension. A 15N-1H TOCSY-HSQC (Marion et al. 1989) was acquired at 600 MHz with spectral widths of 7996 Hz (1H) and 2431 Hz (15N), a mixing time of 60 msec, 130 increments in the indirect 1H dimension and 40 increments for the 15N dimension. Acquisition times of 0.15 sec were used throughout, again with decoupling of the 15N nuclei using a GARP sequence.
Residual dipolar couplings were extracted from 2D IPAP 1H-15N HSQC spectra (Ottiger et al. 1998). Spectra were acquired with 1184 complex points in t2 (148 msec acquisition time) with a spectral width of 8000 Hz, and 270 complex points were collected in t1 (108 msec acquisition time) with a spectral width of 2500 Hz.
All spectra were processed using nmrPipe (Delaglio et al. 1995) and analyzed using XEASY (Bartels et al. 1995). Typically, the acquisition dimension was multiplied by a Gaussian function, and other dimensions with a 90°-shifted sine-bell function. All dimensions were zero-filled at least to the next power of 2. Chemical shifts of 1H resonances were measured from an internal standard dioxane and referenced to 5,5-dimethyl-5-silapentane-2-sulphonate (taken as 3.750 ppm from dioxane) (Wishart et al. 1995), 15N chemical shifts are referenced to liquid NH3 using the frequency ratio method (15N/1H = 0.101329118) (Wishart et al. 1995).
Melatonin titration
Melatonin was purchased from Aldrich Chemical Co. Ltd. A stock solution of 13 mM melatonin in 90% H2O/10% D2O was prepared. For each titration point an aliquot of 50 μL of this stock melatonin solution was added to the NMR tube containing the CaM solution and mixed thoroughly. The changes in pH caused by melatonin additions were negligible.
2D 1H-15N HSQC NMR spectra were recorded for 15N-labeled (Ca2+)4-CaM at various melatonin concentrations from [melatonin]/[CaM] molar ratio of 0 up to 9. The signals in the spectra are well-resolved and individual amide NH 1H/15N signals can be followed throughout the titration. The previously assigned 1H and 15N signals for Ca2+-saturated CaM (Ikura et al. 1990) were used to identify signals in the 2D 1H-15N HSQC spectrum for the [melatonin]/[CaM] molar ratio of 0. Any ambiguities in the assignment of amide NH 1H/15N resonances during the melatonin titration were resolved by using three-dimensional TOCSY-HSQC and NOESY-HSQC experiments.
Partial replacement of Ca2+ by Tb3+
A stock solution of 50 mM TbCl3 in 90% H2O/10% D2O was prepared from a standard 1 M TbCl3 solution; 8.64 μL of this stock solution was added to the solution of the CaM:melatonin complex and mixed thoroughly giving a [Tb3+]/[CaM] molar ratio of 0.66 eq of the CaM concentration. The changes in pH caused by TbCl3 additions were negligible.
Analysis of binding curves
The chemical shift variation of individual resonances were fitted using the following equation: δobs = δi + [δf − δi] • [bound melatonin]/[total CaM], where δi and δf are the initial and final CaM chemical shifts, [bound melatonin] is the concentration of bound melatonin, and [total CaM] is the total concentration of CaM. Fits were performed using standard nonlinear least-squares methods. To determine Kd values from the NMR experimental data, we are assuming a linear relation between complexation and chemical shift changes.
Computational tools
Melatonin force field parametrization
Partial charges for melatonin were obtained using the restrained electrostatic fit (RESP) scheme (Breneman and Wiberg 1990). Electrostatic potentials were computed using ab initio HF/6–31G** calculations using the Gaussian 98 package (Frisch et al. 1998). Most of the parameters for melatonin were taken from the AMBER force field (Cornell et al. 1995). The parameters associated with the torsional degrees of freedom of the ethyl amide chain were not available and were obtained by fitting them iteratively to reproduce the energy profiles obtained from HF/6–31G** calculations (Cornell et al. 1995). The atom types and force field parameters for melatonin are given as supplementary material.
MD simulations
MD simulations were performed using the Amber force field for CaM (Cornell et al. 1995), and the TIP3P model for water (Jorgensen et al. 1983). Calcium ions were represented using the parameters derived by Hori and collaborators (Hori et al. 1988), while parameters for melatonin are described above. Apart from the preparatory work, for which we used AMBER6 software package (Case et al. 1999), all simulations were performed using the package NAMD (Kale et al. 1999).
In all cases, the time step was 2 fsec, the van der Waals interactions were smoothly cut off at 9 Å, while electrostatic interactions were accounted for by using the particle mesh Ewald method (Essman et al. 1995). All bonds were kept rigid (Ryckaert et al. 1977). The Berendsen thermostat (Berendsen et al. 1984) was used to maintain the desired temperature.
Simulations of melatonin
The initial coordinates of melatonin were taken from the HF/6–31G** calculations. The temperature was set to 310 K. Aqueous simulations were performed by placing melatonin into a periodic box consisting of 803 TIP3P water molecules. The volume of the box was adjusted to obtain a pressure of 1 atm using the Berendsen method with a relaxation time of 1 psec (Berendsen et al. 1984). Two trajectories, of 1 and 4 nsec each, were respectively obtained for the isolated (SM0) and solvated melatonin (SM1 trajectory).
Simulations of C-CaM
The crystal structure from the Brookhaven Protein Data Bank (PDB) 4CLN (Taylor et al. 1991) was taken as the initial structure of CaM. The initial positions of the hydrogen atoms were generated by the LEAP module of the AMBER package (Case et al. 1999). The C-terminal domain of CaM (residues 81–148) was placed into a periodic box consisting of 3525 TIP3P water molecules. To neutralize the system, eight potassium ions were also included in the simulation. After an equilibration phase we obtained 1-nsec MD trajectory (SC1 trajectory) at 310 K and 1 atm.
Molecular docking of melatonin:C-CaM complex
Docking simulations were performed with the AUTODOCK 3.0 program (Goodsell and Olson 1990; Morris et al. 1998). The CaM and melatonin initial structure were taken from the SC1 and SM1 MD trajectories, respectively. Lennard-Jones parameters and partial charges were assigned according to the atom types, consistently with the AMBER force field (Cornell et al. 1995). A mesh with a spacing of 0.55 Å was defined in a box measuring 50 × 50 × 50 Å3 around C-CaM. The energy of interaction between C-CaM and melatonin were computed in each point of the grid. The torsional modes associated with ethyl amide and the methoxyl moieties of melatonin were left free while the rest of the molecule was considered rigid.
A Monte Carlo simulated annealing scheme was employed to locate relevant minima. The initial temperature was set to 503 K. We performed 50 Monte Carlo runs, each comprising 50 cooling cycles with a scaling factor of 0.9. The final temperature was 2.6 K.
MD simulations of melatonin:C-CaM complex
MD complex simulations were performed, starting with the most stable structure found with the docking protocol described above. The complex was neutralized with eight potassium ions, and was placed in a periodic box consisting of 3128 water molecules at 300 K and 1 atm. The simulation consisted of a 2.8-nsec trajectory (SMC1).
The theoretical melatonin:C-CaM binding constant was estimated using the free energy perturbation method (Kollman et al. 1993) and the so-called double annihilation process (Jorgensen et al. 1988). The free energy change associated with the binding process was computed by simulating the transfer of melatonin from the complex and from solution to the gas phase (simulations SMC2 and SM2, respectively).
Acknowledgments
We thank Tom Frenkiel, Geoff Kelly, and Fred Muskett for their help and advice with the NMR experiments. We are grateful to Kathy Beckingham (Rice University, Houston, TX) for supplying the original pOTSNco12 vector containing the cDNA coding for Drosophila melanogaster CaM, and to Peter Browne for the original cloning of the wild-type CaM. The NMR experiments were carried out at the MRC Biomedical NMR Centre, Mill Hill. We also thank Fabrizio Giambertone for help with the cluster of PC. D.A.E., R.E.R., and A.G.T. acknowledge financial support from Fundación Antorchas, Universidad de Buenos Aires, ANPCYT, and the L.B.P. Endowment for the Sciences and Arts. D.A.E. acknowledges Pablo de Grande and Microsoft Argentina for a generous donation. D.A.E. and R.E.R. are staff members of CONICET (Argentina). A.G.T. is a CONICET fellow.
Abbreviations
CaM, calmodulin
C-CaM, C-domain of calmodulin
NMR, nuclear magnetic resonance
HSQC, heteronuclear single quantum coherence
NOESY, nuclear Overhauser effect spectroscopy
TOCSY, total correlation spectroscopy
TFP, trifluoperazine
J-8, N-(8-aminooctyl)-5-iodonaphthalene-1-sulfonamide
W-7, N-(6-aminhexyl)-5-chloro-1-naphthalenesulfonamide
AAA, N-(3, 3-diphenylpropyl)-N′-[1-R-(3, 4-bis-butoxyphenyl)ethyl]-propylene-diamine
Mel, melatonin
MD, molecular dynamics
FEP, free energy perturbation
SM0, MD run of isolated Mel
SM1, MD run of Mel in solution
SC1, MD run of C-CaM in solution
SMC1, MD run of the C-CaM-Mel complex
SMC2, FEP run of the C-CaM-Mel complex
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.04611404.
Supplemental material: see www.proteinscience.org
References
- Babu, Y.S., Bugg, C.E., and Cook, W.J. 1988. Structure of calmodulin refined at 2.2 Å resolution. J. Mol. Biol. 204 191–204. [DOI] [PubMed] [Google Scholar]
- Barbato, G., Ikura, M., Kay, L.E., Pastor, R.W., and Bax, A. 1992. Backbone dynamics of calmodulin studied by 15N relaxation using inverse detected two-dimensional NMR spectroscopy: The central helix is flexible. Biochemistry 31 5269–5278. [DOI] [PubMed] [Google Scholar]
- Bartels, C., Xia, T., Billeter, M., Güntert, P., and Wüthrich, K. 1995. The program XEASY for computer-supported NMR spectral analysis of biological macromolecules. J. Biomol. NMR 5 1–10. [DOI] [PubMed] [Google Scholar]
- Benitez-King, G., Huerto-Delgadillo, L., and Anton-Tay, F. 1993. Binding of 3H-melatonin to calmodulin. Life Sci. 53 201–207. [DOI] [PubMed] [Google Scholar]
- Benitez-King, G., Ríos, A., Martine, S., and Anton-Tay, F. 1996. In vitro inhibition of Ca2+/calmodulin-dependent kinase II activity by melatonin. Biochim. Biophys. Acta 1290 191–196. [DOI] [PubMed] [Google Scholar]
- Berendsen, H.J.C., Postma, J.P.M., van Gunsteren, W.F., Di Nola, A., and Haak, J.R. 1984. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 81 3684–3690. [Google Scholar]
- Berridge, M.J., Bootman, M.D., and Lipp, P. 1998. Calcium—A life and death signal. Nature 395 645–648. [DOI] [PubMed] [Google Scholar]
- Biekofsky, R.R., Martin, S.R., Browne, J.P., Bayley, P.M., and Feeney, J. 1998. Ca2+ coordination to backbone carbonyl oxygen atoms in calmodulin and other EF-hand proteins: 15N chemical shifts as probes for monitoring individual site Ca2+ coordination. Biochemistry 37 7617–7629. [DOI] [PubMed] [Google Scholar]
- Biekofsky, R.R., Muskett, F.W., Schmidt, J.M., Martin, S.R., Browne, J.P., Bayley, P.M., and Feeney, J. 1999. NMR approaches for monitoring domain orientations in calcium-binding proteins in solution using partial replacement of Ca2+ by Tb3+. FEBS Lett. 460 519–526. [DOI] [PubMed] [Google Scholar]
- Blask, D.E., Sauer, L.A., and Dauchy, R.T. 2002. Melatonin as a chronobiotic/anticancer agent: Cellular, biochemical, and molecular mechanisms of action and their implications for circadian-based cancer therapy. Curr. Top. Med. Chem. 2 113–132. [DOI] [PubMed] [Google Scholar]
- Bodenhausen, G. and Ruben, D.J. 1980. Natural abundance nitrogen-15 NMR by enhanced heteronuclear spectroscopy. Chem. Phys. Lett. 69 185–189. [Google Scholar]
- Breneman, C.M. and Wiberg, K.B. 1990. Determining atom-centered monopoles from molecular electrostatic potential: The need for high sampling density in formamide conformational analysis. J. Comput. Chem. 11 361–373. [Google Scholar]
- Cardinali, D.P., Golombek, D.A., Rosenstein, R.E., Cutrera, R.A., and Esquifino, A.I. 1997. Melatonin site and mechanism of action: Single or multiple? J. Pineal Res. 23 32–39. [DOI] [PubMed] [Google Scholar]
- Case, D.A., Pearlman, D.A., Caldwell, J.W., Cheatham III, T.E., Ross, W.S., Simmerling, C.L., Darden, T.A., Merz, K.M., Stanton, R.V., Cheng, A.L., et al. 1999. AMBER 6. University of California, San Francisco.
- Cook, W.J., Walter, L.J., and Walter, M.R. 1994. Drug binding by calmodulin: Crystal structure of a calmodulin–trifluoperazine complex. Biochemistry 33 15259–15265. [DOI] [PubMed] [Google Scholar]
- Cornell, W.D., Cieplak, P., Bayly, C.I., Gould, I.R., Merz Jr., K.M., Ferguson, D.M., Spellmeyer, D.C., Fox, T., Caldwell, J.W., and Kollman, P.A. 1995. A second generation force field for the simulation of proteins, nucleic acids, and organic molecules. J. Am. Chem. Soc. 117 5179–5197. [Google Scholar]
- Craven, C.J., Whitehead, B., Jones, S.K., Thulin, E., Blackburn, G.M., and Waltho, J.P. 1996. Complexes formed between calmodulin and the antagonists J-8 and TFP in solution. Biochemistry 35 10287–10299. [DOI] [PubMed] [Google Scholar]
- Crivici, A. and Ikura, M. 1995. Molecular and structural basis of target recognition by calmodulin. Annu. Rev. Biophys. Biomol. Struct. 24 85–116. [DOI] [PubMed] [Google Scholar]
- Dai, J., Inscho, E.W., Yuan, L., and Hill, S.M. 2002. Modulation of intracellular calcium and calmodulin by melatonin in MCF-7 human breast cancer cells. J. Pineal Res. 32 112–119. [DOI] [PubMed] [Google Scholar]
- Delaglio, F., Grzesiek, S., Vuister, G., Zhu, G., Pfeifer, J., and Bax, A. 1995. NMRPipe: A multidimensional spectral processing system based on unix pipes. J. Biomol. NMR 6 277–293. [DOI] [PubMed] [Google Scholar]
- Essman, U., Perera, L., Berkowitz, M.L., Darden, T., Lee, H., and Pedersen, L.G. 1995. A smooth particle mesh Ewald method. J. Chem. Phys. 103 8577–8593. [Google Scholar]
- Ford, J.M. and Hait, W.N. 1990. Pharmacology of drugs that alter multidrug resistance in cancer. Pharmacol. Rev. 42 155–199. [PubMed] [Google Scholar]
- Frisch, M.J., Trucks, G.W., Schlegel, H.B., Scuseria, G.E., Robb, M.A., Cheeseman, J.R., Zakrzewski, V.G., Montgomery Jr., J.A., Stratmann, R., Burant, J., et al. 1998. Gaussian 98, Rev. A7. Gaussian, Inc., Pittsburgh, PA.
- Gilson, M.K., Given, J.S., Bush, B.L., and McCammon, J.A. 1997. The statistical-thermodynamic basis for computation of binding affinities: A critical review. Biophys. J. 72 1047–1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodsell, D.S. and Olson, A.J. 1990. Automated docking of substrates to proteins by simulated annealing. Proteins 8 195–202. [DOI] [PubMed] [Google Scholar]
- Harmat, V., Bocskei, Z., Naray-Szabo, G., Bata, I., Csutor, A.S., Hermecz, I., Aranyi, P., Szabo, B., Liliom, K., Vertessy, B.G., et al. 2000. A new potent calmodulin antagonist with arylalkylamine structure: Crystallographic, spectroscopic and functional studies. J. Mol. Biol. 297 747–755. [DOI] [PubMed] [Google Scholar]
- Hickie, R.A., Wei, J.W., Blyth, L.M., Wong, D.Y., and Klaassen, D.J. 1983. Cations and calmodulin in normal and neoplastic cell growth regulation. Can. J. Biochem. Cell Biol. 61 934–941. [DOI] [PubMed] [Google Scholar]
- Hoeflich, K.P. and Ikura, M. 2002. Calmodulin in action: Diversity in target recognition and activation mechanisms. Cell 108 739–742. [DOI] [PubMed] [Google Scholar]
- Hori, K., Kushick, J.N., and Weinstein, H. 1988. Structural and energetic parameters of Ca2+ binding to peptides and proteins. Biopolymers 27 1865–1886. [DOI] [PubMed] [Google Scholar]
- Huerto-Delgadillo, L., Anton-Tay, F., and Benitez-King, G. 1994. Effects of melatonin on microtubule assembly depend on hormone concentration: Role of melatonin as a calmodulin antagonist. J. Pineal Res. 17 55–62. [DOI] [PubMed] [Google Scholar]
- Ikura, M., Kay, L.E., and Bax, A. 1990. A novel approach for sequential assignment of 1H, 13C, and 15N spectra of larger proteins: Heteronuclear triple-resonance three-dimensional NMR spectroscopy: Application to calmodulin. Biochemistry 29 4659–4667. [DOI] [PubMed] [Google Scholar]
- Jorgensen, W.L., Chandreskhar, J., Madura, J.D., Impey, R.W., and Klein, M.L. 1983. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79 926–935. [Google Scholar]
- Jorgensen, W.L., Buckner, J.K., Boudon, S., and Tirado-Rives, J. 1988. Efficient computation of absolute free energies of binding by computer simulations. Application to the methane dimer in water. J. Chem. Phys. 89 3742–3746. [Google Scholar]
- Kale, L., Skeel, R., Bhandarkar, M., Brunner, R., Gursoy, A., Krawetz, N., Phillips, J., Shinozaki, A., Varadarajan, K., and Schulten, K. 1999. NAMD2: Greater scalability for parallel molecular dynamics. J. Comp. Phys. 151 283–312. [Google Scholar]
- Karasek, M., Reiter, R.J., Cardinali, D.P., and Pawlikowski, M. 2002. Future of melatonin as a therapeutic agent. Neuroendocrinol. Lett. 1 118–121. [PubMed] [Google Scholar]
- Kollman, P.A. 1993. Free energy calculations: Applications to chemical and biochemical problems. Chem. Rev. 93 2395–2417. [Google Scholar]
- León, J., Macias, M., Escames, G., Camacho, E., Khaldy, H., Martin, M., Espinosa, A., Gallo, M.A., and Acuña-Castroviejo, D. 2000. Structure-related inhibition of calmodulin-dependent neuronal nitric-oxide synthase activity by melatonin and synthetic kynurenines. Mol. Pharmacol. 58 967–975. [DOI] [PubMed] [Google Scholar]
- Liu, J., Qi, S., Zhu, H., Zhang, J., Li, Z., and Wang, T. 2002. The effect of calmodulin antagonist berbaminederivative-EBB on hepatoma in vitro and in vivo. Chin. Med. J. (Engl.) 115 759–762. [PubMed] [Google Scholar]
- Luo, H. and Sharp, K. 2002. On the calculation of absolute macromolecular binding free energies. Proc. Natl. Acad Sci. 99 10399–10404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacNeil, S., Griffin, M., Cooke, A.M., Pettett, N.J., Dawson, R.A., Owen, R., and Blackburn, G.M. 1988. Calmodulin antagonists of improved potency and specificity for use in the study of calmodulin biochemistry. Biochem. Pharmacol. 37 1717–1723. [DOI] [PubMed] [Google Scholar]
- Marion, D., Driscoll, P.C., Kay, L.E., Wingfield, P.T., Bax, A., Gronenborn, A.M., and Clore, G.M. 1989. Overcoming the overlap problem in the assignment of proton NMR spectra of larger proteins by use of three dimensional heteronuclear proton-nitrogen-15 Hartmann-Hahn multiple quantum coherence and nuclear Overhauser-multiple quantum coherence spectroscopy: Application to interleukin. Biochemistry 28 6150–6156. [DOI] [PubMed] [Google Scholar]
- Meador, W.E., Means, A.R., and Quiocho, F.A. 1992. Target enzyme recognition by calmodulin. 2.4 Å structure of a calmodulin–peptide complex. Science 257 1251–1255. [DOI] [PubMed] [Google Scholar]
- Menéndez-Pelaez, A., Poeggeler, B., Reiter, R.J., Barlon-Walden, L.R., Pablos, M.I., and Tan, X. 1993. Nuclear localization of melatonin in different mammalian tissues. Immunochemical and radioimmunoassay evidence. J. Cell Biochem. 53 373–382. [DOI] [PubMed] [Google Scholar]
- Mori, S., Abeygunawardana, C., Johnson, M.O., and van Zijl, P.C. 1995. Improved sensitivity of HSQC spectra of exchanging protons at short interscan delays using a new fast HSQC (FHSQC) detection scheme that avoids water saturation. J. Magn. Reson. B 108 94–98. [DOI] [PubMed] [Google Scholar]
- Morris, G.M., Goodsell, D.S., Halliday, R.S., Huey, R., Hart, W.E., Belew, R.K., and Olson, A.J. 1998. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comp. Chem. 19 1639–1662. [Google Scholar]
- Nojima, H. 1989. Structural organization of multiple rat calmodulin genes. J. Mol. Biol. 208 269–282. [DOI] [PubMed] [Google Scholar]
- Osawa, M., Swindells, M.B., Tanikawa, J., Tanaka, T., Mase, T., Furuya, T., and Ikura, M. 1998. Solution structure of calmodulin-W-7 complex: The basis of diversity in molecular recognition. J. Mol. Biol. 276 165–176. [DOI] [PubMed] [Google Scholar]
- Osawa, M., Kuwamoto, S., Izumi, Y., Yap, K.L., Ikura, M., Shibanuma, T., Yokokura, H., Hidaka, H., and Matsushima, N. 1999. Evidence for calmodulin inter-domain compaction in solution induced by W-7 binding. FEBS Lett. 442 173–177. [DOI] [PubMed] [Google Scholar]
- Ottiger, M., Delaglio, F., and Bax, A. 1998. Measurement of J and dipolar couplings from simplified two-dimensional NMR spectra. J. Magn. Reson. 131 373–378. [DOI] [PubMed] [Google Scholar]
- Ouyang, H. and Vogel, H.J. 1998. Melatonin and serotonin interactions with calmodulin: NMR, spectroscopic and biochemical studies. Biochim. Biophys. Acta 1383 37–47. [DOI] [PubMed] [Google Scholar]
- Piotto, M., Saudek, V., and Sklenár, V. 1992. Gradient tailored excitation for single-quantum NMR spectroscopy of aqueous solutions. J. Biomol. NMR 2 661–665. [DOI] [PubMed] [Google Scholar]
- Pozo, D., Reiter, R.J., Calvo, J.R., and Guerrero, J.M. 1997. Inhibition of cerebellar nitric oxide synthase and cyclic GMP production by melatonin via complex formation with calmodulin. J. Cell. Biochem. 65 430–442. [DOI] [PubMed] [Google Scholar]
- Prozialeck, W.C. and Weiss, B. 1982. Inhibition of calmodulin by phenothiazines and related drugs: Structure–activity relationships. J. Pharmacol. Exp. Ther. 222 509–516. [PubMed] [Google Scholar]
- Ryckaert, J.P., Ciccotti, G., and Berendsen, H.J.C. 1977. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comp. Phys. 23 327–341. [Google Scholar]
- Saénz, D.A., Turjanski, A.G., Sacca, G.B., Martí, M., Doctorovich, F., Keller-Sarmiento, M.I., Estrin, D.A., and Rosenstein, R.E. 2002. Physiological concentrations of melatonin inhibit the nitridergic pathway in the Syrian hamster retina. J. Pineal Res. 33 31–37. [DOI] [PubMed] [Google Scholar]
- Shaka, A.J., Barker, P.B., and Freeman, R. 1985. Computer optimized decoupling scheme for wideband applications and low-level operation. J. Magn. Reson. 64 547–552. [Google Scholar]
- Sklenár, V., Piotto, M., Leppik, R., and Saudek, V. 1993. Gradient-tailored water suppression for 1H-15N HSQC experiments optimized to retain full sensitivity. J. Magn. Reson. Series A 102 241–245. [Google Scholar]
- Taylor, D.A., Sack, J.S., Maune, J.F., Beckingham, K., and Quiocho, F.A. 1991. Structure of a recombinant calmodulin from Drosophila melanogaster refined at 2.2-Å resolution. J. Biol. Chem. 266 21375–21380. [DOI] [PubMed] [Google Scholar]
- Vandonselaar, M., Hickie, R.A., Quail, J.W., and Delbaere, L.T. 1994. Trifluoperazine-induced conformational change in Ca2+-calmodulin. Nat. Struct. Biol. 1 795–801. [DOI] [PubMed] [Google Scholar]
- Vertessy, B.G., Harmat, V., Böcskei, Z., Náray-Szabó, G., Orosz, F., and Ovádi, J. 1988. Simultaneous binding of drugs with different chemical structures to Ca2+-calmodulin: Crystallographic and spectroscopic studies. Biochemistry 37 15300–15310. [DOI] [PubMed] [Google Scholar]
- Weiss, B., Prozialeck, W.C., and Wallace, T.L. 1982. Interaction of drugs with calmodulin. Biochemical, pharmacological and clinical implications. Biochem. Pharmacol. 31 2217–2226. [DOI] [PubMed] [Google Scholar]
- Weiss, B., Sellinger-Barnette, M., Winkler, J.D., and Schechter, L.E. 1985. Calmodulin antagonists: Structure–activity relationships. In Calmodulin antagonists and cellular biology (eds. H. Hidaka and D.J. Hartshorne) pp. 45–62. Academic, Orlando, FL.
- Wishart, D.S., Bigam, C.G., Yao, J., Abildgaard, F., Dyson, H.J., Oldfield, E., Markley, J.L., and Sykes, B.D. 1995. 1H, 13C, and 15N chemical shift references in biomolecular NMR. J. Biomol. NMR 6 135–140. [DOI] [PubMed] [Google Scholar]
- Wolfler, A., Schauenstein, K., and Liebmann, P.M. 1998. Lack of calmodulin antagonism of melatonin in T-lymphocyte activation. Life Sci. 63 835–842. [DOI] [PubMed] [Google Scholar]
- Wriggers, W., Mehler, E., Pitici, F., Weinstein, H., and Schulten, K. 1998. Structure and dynamics of calmodulin in solution. Biophys. J. 74 1622–1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaotsu, N., Suga, M., and Hirono, S. 2001. Molecular dynamics simulation of the calmodulin–trifluoperazine complex in aqueos solution. Biopolymers 58 410–421. [DOI] [PubMed] [Google Scholar]
- Zhang, S.P., Prozialeck, W.C., and Weiss, B. 1990. Differential inhibition of calcium-dependent and calmodulin-dependent enzymes by drug-calmodulin adducts. Mol. Pharmacol. 38 698–704. [PubMed] [Google Scholar]
- Zuiderweg, E.R.P. 2002. Mapping protein–protein interactions in solution by NMR spectroscopy. Biochemistry 41 1–7. [DOI] [PubMed] [Google Scholar]
- Zuiderweg, E.R.P. and Fesik, S.W. 1989. Heteronuclear three dimensional NMR spectroscopy of the inflammatory protein C5a. Biochemistry 28 2387–2391. [DOI] [PubMed] [Google Scholar]