

Since the publication of the classical studies of the Dutch scientist J.D. van der Waals in 1873, physical scientists have known that many—if not most—properties of matter can be rationalized by the strength and direction of the forces that molecules exert on each other. Even strictly macroscopic phenomena, such as the elasticity and the melting points of solids, the viscosity and boiling points of liquids, or the compressibility of gases, are macroscopic manifestations of the myriad of small interactions between molecules, and of the forces that molecules exert on one another. Moreover, in 1889, Svante Arrhenius proposed that reactions between molecular species follow pathways that involve the formation of some type of strained, largely unstable, high-energy transition state whose accessibility along the reaction coordinate controls the rate of the reaction. Since then, chemists have recognized that chemical affinity results from the attractive interactions between chemical entities and that stresses and strains often develop within and between these entities in the course of chemical reactions.
Until very recently, chemists and biochemists have had to rely on bulk methods to investigate the properties of molecules and their reactions. These methods did not make it possible to directly investigate the nature, strength, and direction of intermolecular forces and torques. During the last few years, however, the advent of novel methods of single-molecule manipulation have begun to offer researchers, for the first time, the opportunity to measure directly the forces holding molecular structures together, to measure the stresses and strains generated in the course of chemical and biochemical reactions, to exert external forces to alter the fate of these reactions, and to reveal the rules that govern the interconversion of mechanical and chemical energy in these reactions. This area of research can be rightly called mechanochemistry.
Biochemical processes as diverse as protein folding, DNA elasticity, the protein-induced bending of DNA, the stress-induced catalysis of enzymes, the mechanical properties of protein motors, and even the ubiquitous process of induced-fit molecular recognition of proteins for their ligands, are all examples in which stresses and strains develop in molecules as they move along a reaction coordinate. In 1992, we published the results of the first experiments in which a single DNA molecule was mechanically manipulated and stretched to determine its bending and extensional properties (Smith et al. 1992). This work was the basis of a number of assays of DNA-dependent protein machines and laid the experimental and conceptual groundwork for studies of enzyme mechanism at the single molecule level. In what follows, I describe some of the recent and ongoing projects currently being carried out in our laboratory using these novel methods.
I. Mechanical properties of macromolecules
Torque measurements on single DNA molecules1
The physical properties of the DNA double helix are unlike those of any other natural or synthetic polymer. The molecule’s characteristic base stacking and braided architecture lend it unusual stiffness: It takes about 50 times more energy to bend a double-stranded DNA molecule into a circle than to perform the same operation on single-stranded DNA. Moreover, the phosphates in DNA’s backbone make it one of the most highly charged polymers known. The protein machinery involved in copying, transcribing, and packaging DNA has adapted to exploit these unique physical properties. For example, RNA polymerases and heli-cases (which unwind the double helix to provide single-stranded templates for polymerases) have evolved as motors capable of moving along torsionally constrained DNA molecules and of developing sufficient forces and torques to unwind the molecule. Determination of the elastic properties of DNA is, therefore, essential to understanding DNA: protein interactions and the structural dynamics of cellular processes such as replication, transcription, and chromosome condensation.
A complete description of DNA mechanics must also, however, consider the effects of torque, a quantity that heretofore has not been directly measured in micromanipulation experiments. We have recently performed experiments based on optical trapping and video microscopy to measure torque as a function of twist for stretched single DNA molecules (Bryant et al. 2003). In these experiments, torsional strain in over- or underwound molecules was used to power the rotation of submicron beads attached to the sides of the DNA molecule and serving as calibrated viscous loads.
To perform dynamic torque measurements on single DNA molecules, we generated molecular constructs. The use of three distinct chemical modifications of DNA allows for oriented tethering of the ends of the molecule and the subsequent attachment of a rotor to a third, internal position (Fig. 1 ▶). A site-specific nick in the duplex DNA is engineered adjacent to the rotor attachment point; this design allows covalent bonds in the intact strand to serve as free swivels, preventing torque from accumulating in the “lower” DNA segment. Thus, torque stored in the “upper” segment can drive the rotation of a submicron object on a low-friction molecular bearing. At low Reynolds numbers, the magnitude of the torque can be measured by multiplying the observed angular velocity by the rotational drag of the rotor.
Figure 1.

Experimental design. (a) The molecular construct contains three distinct attachment sites and a site-specific nick (asterisk), which acts as a swivel. (b) Each molecule was stretched between two antibody-coated beads using a dual-beam optical trap. A rotor bead was then attached to the central biotinylated patch. The rotor was held fixed by applying a fluid flow, and the micropipette was twisted to build up torsional strain in the upper segment of the molecule. (c) Upon releasing the flow, the central bead rotated to relieve the torsional strain. (d) Video of the rotating bead (Supplementary Movies 2, 3) was analyzed to track cumulative changes in angle. Horizontal sections (red dashed line) of successive video frames can be stacked (e) to allow visualization of the helical path of the bead in space and time. (Left to right) Traces of 920 nm, 760 nm and 520 nm rotor beads.
We thus tested the linearity of DNA’s twist elasticity, directly measured the torsional modulus (Fig. 2 ▶) (finding a value of 430 ± 20 pNnm2, a figure 30% higher than the figure generally accepted), characterized torque-induced structural transitions, and established a framework for future assays of torque and twist generation by DNA-dependent enzymes. In addition, our observations of continuous DNA-powered rotation have implications for the construction of nanomechanical devices: We have demonstrated that cooperative structural transitions in DNA can be exploited to construct either a constant-torque wind-up motor or a force-torque converter.
Figure 2.

Analysis of bead rotation. A tether was overwound by 1200 turns before releasing the 760 nm rotor bead. Red, cumulative rotor angle (Δtwist); green, angular velocity; blue, molecular extension (Δx) vs. time under constant tension (45 pN). The data support a model in which overwinding the molecule converts a fraction of the DNA into P-form. P-DNA converts back into B-DNA at constant torque, after which the molecule regains its B-form extension and the remaining torque decays to 0. (Inset) A molecule was completely converted to P-DNA by introducing 4800 turns before releasing the 920 nm rotor bead. Torsional relaxation of hyper-wound P-DNA preceded the long (~4000 turns) P–B transition. During the gap at ~50 min, rotation was paused by turning on flow.
II. Mechanical unfolding studies
Identifying the kinetic barriers to the mechanical unfolding of T. thermophila ribozyme2
Many regulatory biological processes in the cell such as mitochondrial import, progress of a messenger RNA through the ribosome, the action of RNA helicases in initiation of translation, and spliceosome activity all involve controlled mechanical deformation and unfolding of RNA. In such reactions, chemical energy is converted to mechanical work through the generation of forces that are applied locally to the RNA molecule, leading to its progressive unfolding. Although the structures of RNAs and their folding thermodynamics and kinetics have been the focus of considerable scientific effort, it has proven difficult to investigate their molecular responses to locally applied mechanical forces. What are the magnitudes of the kinetic barriers encountered by the cell machinery that unfolds RNA molecules in vivo? How do they depend on unfolding rates? Where are the barriers located in RNA molecule, and what structures or interactions generate the barriers?
We have recently designed an experiment to determine the strength and location of kinetic barriers opposing the unfolding of single molecules of the L-21 derivative of the Tetrahymena thermophila ribozyme, a 390 nucleotide catalytic RNA (Fig. 3 ▶) whose three-dimensional structure, independently folding domains, intra- and interdomain contacts, and Mg2+-mediated tertiary interactions are well established (Onoa et al. 2003). Thermal unfolding of group I introns has revealed that the helices of the central pseudo-knot (P3 and P7) and their tertiary interactions melt before the rest of the molecule. Although such studies provide important physical insight into the forces that hold together the structure, they do not address the factors that control the unfolding of these structures in vivo. Unfolding of complex RNA molecules such as L21 in the cell is accomplished by helicases that operate as molecular motors and utilize chemical energy to exert mechanical force on their substrates. How does this mechanical unfolding take place, and what molecular factors determine the order of unfolding of the different RNA motifs?
Figure 3.
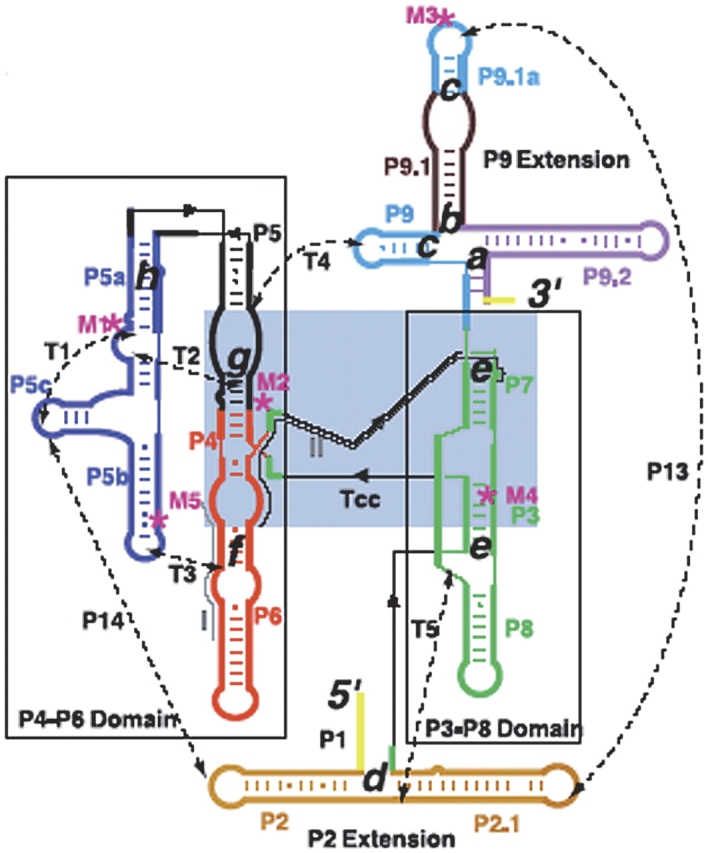
Secondary structure of the L-21 ribozyme. The two main domains, P4-P6 and P3-P8, are boxed; a light blue box indicates the catalytic core. Gray lines label sequences targeted by complementary DNA oligo-nucleotides; dashed lines are tertiary contacts (T) and base paired regions (P); “M” labels are site-directed mutations (M1, A to U at site 186; M2, C to G at 260; M3, UGC to ACG at 348 to 350; M4, U to A at 273; M5, cyclic permutation at 148); cc is the catalytic core. The letters a–h indicate the proposed positions of the kinetic barriers.
Seeking answers to all these questions, we attached L21 T. thermophila ribozyme molecules to polystyrene beads with RNA•DNA hybrid handles, and used optical tweezers to consecutively unfold and refold the molecules under tension. During each experiment, we applied forces between 2 and 30 pN to the molecules at rates of about 3–5 pN s−1 and simultaneously monitored nanometer (nm) changes in their molecular length. We found that mechanical unfolding trajectories for individual molecules display eight intermediates (Fig. 4 ▶) corresponding to discrete kinetic barriers opposing mechanical unfolding with lifetimes of seconds and rupture forces between 10 and 30 pN. Barriers are Mg2+-dependent and we were able to identify the location of these barriers in the molecule and determine the intra- and inter-domain interactions that they involve. Several barrier structures are “brittle”; that is, breaking them requires application of high forces but small (1–3 nm) deformations of the structure, a signature of the mechanical response of tertiary interactions. Moreover, barrier crossing is a thermally activated process and therefore it takes place stochastically, leading to variable unfolding paths. By pulling the molecule many times, we have been able to determine the relative frequency of the various unfolding paths.
Figure 4.

Representative unfolding (black) and refolding (pink) force/extension curves of the L-21 RNA displaying six unfolding events (rips). The RNA is attached by RNA DNA handles to polystyrene beads, which are manipulated by the laser tweezers. Experiments were done at 298 ± 2 K in 10 mM Tris (pH 7), 250 mM NaCl, and 10 mM MgCl2. Letters and arrows correspond to the positions assigned to the kinetic barriers as described in Figure 3 ▶. The unfolding curve chosen here does not display barriers d and g, indicated by the dashed arrows.
We expect that the response of complex RNA structures to locally applied mechanical forces may be similar to the responses of RNA molecules during translation, messenger RNA export from the nucleus and viral replication. Our results indicate that local kinetic barriers imposed by tertiary interactions control the mechanical unfolding of RNAs in these cellular processes.
III. The mechanochemistry of DNA binding molecular motors
Our laboratory is also involved in characterizing several of the fundamental nuclear processes in the cell, including the control of DNA topology and DNA storage, replication, and transcription. These processes are carried out by specialized machinery that function as molecular motors, being able to convert chemical into mechanical energy, movement, and the generation of force. The mechanochemistry of these motor molecules is still poorly understood, in part, because of the inherent complexity of the systems, but also because of the lack of appropriate methods to characterize the structure and function of complex molecular machines. Here again, single molecule methods make it possible to address the structural dynamics and the mechanochemical conversion performed by these tiny machines in real time. Below, I describe two of our recent studies in this area (Smith et al. 2001; Stone et al. 2003).
Chirality sensing by E. colitopoisomerase IV and the mechanism of type-II topoisomerases3
Escherichia coli topoisomerase IV (topo IV) is an essential type-II topoisomerase that removes DNA entanglements created during DNA replication. Topo IV relaxes (+) super-coils much faster than (−) supercoils, promoting replication while sparing the essential (−) supercoils. The question of how is it that the enzyme can discriminate between positive and negative supercoils has been a subject of much discussion. It is possible to imagine that topoIV either recognizes the overall writhe of the supercoiled plectoneme, that the enzyme is able to detect a difference in DNA twist that accompanies the formation of (+) and (−) supercoils in closed intact circular DNA molecules, or that the active site of the enzyme recognizes the chirality of the double stranded DNA crossing of supercoils of opposite sign. It is not easy to distinguish among these different models using bulk methods, because changes in twist and writhe are almost impossible to separate in supercoiled circular molecules. Thus, to investigate the chiral mechanism underlying the substrate preference of the enzyme, we adapted the experimental set up described above for the torsional elasticity of DNA (Stone et al. 2003). The idea was to present the enzyme with a substrate in which it could control the writhe with out modifying its twist. To this end, two nicked double stranded DNA molecules were tethered between two beads and were braided by twisting one of the beads by rotating the micropipette to which it was attached (Fig. 5 ▶). In this way it was possible to present the enzyme with (+) or (−) writhe without affecting the twist of the molecules involved in the braid. These experiments unequivocally showed that topo IV recognizes the chiral crossings imposed by the left-handed superhelix of a (+) supercoiled DNA (Fig. 6 ▶), rather than global topology, twist deformation, or local writhe. Monte Carlo simulations of braid, supercoil, and catenane configurations demonstrate how a preference for a single crossing geometry can allow topo IV to perform its physiological functions. Single-enzyme braid relaxation experiments also provided a direct measure of the processivity of the enzyme and offered insight into its mechanochemical cycle.
Figure 5.

Characterization of a single-molecule DNA braiding system. Two nicked DNA molecules (solid black lines) are oriented between one bead on a rotating micropipette and another bead held in an optical trap. As braiding density, |σbr|, increases, the length of the braid, Z, diminishes gradually until a critical braiding density is reached, | σbr crit|, at which point the braid buckles, forms a second-order plectonemic superhelix, and shortens much more dramatically with each introduced turn.
Figure 6.

Topo IV unbraiding of single DNA braids. Preferential relaxation of left-handed braids. Topo IV activity was assayed by monitoring the extension of the braids held under constant tension (1 pN). Raw data are plotted in green (left-handed braid) and red (right-handed braid), and a 1-sec moving average is shown as a solid black line. (Inset) Histogrammed rates of unbraiding were calculated as ΔZ/Δt for successive 5-sec windows in 10 independent runs each on right- and left-handed braids (|σbr| = 0.05). To avoid effects of limiting substrate, only those portions of the traces where |σbr| >0.02 were included. The mean unbraiding rate was 9 ± 1.4 nm/sec for left-handed braids (green) and 0.3 ± 0.3 nm/sec for right-handed braids (red).
Studies of DNA packaging by single φ29 bacteriophage particles using optical tweezers4
As part of their infection cycle, many viruses must package their newly replicated genomes inside a protein capsid to insure its proper transport and delivery to other host cells. Bacteriophage φ29 packages its 6.6 mm long double-stranded DNA into a 42 nm dia. × 54 nm high capsid via a portal complex that hydrolyzes ATP. This process is remarkable because entropic, electrostatic, and bending energies of the DNA must be overcome to package the DNA to near-crystalline density. We have used the optical tweezers instrument built in our laboratory to pull on single DNA molecules as they are packaged by individual particles of bacteriophage φ29, thus demonstrating that the portal complex is a force generating motor (Smith et al. 2001). Moreover, we find that this motor can work against loads of up to ~57 pN on average, making it one of the strongest molecular motors ever described. Movements of over 5 mm are observed, indicating high processivity during the packaging reaction. Pauses and slips also occur, particularly at higher forces. We establish the force–velocity relationship of the motor and find that the rate-limiting step of the motor’s cycle is a force dependent step even at low loads and associated with the translocation of the DNA molecule. Interestingly, the packaging rate begins to decrease when the prohead has been filled with just about 50% of the total genome of the virus, indicating that, at this point in the process, an internal pressure builds up due to DNA compression inside the capsid. We estimate that at the end of the packaging an internal force of nearly 50 pN opposes the packaging of the DNA inside the capsid. This force corresponds to a pressure of ~6 MegaPascals or 60 atmospheres. It is valid to ask, why would nature design such a powerful motor and spend all this ATP energy just to package the viral genome. This question is especially appropriate given that if making the head twice as long could have avoided the enormous pressures developed and large energies utilized in the process. The phage strategy, however, makes perfect sense. To package its DNA, the phage uses a lot of ATP, but it is the host’s ATP and it uses it where it can find it in abundance, i.e., in the interior of the host cell. But the phage must solve an additional problem, how to inject its DNA into a new host cell at the beginning of the next infectious cycle when it is sitting in the extra-cellular medium where there is no ATP. The solution is indeed mechanical. The solution is to convert the energy from ATP hydrolysis used during the packaging into mechanical potential energy stored in the DNA in a loaded spring-like fashion. This potential energy can then be utilized to initiate the process of DNA injection during infection, where the virus cannot use an active, motor-driven mechanism of injection.
Article and publication are at http://www.proteinsci.org/cgi/doi/10.1110/ps.041064504.
Footnotes
References
- Bryant, Z., Stone, M.D., Gore, J., Smith, S.B., Cozzarelli, N.R., and Busta-mante, C. 2003. Structural transitions and elasticity from torque measurements on DNA. Nature 424 338–341. [DOI] [PubMed] [Google Scholar]
- Onoa, B., Dumont, S., Liphardt, J., Smith, S.B., Tinoco Jr., N., and Bustamante, C. 2003. Identifying the kinetic barriers to mechanical unfolding of the T. thermophila ribozyme. Science 299 1892–1895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith, S.B., Finizi, L., and Bustamante, C. 1992. Direct mechanical measurements of the elasticity of single DNA molecules by using magnetic beads. Science 258 1122–1126. [DOI] [PubMed] [Google Scholar]
- Smith, D.E., Tans, S.I., Smith, S.B., Grimes, S., Anderson, D.E., and Busta-mante, C. 2001. The bacteriophage 29 portal motor can package DNA against a large internal force. Nature 413 748. [DOI] [PubMed] [Google Scholar]
- Stone, M.D., Bryant, Z., Crisona, N.J., Smith, S.B., Vologodskii, A., Busta-mante, C., and Cozzarelli, N.R. 2003. Chirality sensing by Escherichia coli topoisomerase IV and the mechanism of type II topoisomerases. Proc. Natl. Acad. Sci. 100 8654–8659. [DOI] [PMC free article] [PubMed] [Google Scholar]