

Abstract
The crystal structure of the flavoprotein Pad1 from Escherichia coli O157:H7 complexed with the cofactor FMN has been determined by the multiple anomalous diffraction method and refined at 2.0 Å resolution. This protein is a paralog of UbiX (3-octaprenyl-4-hydroxybenzoate carboxylyase, 51% sequence identity) that catalyzes the third step in ubiquinone biosynthesis and to Saccharomyces cerevisiae Pad1 (54% identity), an enzyme that confers resistance to the antimicrobial compounds phenylacrylic acids through decarbox-ylation of these compounds. Each Pad1 monomer consists of a typical Rossmann fold containing a non–covalently bound molecule of FMN. The fold of Pad1 is similar to MrsD, an enzyme associated with lantibiotic synthesis; EpiD, a peptidyl-cysteine decarboxylase; and AtHAL3a, the enzyme, which decarboxylates 4′-phosphopantothenoylcysteine to 4′-phosphopantetheine during coenzyme A biosynthesis, all with a similar location of the FMN binding site at the interface between two monomers, yet each having little sequence similarity to one another. All of these proteins associate into oligomers, with a trimer forming the common structural unit in each case. In MrsD and EpiD, which belong to the homo-dodecameric flavin-containing cysteine decarboxylase (HFCD) family, these trimers associate further into dodecamers. Pad1 also forms dodecamers, although the association of the trimers is completely different, resulting in exposure of a different side of the trimer unit to the solvent. This exposure affects the location of the substrate binding site and, specifically, its access to the FMN cofactor. Therefore, Pad1 forms a separate family, distinguishable from the HFCD family.
Keywords: flavin mononucleotide, decarboxylase, UbiX, crystal structure
Decarboxylation is a common reaction in living organisms, occurs in various catabolic and anabolic processes, and is carried out by a wide variety of enzymes. Within the IUPAC classification scheme, the enzymes catalyzing these reactions are presently divided into 76 different classes (E.C.4.1.1.–). Some of these decarboxylases use flavin as a cofactor. In Escherichia coli, flavin-dependent decarboxylases are involved in ubiquinone biosynthesis. All organisms synthesize ubiquinone using similar pathways, although some steps in these pathways differ (Jonassen and Clarke 2000; Meganathan 2001). The biosynthesis of ubiquinone commences with the conversion of chorismate to 4-hydroxybenzoate by UbiC, followed by a transfer of the aliphatic chain of farnesylfarnesylgeranyl-PP to the hydroxybenzoate by UbiA. These two genes are transcriptionally coregulated (Soballe and Poole 1997). In the third step in E. coli and Salmonella enterica, the hydroxybenzoate head group is decarboxylated to a phenol by the action of a carboxylyase. In yeast, this reaction occurs in the fifth step.
There are two isofunctional enzymes in the K-12 strain of E. coli: UbiD (Leppik et al. 1976), and UbiX (Howlett and Bar-Tana 1980), which can catalyze this reaction (Cox et al. 1969; Meganathan 2001). Their amino acid sequences share no similarity. UbiX, a 21-kDa protein, may require a flavin nucleotide as a cofactor (Breinig et al. 2000), whereas UbiD is a 55-kDa protein requiring divalent metals for activity (Leppik et al. 1976; Zhang and Javor 2000). Of the two enzymes, UbiD accounts for almost 80% of the total activity. The regulation of these two genes was also recently studied (Zhang and Javor 2003). There is very limited biochemical data available for either of these enzymes, and it is not clear why E. coli and some other bacteria require both of these enzymes.
Several other E. coli strains, including the enterohaem-orrhagic O157:H7 strain, also contain in addition to UbiX a second paralog named Pad1. Its amino acid sequence shows 52% identity to UbiX and slightly higher sequence identity to Saccharomyces cerevisiae phenylacrylic acid decarboxylase Pad1 (Clausen et al. 1994). The E. coli Pad1 has not been biochemically characterized and is annotated in various databases as a putative phenylacrylic acid decarboxylase based solely on sequence similarity to the yeast enzyme (Perna et al. 2001). Its biological role is unknown at present. Together with UbiX, Pad1 is classified in the InterPro database as a member of the phenylacrylic acid decarboxylase, 3-octaprenyl-4-hydroxybenzoate carboxylyase family IPR004507. Multiple sequence alignment using hidden Markov models indicates that this family belongs to the flavoprotein superfamily IPR003382, which contains mono-and bifunctional enzymes. There are no known three-dimensional structures for enzymes from the UbiX family. However, structures of three enzymes from the flavoprotein superfamily are presently known; namely, Arabidopsis thaliana phosphopantothenoylcysteine decarboxylase Athal3a (PDB code 1MVL; Steinbacher et al. 2003), Staphylococcus epidermidis peptidyl-cysteine decarboxylase EpiD (PDB code 1G63; Blaesse et al. 2000), and Bacillus sp. MrsD (EpiD family, PDB code 1P3Y; Blaesse et al. 2003). EpiD, AtHAL3a, and MrsD are cysteine decarboxylases active on similar substrates, but whereas the first two use flavin mononucleotide (FMN) as a cofactor, MrsD uses flavin adenine dinucleotide (FAD). Pairwise sequence alignment of Pad1 from E. coli O157:H7 with each of these three enzymes using BLAST (Tatusova and Madden 1999) finds no detectable similarities between them.
We describe here the structure of Pad1 from E. coli O157:H7 at 2.0 Å resolution, the first representative of the UbiX family, and compare its substrate specificity with yeast Pad1, showing that despite high sequence similarity, they belong to different classes in the E.C. classification.
Results and Discussion
PAD1 was purified to homogeneity, as judged by SDS-PAGE electrophoresis. The molecular weight of purified SeMet-labeled protein was measured by electron spray ion-ization mass spectrometry and gave a value of 23,175 ± 5 Da, which is very close to the expected MW = 23183 Da for the His-tagged SeMet protein. Dynamic light scattering measurements of the purified protein showed that Pad1 is monodispersed in solution with a molecular weight of ~260 kDa, consistent with the presence of dodecamers. This situation was further confirmed by gel filtration on a Superose 12 column calibrated with standards in which the protein was eluted as a single peak at a volume corresponding to a molecular weight of ~295 kDa. The purified protein had a yellow color, indicating the presence of a cofactor. Indeed, the absorption spectrum of the protein solution had maxima at 382 and 460 nm, characteristic of FMN.
Monomer structure
There are four independent molecules in the asymmetric unit. Each monomer of E. coli Pad1 consists of a single domain with a three-layered α/β/α structure (Fig. 1A ▶). The topology of the secondary structure elements corresponds to that of the Rossmann fold (Rossmann et al. 1974), consistent with the prediction based on sequence analysis that Pad1 is a flavin-binding protein. The central β-sheet is composed of six parallel β-strands (β1–β6) in the order of 3–2–1–4–5–6. The sheet is flanked on either side by three β-helices approximately parallel to the β-strands. There is an additional α-helix (α2) following strand β2, which is oriented perpendicularly to the strands (Fig. 1A ▶).
Figure 1.
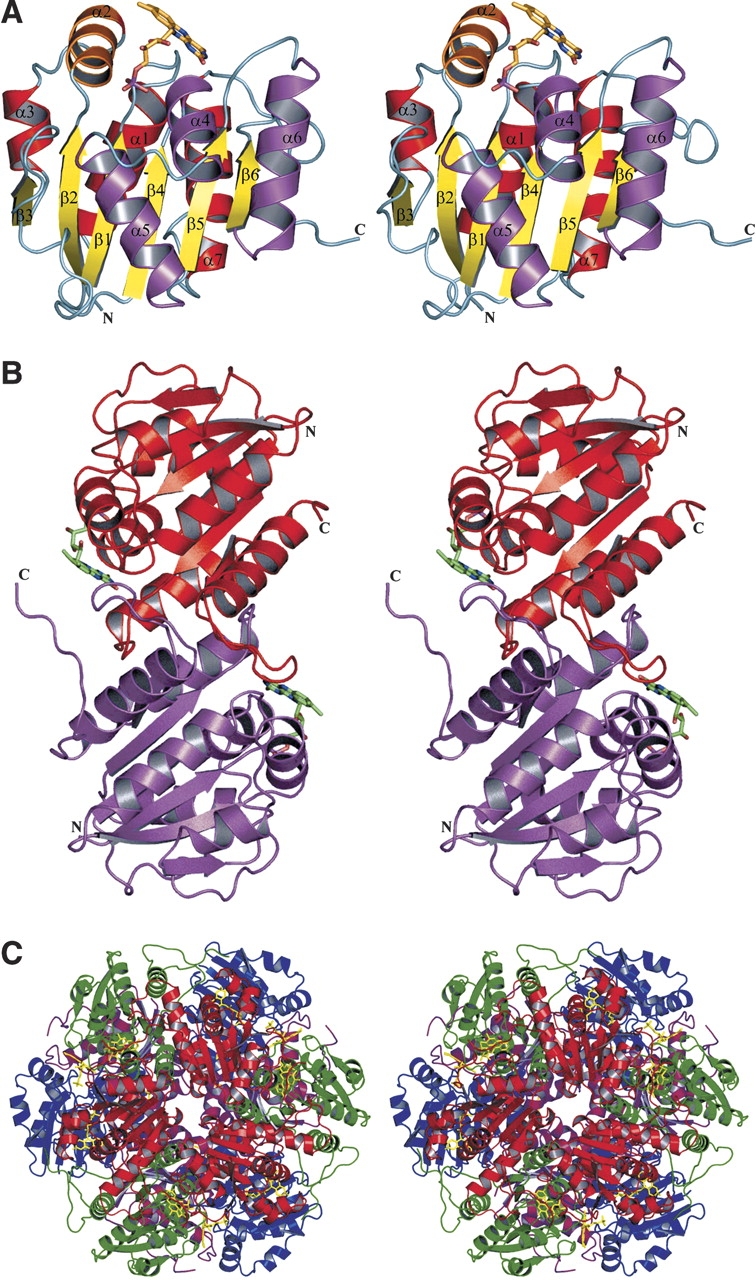
Structure of Pad1 decarboxylase. (A) Monomer: The central β-sheet is colored in yellow and the helices on one side of the β-sheet are shown in red, while on the other side they are in magenta. The helix on the top of the sheet and perpendicular to the strands is shown in orange. The FMN cofactor is shown as a stick model. (B) Pad1 dimer: The dimers associate tightly through interactions of the loops 121–127 and 146–158, helix α6 (128–140) and strand β6 (143–145) in one molecule with their counterparts in the second molecule. (C) Pad1 dodecamer: The four molecules within the asymmetric unit are colored blue, magenta, green, and red. This figure was prepared using the program PyMOL (http://www.pymol.org).
The C-terminal ~15 residues are flexible and are disordered to various extents in each of the four independent molecules. The best-ordered C terminus is in molecule D, where it extends toward the FMN cofactor bound to a neighboring molecule. The proximity of the C terminus to the active site, as well as its flexibility, together suggest it may play an important role in controlling substrate accessibility (see below). The four copies of Pad1 in the asymmetric unit are very similar and can be superimposed on each other with a root-means-squares deviation of ~0.25 Å for 177 Cα atoms.
Oligomeric state and crystal packing
Dynamic light scattering and gel filtration experiments indicated that Pad1 forms large oligomers, most likely dodecamers. The crystal structure showed that Pad1 molecules are indeed assembled into dodecamers in the crystals. The basic repeating unit of the dodecamer is a dimer, with the two molecules related by two-fold noncrystallographic symmetry (Fig. 1B ▶). There are two such dimers in the asymmetric unit, and they can be superimposed with a root-means-square (rms) deviation of 0.21 Å, indicating that the independent dimers are virtually identical. The dimers associate tightly through interactions of the loops 121–127 and 146–158, helix β6 (128–139), and strand α6 (143–145) in one molecule with their counterparts in the second molecule. The surface area of the monomer that becomes buried by dimerization is ~3170 Å2, which accounts for 16% of the total surface area of the monomer (calculated using the program GRASP with a probe radius of 1.4 Å; Nicholls et al. 1991). Six such dimers (two are crystallographically independent, together with their copies related by crystallographic three-fold symmetry) form a dodecamer, with a roughly spherical shape and with dimensions of 96 × 93 × 87 Å3 (Fig. 1C ▶). The dodecamer has approximate 32-point symmetry, with the molecules around the three-fold axis forming distinct trimers (see following). The dodecamer that exists in solution and constitutes the biologically functional unit is most likely the same as that observed in the crystal.
Comparison with other structures
Structural neighbors
Pad1 is grouped with ~250 other proteins in the flavo-protein superfamily IPR003382 (InterPro database; Apweiler et al. 2001) or family PF02441 in PFAM (Bateman et al. 2002), encompassing an ~120 amino acid–long segment. The proteins in this superfamily include two families: that of UbiX (phenylacrylic acid decarboxylase, 3-octaprenyl-4-hydroxybenzoate carboxylyase, IPR004507) with only a small number of known members, and that of a larger family that comprises the N-terminal domain of Dfp (DNA/pantothenate metabolism flavoprotein, IPR005252). The three-dimensional structures of three enzymes from the latter family are presently known: that of the MrsD from Bacillus sp. HIL-Y85/54728 (PDB code 1P3Y; Blaesse et al. 2003), the halotolerance protein AtHal3a from Arabidopsis thaliana (PDB code 1E20; Steinbacher et al. 2003), and a peptidyl-cysteine decarboxylase EpiD from Staphylococcus epidermidis Tü3298 (PDB code 1G5Q; Blaesse et al. 2000). The structure of Pad1 presented here is, however, the first representative of the UbiX family. In accordance with the assignment of all of these proteins to the same superfamily, a search for structural homologs using the DALI server (Holm and Sander 1995) showed that these three proteins are indeed closely structurally related to Pad1, each having Z-scores of ~17. The structure-based alignment of proteins from the Dfp family shows that they are rather distantly related to E. coli Pad1, sharing only ~15% sequence identity with it.
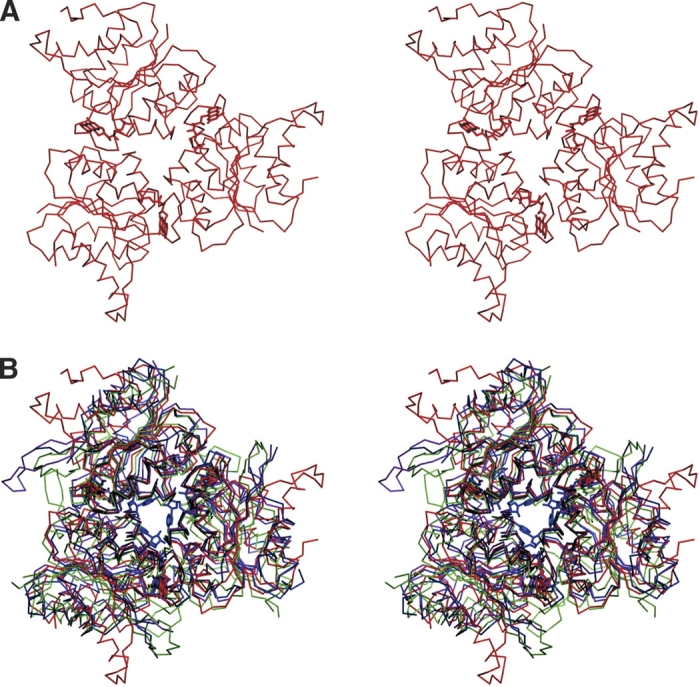
The superposition of Pad1, EpiD, MrsD, and AtHal3a monomers is shown in Figure 2 ▶. Despite the low identity between their amino acid sequences, the monomers’ three-dimensional structures are quite similar, with an rmsd of 1.60 Å for 128 Cα atoms, 1.66 Å for 130 Cα atoms, and 1.60 Å for 128 α atoms, respectively. There are two regions of variability among these four proteins, encompassing residues 62–76 and 146–158, corresponding to Pad1 numbering. The Pad1 region that forms the 62–76 loop is shorter than in the other proteins and contains no secondary structure, whereas the other proteins contain a short α-helix within this region (Fig. 2 ▶). The second region, 146–158 in Pad1, forms a long extension, whereas the corresponding regions of polypeptide in the other proteins adopt quite different conformations. This loop is well ordered in EpiD (Blaesse et al. 2000) as well as in the Pad1 structure presented here. In contrast, the corresponding polypeptide segment in MrsD and in Hal3A proteins is disordered in the absence of substrate. In the structure of Hal3A complexed with substrate, this loop becomes well ordered and forms a substrate binding clamp (Steinbacher et al. 2003).
Figure 2.

Superposition of Pad1 and related structures. A stereo view of the superposition of the Cα traces of Pad1 (thin solid lines) with EpiD (dotted lines; PDB code 1G5Q), Hal3A (thin dashed lines; PDB code 1E20), and MrsA (thick dashed lines; PDB code 1P3Y). The regions 62–76 and 143–164 in Pad1, where the largest differences between the structures exist, are indicated by thicker lines. Figure prepared using Swiss PDB Viewer (Guex and Peitsch 1997) and Povscript (Fenn et al. 2003) and rendered in Povray (http://www.povray.org/).
Oligomers
The formation of oligomers is a common feature of the three other proteins from this superfamily with known three-dimensional structures. AtHal3 forms trimers in the crystal, and MrsD and EpiD form dodecamers. However, the basic dimeric unit observed in Pad1 is unique to this protein, as it results from the crucial involvement of the β-strand 143–145 (β6) and loop 146–158 in Pad1 dimerization, a loop that has very different conformations in the other enzymes. In contrast, the association into trimers, observed in AtHal3 (Albert et al. 2000), is common to all the other structures (Fig. 3 ▶). These trimers are formed through the contacts involving mainly three helices—α4, α5, and α6—on one side of the β-sheet. These helices are structurally well conserved among these proteins, as are the trimers themselves (Fig. 3B ▶). The trimers have an overall shape of thick triangular-shaped boxes with two relatively flat surfaces. In each case, the cofactors are bound on one side and near the center of the trimer.
Figure 3.
Trimeric arrangement of Pad1 monomers. Stereo views of (A) trimer of Pad1 made of monomers related by the crystallographic threefold axis; (B) superposition of the trimers of Pad1 in red, EpiD in magenta, AtHal3a in green, and MrsD in blue.
In MrsD and EpiD these trimers associate further into dodecamers, which form the biologically functional unit of these enzymes (Blaesse et al. 2000, 2003). In these enzymes, the side of the trimer where the cofactor binds is directed toward the center of the dodecamer, and the opposite face is exposed to the solvent. The biological unit of Pad1 is also a dodecamer. Though dodecamer formation is through the association of six dimers, the monomers from three adjacent dimers also result in the formation of two different kinds of trimers that differ in their mutual interactions. The first type of trimer is one that is formed by monomers B, C, and Dsym (crystallographically related molecule of D). The buried surface area in this arrangement of trimers is ~2071 Å2, which corresponds to 7% (690 Å2) of the total surface area of each monomer. The second type of trimer is formed by the monomers C, D, and Asym (crystal-lographically related molecule of A; Fig. 3A ▶) and has a buried surface area of ~4218 Å2, or 14% (1406 Å2) of the total surface area of each monomer. Of the two types of trimers, the latter one is similar to that found in EpiD, MrsD, and Athal3a. Unlike in MrsD and EpiD, however, neither of the two possible trimers of PAD1 forms the repeating unit of the dodecamer. The formation of a dodecamer is mediated through the tight dimeric association (see above) of monomers from different trimers and leads to solvent exposure of the face of the trimer that contains bound FMN (Fig. 4 ▶). The relationship between these two types of dodecamers can be described as an inside-out flipping. Consequently, access to the cofactor and substrate binding sites is different in Pad1 from that in MrsD/EpiD. Inspection of the MrsD/EpiD dodecamers shows larger tunnels leading toward the center of the dodecamer, whereas the Pad1 dodecamer is more compact (Fig. 4 ▶). This is consistent with the nature of the peptide substrate used by EpiD, MrsD, and Athal3a, which could be accommodated by the central cavity.
Figure 4.
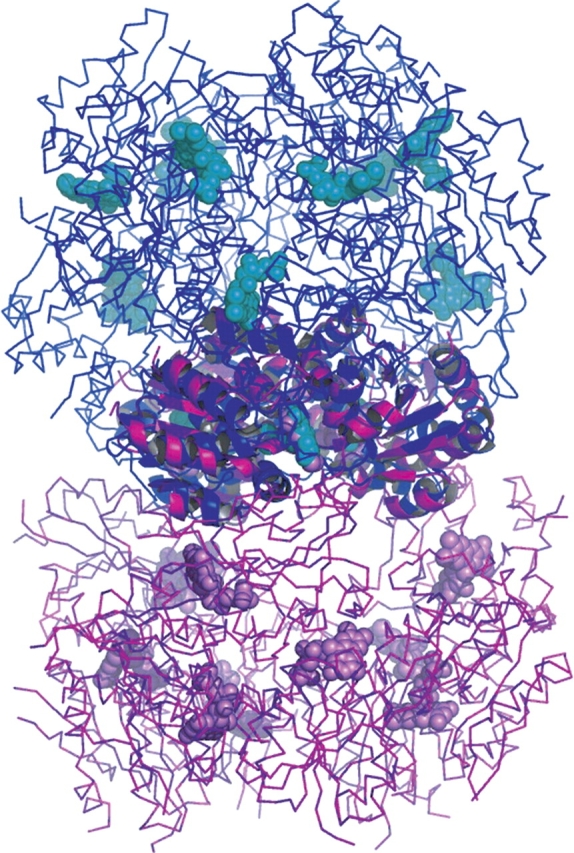
The dodecamers of Pad1 and MrsD. The superposition was done in such a way that the corresponding trimers were superimposed first and then the rest of the dodecamer was drawn from the initial trimers. Although the trimers have a common structure, the interactions between the trimers differs in Pad1 and MrsD/EpiD, resulting in an “inside-out” relationship of these dodecamers. The cofactors are on the outside of the dodecamer in Pad1, and they are inside the dodecamer in MrsD/EpiD.
Although the trimeric quaternary structure is common to all proteins from this superfamily, many proteins associate into larger oligomers. If additional favorable contacts between the monomers can be formed, as is the case of Pad1, MrsD, and EpiD, larger oligomeric structures arise, with this association occurring in several distinct ways. As EpiD, MrsD, and AtHal3a show significant sequence similarity and exhibit a similar catalytic mechanism of decarboxylation, they have been grouped within the homo-oligomeric flavin-binding cysteine decarboxylase (HFCD) family of proteins. The UbiX-like proteins represent a novel dodecameric organization and form a separate family, which differs from the HFCD family at the level of quaternary structure. With respect to ligand binding, E. coli Pad1 shares neither overall sequence similarity nor the substrate-binding motif common to the HFCD family.
Interaction with FMN
A strong feature was identified in the difference electron density map near the C-terminal ends of the central strands of the β-sheet, which indicated the presence of a bound cofactor. Based on the shape of this electron density the cofactor was interpreted as a molecule of FMN (Fig. 5A ▶), in agreement with the absorption spectrum of the protein sample. Each of the four independent Pad1 molecules in the asymmetric unit contains a FMN cofactor bound in a similar way. The FMN molecule is located in a tunnel formed at the interface between two monomers of a trimeric unit (see above). The flavin and the phosphate group are partially exposed to the solvent at the two opposite ends of this tunnel. The flavin-occupied end is partially closed off by the 146–158 loop from a third Pad1 monomer belonging to a different trimer. The majority of the interactions are with one monomer (monomer A) and include contacts of the si side of the FMN aromatic ring system and the ribityl chain, with the surface created by the secondary structural elements β1, α1, β4, and α4 (residues Thr8A–Thr11A, Ser36A–Trp38A, Ser87A–Thr90A, and Arg122A–Glu123A). The terminal phosphate group forms five direct hydrogen bonds to the backbone and side chain atoms and several more through bridging water molecules (Fig. 5B ▶). One of these water molecules (WAT25) not only is conserved in the four independent copies of Pad1 in the asymmetric unit but is also present in EpiD (Blaesse et al. 2000), MrsD (Blaesse et al. 2003), and AtHal3 (Albert et al. 2000). The re side of the flavin ring is flanked by the side chain of Arg105B from the second monomer of the trimer. This arginine forms a salt bridge with Glu123A of monomer A. In addition, Gln67B and Ala99B are hydrogen bonded to the hydroxyl group of the ribityl chain.
Figure 5.
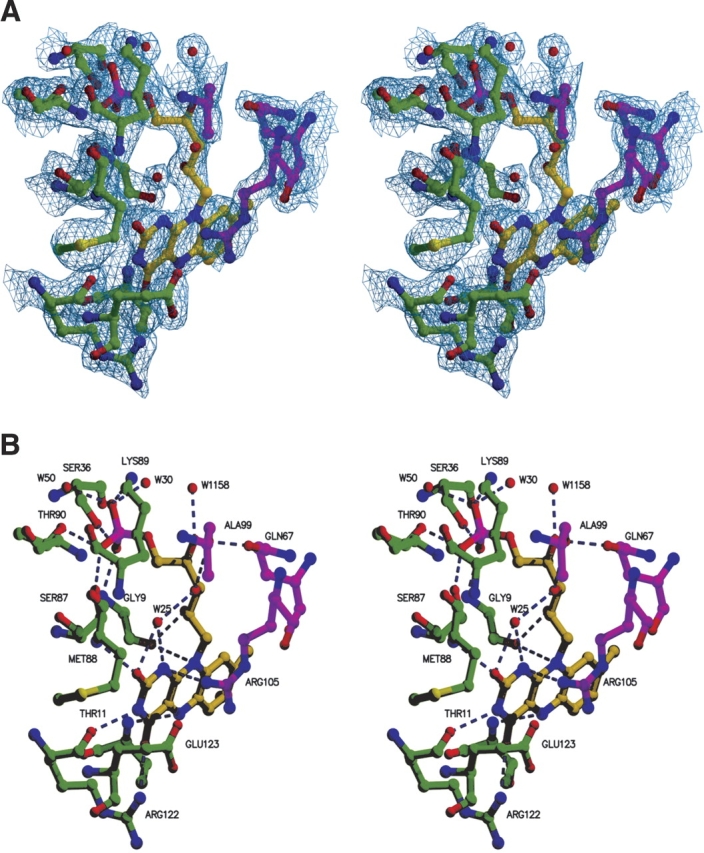
FMN binding site. (A) Simulated-annealing 2Fo-Fc omit map of the Pad1 active site region. The residues depicted and the FMN molecule both were omitted before refinement. The map is contoured at a level of 1σ. (B) FMN binding site showing hydrogen bonding interactions (dashed lines) with two different Pad1 monomers.
The isoalloxazine ring of FMN is properly oriented not only by being sandwiched between Gly9A–Ala10A–Thr11A on one side and by Glu123A and Arg105B on the other side but also through hydrogen bonds to the two-ring carbonyl oxygens (backbone of Met88A and NH1 of Arg122A) and to the flavin N3 atom between them (Arg122A backbone carbonyl) (Fig. 5B ▶). The group directly involved in the oxidation reaction and that controls the redox potential of FMN has to be localized close to the flavin N5 atom. This role in Pad1 is likely played by the backbone amide of Ser11 (Ile13 in EpiD), which acts as a hydrogen bond donor and is situated 3.2 Å from N5, which is within the range of 2.8–3.3 Å that is usually observed (Fraaije and Mattevi 2000).
Sequence alignment
A search with PSI-BLAST (Altschul et al. 1997) for related sequences finds several hundred sequences sharing significant similarity with Pad1. We have analyzed more closely the top 160 sequences, which all had lengths of ~200 residues and had over 30% sequence identity to Pad1. The majority of these sequences are annotated as (putative) phenylacrylic acid decarboxylases or 3-octaprenyl-4-hydroxybenzoate carboxylyases. A group of proteins with less sequence similarity and with a length of ~400 residues are annotated as DNA/pantothenate metabolism flavoproteins. Although they belong to the same superfamily, they clearly differ in substrate specificity and biological function.
Among the 160 sequences there are ~25 residues conserved in all sequences, as highlighted in a subset of these sequences, shown in Figure 6 ▶. Within the context of a single monomer, these residues seem to be scattered throughout and do not show substantial clustering. However, when the location of these residues is analyzed in the context of a trimer and the entire dodecamer, a different picture emerges (Fig. 6A ▶). Most of the invariant residues cluster around the FMN binding site, or nearby, in the putative substrate binding site. Of these, Gly9, Ser/Thr11, Ser36, Ser/Thr87, and Arg122 are clearly involved in FMN binding. In addition, residue Arg105 is involved in binding the cofactor and most likely in substrate binding. Finally, Ser73, Glu123, Tyr152, and Trp183 are part of the putative substrate-binding site. Of the three other invariant residues, Leu102 and Leu117 are embedded in aliphatic clusters, and Pro125 assumes a cis conformation.
Figure 6.
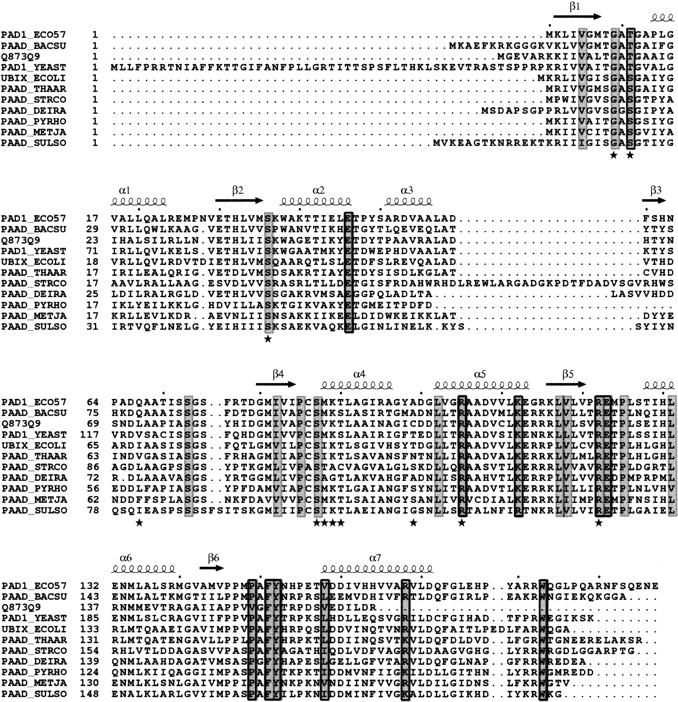
Sequence alignment of Pad1 and selected sequences. Conserved residues are shaded gray, with residues involved in dimerization boxed, and those involved in FMN binding highlighted by a star. Secondary structure elements of Pad1 are depicted above the sequence alignment. Sequences include Escherichia coli O157:H7 Pad 1 (gi15832847), Bacillus subtilis PaaD (gi13124411), S. cerevisiae PaD (gi1709555), Nectria haematococca PaaD (gi29289980), E. coli UbiX (gi2507150), Thauera aromatica PaaD (gi13124408), Streptomyces coelicolor PaD (gi13124421), Deinococcus radiodurans PaaD (gi13124427), Pyrococcus horikoshi PaD (gi13124401), Methanocaldococcus jannaschii PaaD (gi2495798), and Sulfolobus solfataricus PaaD (gi13124432). Sequence alignment was performed using the program ClustalW (Chenna et al. 2003) and formatted using the program ESPript (Gouet et al. 2003).
The clustering of invariant residues around the FMN cofactor when a trimer is considered as one unit provides a strong indication that this association is common to the entire UbiX family (Fig. 6 ▶). Taken together with the observed trimeric association in the HFCD family, a trimeric structure is expected to be common to all proteins from the flavoprotein superfamily IPR3382 (InterPro). As mentioned above, the trimers in many proteins from this superfamily combine to form dodecamers, and two types of dodecameric association have been identified. One type is present in the HFCD family, and a different one is described here for the UbiX family. The association of trimers in Pad1 is driven by the formation of tight dimers (along a pseudo two-fold axis) between two molecules from different trimers. We therefore analyzed the alignment of 160 proteins from the UbiX family for the conservation of residues involved in this dimerization. Two loops are specifically involved in the dimerization, 121–127 and 146–158, which lie on one side of the molecule. Within the prism-shaped trimer, these residues are near the corners of the prism. Nevertheless, several of these residues are highly conserved in type. In the first loop, Pro125 is invariant (cis conformation, see above), and 131 is either leucine or isoleucine. These residues are on the surface of the trimer and interact with a hydrophobic patch on the other monomer. The aliphatic portion of an invariant Arg122 side chain also contributes to the van der Waals interactions, and its polar group interacts with FMN. Within the second loop, there is a stretch of seven hydrophobic residues containing three prolines. This region maintains a hydrophobic character in all aligned sequences and has at least two prolines: Pro146 is highly conserved, replaced in some sequences by an alanine, and the next position is equally likely to be a proline or an alanine. Residues 149–150 are most likely Pro-Ala, and residue 151 is most likely phenylalanine, tyrosine, or tryptophan. In addition, residue 158 is always aliphatic—a leucine, isoleucine, methionine, or valine—and 168 is either lysine or arginine. The conservation of residues forming the dimer interface indicates that all these proteins are likely to form dimers and therefore associate in dodecamers of the same type as Pad1.
Active site and specificity
The E. coli Pad1 has been annotated in ExPASy as a probable aromatic acid decarboxylase and as a ‘phenylacrylic acid decarboxylase, 3-octaprenyl-4-hydroxybenzoate carboxylyase’ in the InterPro database. We have tested the activity of Pad1 on several commercially available phenyl-acrylic acids, including trans-cinnamic acid, p-coumaric acid, caffeic acid, vanillic acid, and ferulic acid. We compared the activity of the purified enzyme to that of the phenolic acid decarboxylase PadA from Bacillus sp BP-7 and yeast Pad1. Decarboxylase activity was measured by the increase in intensity of a new absorption maximum at a wavelength of ~260 nm, corresponding to the vinyl product of these acids. Although high activity for these substrates was measured with PadA (with the exception of trans-cin-namic acid and vanillic acid), no significant activity could be detected using both crude E. coli lyasates containing overexpressed Pad1 and purified E. coli Pad1. It seems, therefore, that by analogy to the UbiX protein, Pad1 may remove the carboxylate group from derivatives of benzoic acid but not from substituted phenolic acids.
This notion is strongly supported by the examination of the amino acid sequence conservation between the E. coli paralogs Pad1 and UbiX. The si face of the isoalloxazine ring of FMN abuts the protein, and most interactions are of the van der Waals type. The residues contacting this face of FMN are not identical in Pad1 and UbiX. However, residues in the vicinity of the re face of FMN, on which side the substrate is expected to bind and where a cavity is observed, are almost all identical in Pad1 and UbiX. The residues forming the cavity come from three different monomers (Fig. 7A ▶). In two of the active sites contained within a trimer, this cavity opens toward the solvent, whereas the third active site is closed off by the only ordered C terminus (aa 178–183) among the three monomers. It is likely that the C termini play the role of gatekeepers for accessing the active site. The strictly conserved residues of the UbiX family likely involved in substrate binding are Ser73A, Arg105A, Lys112A, Arg122B, Glu123B, Pro125B, Tyr152C, Arg168C, and Trp183C (Fig. 7B ▶). Residues equivalent to Glu123 and Pro125 (i.e., the EXP motif) have previously been suggested to play a role in either substrate binding or catalysis (Breinig et al. 2000).
Figure 7.
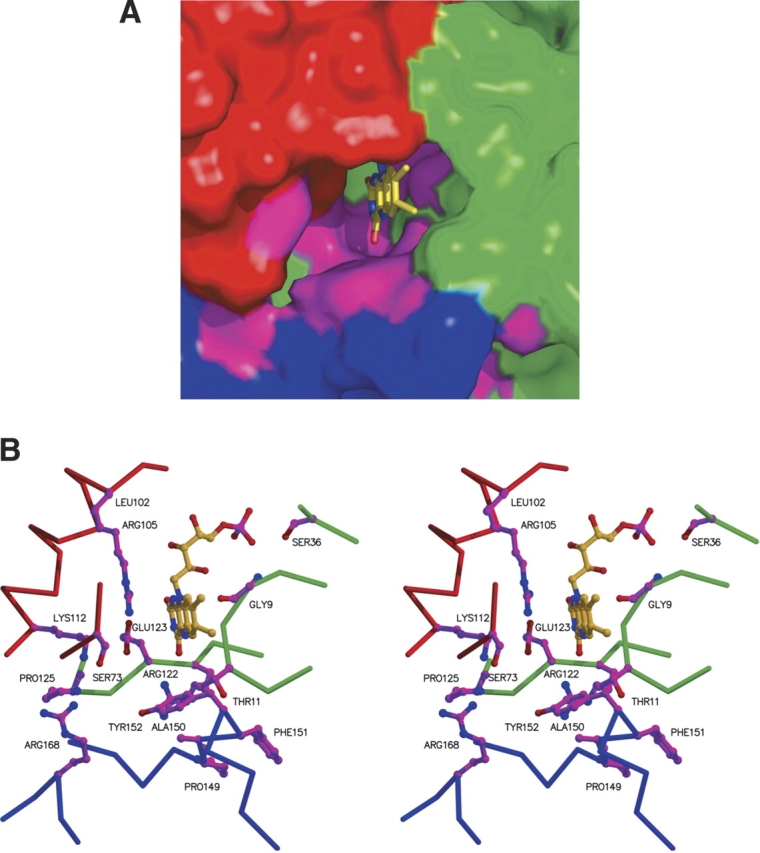
SPad1 active site region. (A) Molecular surface of the active site region formed at the interface of three PadA monomers (blue, red, and green) with highly conserved residues shown in magenta. Figure prepared using PyMol. (B) Same orientation as in A, but showing highly conserved side chains from three different Pad1 monomers, in magenta. Main chain atoms of the three monomers are shown in blue, red, and green, respectively.
Materials and methods
Cloning, expression, and purification
The full-length Pad1 gene was amplified by PCR from E. coli strain O157:H7 genomic DNA (Perna et al. 2001), using recombinant PfU polymerase (Stratagene). Oligonucleotide primers were obtained from integrated DNA Technologies (IDT). The pad1 gene was cloned into a modified pET15b vector (Amersham-Phar-macia) and expressed in E. coli BL21(DE3) as a fusion with a noncleavable N-terminal (His)6 tag. For production of selenomethionine-labeled protein, the expression plasmid was transformed into the E. coli methionine auxotroph strain DL41 (DE3) (Hendrickson et al. 1990).
For production of unlabeled protein, bacterial cultures were grown in Circle Grow medium (BIO101 Inc.), whereas selenome-thionine-labeled protein was produced using LeMaster medium (Hendrickson et al. 1990). Protein expression was induced by addition of 100 μM IPTG followed by 6 h incubation at room temperature (RT). Cells were harvested by centrifugation (5000g, 20 min, 4°C) and solubilized by lysis in 50 mM Tris-Cl (pH 7.5), containing 0.4 M NaCl, 20 mM imidazole, 5% (v/v) glycerol, 10 mM β-mercaptoethanol, 0.7 mg lysozyme, 10 U/mL benzonase nuclease (Novagen), 1× Bugbuster detergent solution (Novagen), and 1 tablet of Complete EDTA-free protease inhibitor cocktail (Roche Molecular Biologicals). The cell lysate was clarified by ultracentrifugation (100,000g, 40 min, 4°C). Soluble protein was incubated gently with 2 mL of DEAE-Sepharose (Amersham-Pharmacia) equilibrated in a buffer (50 mM Tris-Cl [pH 7.5], 0.4 M NaCl, 20 mM imidazole, 5% glycerol, and 10 mM β-mercap-toethanol) for 30 minutes at RT to remove nucleic acid fragments. The flow-through was loaded onto 2-mL (bed volume) of Ni-NTA resin (Qiagen) and incubated with gentle shaking for 30 min at RT. The resin was then poured into a column and washed with 60 mL of 50 mM Tris-Cl buffer (pH 7.5), 0.4 M NaCl, 5% (v/v) glycerol, 40 mM imidazole, and 10 mM β-mercaptoethanol. His-tagged Pad1 was eluted with 15 mL of the above buffer containing 200 mM imidazole. The eluted protein was concentrated by ultrafiltration to 8 mg/mL in 50 mM Tris-Cl buffer (pH 7.5), 0.4 M NaCl, 5% (v/v) glycerol, and 5 mM DTT. The expressed protein had the N-terminal sequence MGSSHHHHHHGS-Met(1). Selenomethionine-labeled protein was purified in a similar manner.
Gel filtration chromatography was carried out on purified Pad1 using a Superose 12 HR10/30 column on an ÄKTA Purifier FPLC system (Amersham Pharmacia, Uppsala, Sweden). Purified Pad1 enzyme (200 μg was applied to the column preequilibrated with buffer (20 mM Tris-Cl [pH 7.5], 0.4 M NaCl, 5% glycerol and 5 mM DTT) and protein elution monitored by UV absorption at λ = 280 nm. Chromatograms were analyzed with the Unicorn 3.10.11 software, provided with the ÄKTA purifier system. Molecular masses were estimated by comparison with the elution profile of molecular mass standards (Sigma) albumin (Mr 67,000), aldolase (Mr 152,000), catalase (Mr 213,000), and ferritin (MMr 300,000).
Dynamic light-scattering measurements were done on a DynaPro 801 molecular-sizing instrument (Protein Solutions) at room temperature, using a protein concentration of 7.5 mg mL−1 in 50 mM Tris-Cl buffer (pH 7.5), 0.4 M NaCl, 5% (v/v) glycerol, and 5 mM DTT.
The ultraviolet-visible spectrum of Pad1 was recorded on a Cary 3E UV-VIS spectrophotometer using the wavelength range of 600–250 nm. The protein was in a buffer consisting of 20 mM Tris-Cl (pH 7.5), 0.4 M NaCl, 5% glycerol, and 5 mM DTT. Electron spray ionization mass spectrometry (ESI-MS) was performed on an Agilent LC-MS mass spectrometer (1100 Series LC/MSD). A sample of protein in buffer was diluted to a final concentration of 0.4 mg/mL in 20% (v/v) acetonitrile and 0.1% (v/v) formic acid and was ionized by direct injection. For the characterization of potential Pad1 enzymatic activity, a number of substrates were employed. These included trans-cinnamic acid and p-coumaric acid (Sigma Chemical Co.), caffeic acid, ferulic acid, and vanillic acid (Fluka Chemical Co.). As a positive control for the various activities, a crude E. coli lysate containing overex-pressed, recombinant PadA enzyme from Bacillus sp. BP-7 (Prim et al. 2003) was used. Expression of PadA was confirmed by SDS-PAGE. Enzymatic activity measurements were performed according to the procedure of Cavin et al. (1997). Briefly, enzyme and substrate (at a final concentration of 2 mM) were incubated in buffer (50 mM sodium acetate [pH 5], 50 mM Tris-Cl [pH 7 or pH 8]) in a final volume of 50 μL at 25°C for as long as 48 h or at 30°C for 2 h. Reactions were terminated by adding 950 μL of 25 mM Tris-Cl (pH 8), 0.3% [w/v] SDS. The corresponding vinyl products were identified by either ultraviolet-visible spectrophotometry (by scanning the absorption range 230–350 nm) or by thin layer chromatography (TLC), on a thin-layer silica gel, using a solvent system of toluene: acetone 15:2 (v/v) and visualized under ultraviolet light.
Crystallization
Initial crystallization conditions were identified utilizing hanging drop vapor diffusion using sparse matrix screens (Hampton Research). The best Pad1 crystals were obtained by equilibrating a 1-μL drop of protein (7.5 mg/mL) in buffer (50 mM Tris-Cl [pH 7.5], 5 mM DTT, 0.4 M NaCl and 20% (v/v) glycerol, 5 mM FMN), mixed with 1 μL of reservoir solution containing 15% (w/v) PEG 4000, 0.2 M LiSO4, and 0.1 M Hepes buffer (pH 7.0) and suspended over 1 mL of reservoir solution. Crystals grew to a size of ~0.1 × 0.1 × 0.06 mm in 2 d at 21°C. For data collection, crystals were transferred for 1 min to a cryo-protectant solution containing reservoir solution supplemented with 20% (v/v) glycerol, picked up in a nylon loop and flash cooled in a N2 cold stream (Oxford Cryosystem). Crystals of Pad1 belong to the trigonal system, space group R3 with unit cell dimensions a = b = 95.4, c = 217.5 Å and β = 120°. The crystals contain four molecules in the asymmetric unit (Z = 36) corresponding to Vm = 2.22 Å3 Da−1 and a solvent content of 43% (Matthews 1968).
Data collection, structure solution, and refinement
Diffraction data from a SeMet-labeled crystal of Pad1 were collected using a three-wavelength MAD regime on a Quantum-4 CCD detector (Area Detector Systems Corp.) at beamline X8C at the NSLS, Brookhaven National Laboratory (Table 1). Data processing and scaling were performed with HKL2000 (Otwinowski and Minor 1997; Table 1). The structure was determined using the program SOLVE (Terwilliger and Berendzen 1999). Data to 2.2 Å resolution were used to locate 40 out of 44 expected Se atoms in the asymmetric unit, resulting in a figure of merit FOM = 0.56. Density modification with the program RESOLVE (Terwilliger 2003) improved the quality of the map (FOM = 0.66) and allowed for automated model building of ~75% of the residues.
Table 1.
Data collection and refinement statistics
| Data collection | ||||
| Data set | Inflection | Peak | Hard remote | Soft remote |
| Space group | H3 | H3 | H3 | H3 |
| Unit Cell (Å) | a = 95.4, c = 17.5 | a = 95.4, c = 17.5 | a = 95.4, c = 17.5 | a = 95.4, c = 217.5 |
| Resolution range (Å) | 50–2.2 | 50–2.2 | 50–2.2 | 50–2.0 |
| Wavelength (Å) | 0.9792 | 0.9786 | 0.9643 | 0.9950 |
| Observed reflections | 142,284 | 142,182 | 143,286 | 155,447 |
| Unique reflections | 37,368 | 37,327 | 37,240 | 46,319 |
| Completeness (%) | 99.8 (97.9) | 99.8 (98.0) | 99.9 (99.4) | 96.5 (80.1) |
| Overall (I/σI) | 12.4 (4.4) | 11.9 (4.2) | 10.8 (3.6) | 12.9 (1.99) |
| Rsym (%)a | 0.116 (0.35) | 0.105 (0.348) | 0.099 (0.431) | 0.062 (0.451) |
| Refinement and model quality | ||||
| Resolution range (Å)b | 20–2.0 | |||
| Rfree,c no. of reflections | 0.215 (4137) | |||
| Rwork,d no. of reflections | 0.174 (42,181) | |||
| rmsd bond lengths (Å) | 0.011 | |||
| rmsd bond angles (degrees) | 1.14 | |||
| Average B-factors (Å2), number of atoms | ||||
| Protein | 25.3 (6124) | |||
| Main chain atoms | 26.4 (2879) | |||
| Side chain atoms | 27.3 (2663) | |||
| Water | 33.0 (458) | |||
| FMN | 21.7 (124) | |||
| Ramachandran plot | ||||
| % of residues in | ||||
| Most favorable regions | 93.8 | |||
| Additional allowed regions | 6.2 | |||
aRsym = (Σ|Iobs - Iavg|)/ΣIavg.
b Refinement performed using merged data collected at the soft remote wavelength, using all data.
cRwork = (Σ|Fobs - Fcalc|)/ΣFobs.; dRfree = Rwork, but for a random set of 10% of the unique reflections.
d Values in parentheses, for data collection statistics, correspond to the last resolution shell: 2.28–2.20 for inflection, peak, and hard remote, and 2.07–2.00 for soft remote.
Data collected at the soft remote wavelength (λ = 0.9950) with the Bijvoet pairs merged were used for refinement of the model. The partial model obtained from RESOLVE was extended manually with the help of the program O (Jones et al. 1991) and was improved by several cycles of refinement, using the program REFMAC (Murshudov et al. 1997), and model refitting. Noncrystallographic symmetry restraints were not used during refinement. The C termini of each of the four independent molecules were disordered. Out of 197 residues of Pad1, the final model includes residues 1–176 and 179–185 for monomer A, residues 1–177 for monomers B and C, and residues 1–186 for monomer D. Several residues have poor electron density for their side chains, including Arg24A, Glu25A, Lys37A, Arg52A, Arg77A, and Lys116A in monomer A; Arg52B; and Arg77B in monomer B; Arg52C in monomer C; and Asn28D, Lys37D, Arg52D, Arg77D, Glu176D–Arg181D, and Gln184D in monomer D. Two proline residues in each monomer, Pro85 and Pro125, adopt a cis conformation. A strong density feature near each of the four independent molecules in the difference electron density map showed the clear presence of a cofactor and was modeled as FMN. Pad1 is classified in InterPro as a probable flavin-binding protein, and the shapes of the difference density corresponded very well to molecules of FMN and were modeled as such. In addition, 458 water molecules were included in the model. The final model has an R factor of 0.162 and R-free of 0.214 for all data to 2.0 Å resolution (Table 1) and has good stereochemistry, with no outliers in the Ramachandran plot computed using PROCHECK (Laskowski et al. 1993). Coordinates have been deposited in the RCSB Protein Data Bank with accession code 1SBZ.
Acknowledgments
We thank Dr. P. Diaz (University of Barcelona, Spain) for providing an expression clone of the Bacillus Sp. BP-7 PadA enzyme. We also thank Leon Flaks (beamline X8C, NSLS, BNL) for assistance with X-ray data collection and Stephane Raymond for maintenance of the local computer environment. This research was supported by Canadian Institutes for Health Research grant 200103GSP-90094-GMX-CFAA-19924 to M.C.
Article published online ahead of print. Article and publication date are at http://www.proteinscience.org/cgi/doi/10.1110/ps.04953004.
References
- Albert, A., Martinez-Ripoll, M., Espinosa-Ruiz, A., Yenush, L., Culianez-Macia, F.A., and Serrano, R. 2000. The X-ray structure of the FMN-binding protein AtHal3 provides the structural basis for the activity of a regulatory subunit involved in signal transduction. Structure Fold. Des. 8 961–969. [DOI] [PubMed] [Google Scholar]
- Altschul, S.F., Madden, T.L., Schaffer, A.A., Zhang, J., Zhang, Z., Miller, W., and Lipman, D.J. 1997. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 25 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apweiler, R., Attwood, T.K., Bairoch, A., Bateman, A., Birney, E., Biswas, M., Bucher, P., Cerutti, L., Corpet, F., Croning, M.D., et al. 2001. The InterPro database, an integrated documentation resource for protein families, domains and functional sites. Nucleic Acids Res. 29 37–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bateman, A., Birney, E., Cerruti, L., Durbin, R., Etwiller, L., Eddy, S.R., Griffiths-Jones, S., Howe, K.L., Marshall, M., and Sonnhammer, E.L. 2002. The Pfam protein families database. Nucleic Acids Res. 30 276–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaesse, M., Kupke, T., Huber, R., and Steinbacher, S. 2000. Crystal structure of the peptidyl-cysteine decarboxylase EpiD complexed with a pentapeptide substrate. EMBO J. 19 6299–6310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- —–—. 2003. Structure of MrsD, an FAD-binding protein of the HFCD family. Acta Crystallogr. D Biol. Crystallogr. 59 1414–1421. [DOI] [PubMed] [Google Scholar]
- Breinig, S., Schiltz, E., and Fuchs, G. 2000. Genes involved in anaerobic metabolism of phenol in the bacterium Thauera aromatica. J. Bacteriol. 182 5849–5863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavin, J.F., Barthelmebs, L., and Divies, C. 1997. Molecular characterization of an inducible p-coumaric acid decarboxylase from Lactobacillus plantarum: Gene cloning, transcriptional analysis, overexpression in Escherichia coli, purification and characterization. Appl. Environ. Microbiol. 63 1939–1944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chenna, R., Sugawara, H., Koike, T., Lopez, R., Gibson, T.J., Higgins, D.G., and Thompson, J.D. 2003. Multiple sequence alignment with the Clustal series of programs. Nucleic Acids Res. 31 3497–3500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clausen, M., Lamb, C.J., Megnet, R., and Doerner, P.W. 1994. PAD1 encodes phenylacrylic acid decarboxylase which confers resistance to cinnamic acid in Saccharomyces cerevisiae. Gene 142 107–112. [DOI] [PubMed] [Google Scholar]
- Cox, G.B., Young, I.G., McCann, L.M., and Gibson, F. 1969. Biosynthesis of ubiquinone in Escherichia coli K-12: Location of genes affecting the metabolism of 3-octaprenyl-4-hydroxybenzoic acid and 2-octaprenylphenol. J. Bacteriol. 99 450–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenn, T.D., Ringe, D., and Petsko, G.A. 2003. POVScript+: A program for model and data visualization using persistence of vision ray-tracing. J. Appl. Crystallogr. 36 944–947. [Google Scholar]
- Fraaije, M.W. and Mattevi, A. 2000. Flavoenzymes: Diverse catalysts with recurrent features. Trends Biochem. Sci. 25 126–132. [DOI] [PubMed] [Google Scholar]
- Gouet, P., Robert, X., and Courcelle, E. 2003. ESPript/ENDscript: Extracting and rendering sequence and 3D information from atomic structures of proteins. Nucleic Acids Res. 31 3320–3323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guex, N. and Peitsch, M.C. 1997. SWISS-MODEL and the Swiss-PdbViewer: An environment for comparative protein modeling. Electrophoresis 18 2714–2723. [DOI] [PubMed] [Google Scholar]
- Hendrickson, W.A., Horton, J.R., and LeMaster, D.M. 1990. Selenomethionyl proteins produced for analysis by multiwavelength anomalous diffraction (MAD): A vehicle for direct determination of three-dimensional structure. EMBO J. 9 1665–1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holm, L. and Sander, C. 1995. Dali: A network tool for protein structure comparison. Trends Biochem. Sci. 20 478–480. [DOI] [PubMed] [Google Scholar]
- Howlett, B.J. and Bar-Tana, J. 1980. Polyprenyl p-hydroxybenzoate carboxyl-ase in flagellation of Salmonella typhimurium. J. Bacteriol. 143 644–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonassen, T. and Clarke, C.F. 2000. Genetic analysis of coenzyme Q biosynthesis. In Coenzyme Q: Molecular mechanisms in health and disease (eds. V.E. Kagan and J. Quinn), pp. 185–208. CRC Press, Boca Raton, FL.
- Jones, T.A., Zhou, J.Y., Cowan, S.W., and Kjeldgaard, M. 1991. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr. A 47 110–119. [DOI] [PubMed] [Google Scholar]
- Laskowski, R.A., MacArthur, M.W., Moss, D.S., and Thornton, J.M. 1993. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 26 283–291. [Google Scholar]
- Leppik, R.A., Young, I.G., and Gibson, F. 1976. Membrane-associated reactions in ubiquinone biosynthesis in Escherichia coli. 3-Octaprenyl-4-hydroxyben-zoate carboxylyase. Biochim. Biophys. Acta 436 800–810. [DOI] [PubMed] [Google Scholar]
- Matthews, B.W. 1968. Solvent content of protein crystals. J. Mol. Biol. 33 491–497. [DOI] [PubMed] [Google Scholar]
- Meganathan, R. 2001. Ubiquinone biosynthesis in microorganisms. FEMS Microbiol. Lett. 203 131–139. [DOI] [PubMed] [Google Scholar]
- Murshudov, G.N., Vagin, A.A., and Dodson, E.J. 1997. Refinement of macro-molecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53 240–255. [DOI] [PubMed] [Google Scholar]
- Nicholls, A., Sharp, K.A., and Honig, B. 1991. Protein folding and association: Insights from the interfacial and thermodynamic properties of hydrocarbons. Proteins 11 281–296. [DOI] [PubMed] [Google Scholar]
- Otwinowski, Z. and Minor, W. 1997. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276 307–326. [DOI] [PubMed] [Google Scholar]
- Perna, N.T., Plunkett, G.I., Blattner, F.R., Mau, B., and Blattner, F.R. 2001. Genome sequence of enterohaemorrhagic Escherichia coli O157:H7. Nature 409 529–533. [DOI] [PubMed] [Google Scholar]
- Prim, N., Pastor, F.I., and Diaz, P. 2003. Biochemical studies on cloned Bacillus sp. BP-7 phenolic acid decarboxylase PadA. Appl. Microbiol. Biotechnol. 63 51–56. [DOI] [PubMed] [Google Scholar]
- Rossmann, M.G., Moras, D., and Olsen, K.W. 1974. Chemical and biological evolution of nucleotide-binding protein. Nature 250 194–199. [DOI] [PubMed] [Google Scholar]
- Soballe, B. and Poole, R.K. 1997. Aerobic and anaerobic regulation of the ubiCA operon, encoding enzymes for the first two committed steps of ubiquinone biosynthesis in Escherichia coli. FEBS Lett. 414 373–376. [DOI] [PubMed] [Google Scholar]
- Steinbacher, S., Hernandez-Acosta, P., Bieseler, B., Blaesse, M., Huber, R., Culianez-Macia, F.A., and Kupke, T. 2003. Crystal structure of the plant PPC decarboxylase AtHAL3a complexed with an ene-thiol reaction intermediate. J. Mol. Biol. 327 193–202. [DOI] [PubMed] [Google Scholar]
- Tatusova, T.A. and Madden, T.L. 1999. BLAST 2 Sequences, a new tool for comparing protein and nucleotide sequences. FEMS Microbiol. Lett. 174 247–250. [DOI] [PubMed] [Google Scholar]
- Terwilliger, T.C. 2003. Automated main-chain model building by template matching and iterative fragment extension. Acta Crystallogr. D Biol. Crystallogr. 59 38–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terwilliger, T.C. and Berendzen, J. 1999. Automated MAD and MIR structure solution. Acta Crystallogr. D Biol. Crystallogr. 55 849–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, H. and Javor, G.T. 2000. Identification of the ubiD gene on the Escherichia coli chromosome. J. Bacteriol. 182 6243–6246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ———. 2003. Regulation of the isofunctional genes ubiD and ubiX of the ubiquinone biosynthetic pathway of Escherichia coli. FEMS Microbiol. Lett. 223 67–72. [DOI] [PubMed] [Google Scholar]