

Abstract
The interactions between the N-terminal domain of the ɛ (ɛ186) and θ subunits of DNA polymerase III of Escherichia coli were investigated using electrospray ionization mass spectrometry. The ɛ186–θ complex was stable in 9 M ammonium actetate (pH 8), suggesting that hydrophobic interactions have a predominant contribution to the stability of the complex. Addition of primary alkanols to ɛ186–θ in 0.1 M ammonium acetate (pH 8), led to dissociation of the complex, as observed in the mass spectrometer. The concentrations of methanol, ethanol, and 1-propanol required to dissociate 50% of the complex were 8.9 M, 4.8 M, and 1.7 M, respectively. Closer scrutiny of the effect of alkanols on ɛ186, θ, and ɛ186–θ showed that ɛ186 formed soluble aggregates prior to precipitation, and that the association of ɛ186 with θ stabilized ɛ186. In-source collision-induced dissociation experiments and other results suggested that the ɛ186–θ complex dissociated in the mass spectrometer, and that the stability (with respect to dissociation) of the complex in vacuo was dependent on the solution from which it was sampled.
Keywords: electrospray ionization mass spectrometry, noncovalent, DNA polymerase III, hydrophobic interactions
Electrospray ionization mass spectrometry (ESI-MS) has widespread routine use as a tool in proteomics for confirmation of primary structure determination and for characterization of purified proteins (Griffiths et al. 2001). Application of ESI-MS to detection and characterization of non-covalent complexes of biomolecules is not as well established, but there are now many examples of noncovalent interactions that have been studied in the gas phase, including those between protein subunits, proteins and nucleic acids, and enzymes and substrates (Veenstra 1999; Burkitt et al. 2003; Sanglier et al. 2003). The most obvious use of ESI-MS for study of these complexes is in determination of the stoichiometry of binding partners, and this has recently been extended to monitor subunit exchange between small heat shock proteins in real time (Sobott et al. 2002).
The establishment of stoichiometry is a prelude to more detailed structural determination of biomolecules in complexes. The stability of the complex (dissociation constants), the types of noncovalent interactions (e.g., polar vs. nonpolar), and conformational changes in the binding partners upon complex formation are important when considering the mechanism of biological action of biomolecular complexes (e.g., protein–protein, protein–DNA). There is a suite of biophysical techniques that can be applied to study these properties. These range from monitoring of changes in fluorescence or surface plasmon resonance (SPR) for determination of dissociation constants and use of circular dichroism and NMR spectroscopy for following conformational changes, ultimately to determination of complete structures of complexes by NMR, X-ray crystallography, or cryo-electron microscopy.
ESI-MS offers speed and sensitivity in monitoring components of equilibrium mixtures. Consequently, there are increasing numbers of reports of its use for determination of dissociation constants or relative binding affinities of non-covalent complexes (Jorgensen et al. 1998; Kapur et al. 2002; Bligh et al. 2003). Furthermore, there are ESI-MS studies where the stabilities of noncovalent complexes have been assessed by their resistance to dissociation in the mass spectrometer using CID (collision-induced dissociation) or thermal denaturation experiments (Gupta et al. 2001; de Brouwer et al. 2002; Benesch et al. 2003).
Nevertheless, data obtained from ESI-MS studies need to be interpreted with caution. First, the ionization process itself might perturb equilibria (Wang and Agnes 1999). Second, there is a paucity of information about changes in the strength or specificity of noncovalent interactions that occur on transfer from the condensed to the gas phase during the ionization process. The stabilities of complexes between biological macromolecules involve contributions from ionic, hydrogen bonding, hydrophobic, and/or van der Waals interactions. Several ESI-MS studies support the proposal that electrostatic interactions are strengthened in vacuo, while hydrophobic interactions are unaffected or weakened through loss of water during desolvation and/or ionization (Loo 1997). For these reasons, it is important to study the behavior of noncovalent complexes that have been well characterized in solution to enable evaluation of data from ESI-MS experiments.
Recently, as part of a study aimed at investigating the behavior of noncovalent complexes on transferal from solution to the gas phase, we used ESI-MS to study the well-characterized Tus-Ter (protein–DNA) complex that terminates DNA replication in Escherichia coli (Kapur et al. 2002). We showed that relative binding affinities of mutant Tus proteins for double-stranded TerB DNA were the same in the gas phase as in solution, and conversely, that the relative affinities of wild-type Tus for various double-stranded DNA sequences were unchanged on transferal to the gas phase. Both the X-ray structure (Kamada et al. 1996) and SPR studies of the ionic strength dependence of dissociation of the Tus-TerB complex (Neylon et al. 2000) show there are substantial polar and electrostatic contacts between the binding partners. Consistent with this, ESI mass spectra showed that dissociation of the complex required high concentrations of ammonium acetate, in the range of 1–2 M (Kapur et al. 2002). For the present work, we used ESI-MS to investigate the predominantly hydrophobic interactions between two protein subunits of E. coli DNA polymerase III: the θ subunit, and the N-terminal domain (residues 2–186) of the ɛ subunit (ɛ186).
DNA polymerase III is a multisubunit enzyme that is the major replicative polymerase of E. coli (Kelman and O’Donnell 1995; McHenry 2003). Three of the 10 subunits, α, ɛ, and θ, comprise the catalytic core: the large α-subunit contains the polymerase active site, and ɛ contributes the proofreading 3′→5′ exonuclease activity, while the precise function of θ is not known (Studwell-Vaughan and O’Donnell 1993; Kunkel and Bebenek 2000). The ɛ subunit consists of two domains (Perrino et al. 1999; Taft-Benz and Schaaper 1999; Hamdan et al. 2000). The N-terminal domain (ɛ186) contains the exonuclease active site and forms a stable 1:1 complex with θ (Perrino et al. 1999; Hamdan et al. 2002a). The complex forms readily and essentially quantitatively on mixing of the two subunits, and is sufficiently stable that it can be isolated by ion-exchange chromatography (Hamdan et al. 2002a). It is stable for extended periods at 25°C in aqueous solution under conditions required for NMR studies (Pintacuda et al. 2004).
Although there is no high-resolution structure yet available for the ɛ186–θ complex, we have reported the crystal structure of ɛ186 (Hamdan et al. 2002b) and the solution structure of θ (Keniry et al. 2000). NMR chemical shift mapping experiments in the latter study suggested that a series of small hydrophobic residues on the external face of the first helix of θ (residues 21–27, AAAGVAF) are involved in its association with ɛ186, and this has been confirmed in the more recent NMR structure of θ in the ɛ186–θ complex (M. Keniry, pers. comm.). Moreover, recent comparisons of NMR spectra of free ɛ186 and ɛ186–θ have identified hydrophobic residues in ɛ at the θ-binding interface, including Ile31, Val50, Val58, Ile68, Leu74, Ile154, Leu161, Leu165, and Leu166 (DeRose et al. 2003). It appears, therefore, that interaction between the two proteins is mediated largely via aliphatic side chains, and the forces that hold them together are largely hydrophobic in nature.
In the present work, we aimed to use ESI-MS to supply “snapshots” of components of mixtures of ɛ186 and θ. The stability of the ɛ186–θ complex was studied under various solution and instrumental conditions. Its stability at high ionic strength is consistent with a dominant contribution of nonpolar interactions, in contrast with that of the Tus-Ter complexes, where interactions are primarily polar and electrostatic in nature and are disrupted at relatively low ionic strength (Kapur et al. 2002). In addition, ESI-MS experiments suggested that the ɛ subunit protects ɛ186 from aggregation in organic solvent/water mixtures. This is consistent with earlier experiments in which θ was shown to stabilize ɛ186 against thermal inactivation (Hamdan et al. 2002a).
Results and Discussion
Conditions were developed to prepare ɛ186–θ and to acquire high quality ESI mass spectra. The separate subunits were dialyzed against 10 mM ammonium acetate (NH4OAc) at pH 6.8, and then mixed together in equimolar amounts with a small volume of concentrated NH4OAc solution at pH 8, so that the final buffer concentration was 0.1 M. Figure 1A ▶ shows a spectrum obtained with a 2 μM solution of ɛ186–θ under optimal conditions, using a cone voltage of 30 V. These conditions were also optimal for detection of the individual subunits. The most abundant ions in the spectrum are at m/z 2453.9 and 2676.8, which correspond to the [M+12H]12+ and [M+11H]11+ ions of the ɛ186–θ complex (Mr = 29,434), respectively. Ions of low to medium abundance from free ɛ186 (m/z 2059.7, 2288.4, 3432.3, and 4118.2; [M+10H]10+, [M+9H]9+, [M+6H]6+, and [M+5H]5+, respectively), and θ (m/z 1264.8, 1475.4, and 1770.5 from [M+7H]7+, [M+6H]6+, and [M+5H]5+, respectively) were also observed. If we were to assume that the distribution of ions in this spectrum reflects the composition of free and complexed proteins in solution, then >80% of the individual proteins reside in the complex at equilibrium, giving a value for the equilibrium dissociation constant for ɛ186–θ, KD <20 nM under these conditions. This is probably an overestimate of the true value because the complex is likely to dissociate to some extent within the spectrometer (as discussed below).
Figure 1.

Positive ion ESI mass spectra of ɛ186–θ acquired under different conditions. (A) ɛ186–θ in 0.1 M NH4OAc at pH 8; cone 30 V. (B) ɛ186–θ in 0.1 M NH4OAc at pH 8; cone 60 V. (C) ɛ186– θ in 9 M NH4OAc at pH 8; cone 30 V. Ions from: θ (•); ɛ186 (▪); ɛ186–θ (♦).
When the cone voltage was increased, dissociation of ɛ186–θ was observed. The ESI mass spectrum of the mixture obtained with a cone voltage of 60 V (Fig. 1B ▶) shows that under these conditions ɛ186–θ was almost completely dissociated to the free subunits. At 40 V, the complex remained intact, while at 50 V, more than 80% was dissociated, as calculated by expressing the total intensity of all ions from the ɛ186–θ complex as a percentage of the intensities of all ions (ɛ186–θ, ɛ186, and θ) in the ESI mass spectrum (not shown).
In previous work we studied the effect of ionic strength on the Tus-Ter protein–DNA complex. ESI mass spectra showed that it dissociated over the range of NH4OAc concentrations from 1–2 M (pH 8), consistently with disruption at high ionic strength of electrostatic interactions important in maintaining the Tus-Ter interaction (Kapur et al. 2002). If electrostatic interactions do not contribute overall to the stability of ɛ186–θ, changing the ionic strength of the solution would have little impact on the stability of the complex. In contrast, organic solvents that are expected to disrupt hydrophobic interactions might destabilize it.
The effect on the ɛ186–θ complex of increasing ionic strength was therefore examined. The ESI mass spectrum of a sample of ɛ186–θ that had been treated for 1 min with 9 M NH4OAc at pH 8 (Fig. 1C ▶) was essentially the same as that obtained with the complex in 0.1 M NH4OAc (Fig. 1A ▶), except that the predominant ion was [M+11H]11+ instead of [M+12H]12+. Furthermore, ions from free θ and ɛ186 were in lower abundance than in Figure 1A ▶, suggesting that the complex might be stabilized further at high ionic strength. The change in charge state distribution may be the result of a conformational change of the complex. This proposal awaits confirmation as, to date, there have been no solution structural studies that have examined the conformation of ɛ186–θ complex under these conditions. That the mass spectrum was unchanged after treatment of ɛ186–θ with 9 M NH4OAc for 1 h indicates that the complex is stable under these conditions. This stability at such high salt concentrations suggests that electrostatic interactions do not play a major role in the overall stability of ɛ186–θ, and is consistent with NMR spectroscopic studies that indicate that contacts between the two subunits largely involve hydrophobic residues (Keniry et al. 2000; DeRose et al. 2003).
In-source CID (collision-induced dissociation) experiments have been used previously to probe the gas phase stability of noncovalent complexes (Potier et al. 1998; Schnier et al. 1998; Nousiainen et al. 2001). Increasing the cone voltage increases the internal energy of the ions through the intermediate pressure region in the source, such that collisions are more likely to result in dissociation of noncovalent complexes. ESI mass spectra of ɛ186–θ in 0.01, 0.1 or 2 M NH4OAc (pH 8), were acquired using cone voltages in the range 20–60 V (in 5 V increments). The total intensity of ions from the complex was expressed as a percentage of the summed intensities of all ions in the spectrum and plotted against the cone voltage (Fig. 2 ▶). In each of these solutions, the percentage of the intensity of ions from the complex compared to the intensity of all ions in the spectrum was similar (~60%–70%) at 20–30 V, in agreement with the experiments described above. This suggests that these solution conditions had little effect on the stability of the complex (with respect to dissociation). Over the range 30–40 V, the percentage dissociation of the complex was different for the samples of ɛ186–θ treated with the different solutions. When the complex was treated with 0.01, 0.1, or 2 M NH4OAc, the cone voltages required to cause 50% dissociation were 35.8, 37.5, and 40.5, respectively. The data show that a small change in the cone voltage was required for 50% dissociation of the complex in the different solutions, and that ɛ186–θ was marginally more stable in vacuo when it had been electrosprayed from a solution of higher ionic strength. These results suggest that dissociation of ɛ186–θ occurred within electrosprayed droplets during the desolvation process or that fully desolvated ɛ186–θ ions retained “memory” of the solution from which the ions were generated. “Memory” effects have been observed by other workers. Recently, cytochrome c dimers from two solutions where the conformations were different were shown to dissociate to form monomers with different numbers of charges (Jurchen and Williams 2003). When the dimer ions were formed from a denaturing solution, gas-phase dissociation resulted in monomers with different numbers of charges (asymmetric charge-partitioning), whereas when dimer ions were formed from a solution where cytochrome c had a native conformation, the dissociation process was symmetric. This suggested that the dimer ions “remembered” the solution from which they were formed.
Figure 2.

Stability of ɛ186–θ in various solvents as a function of cone voltage. The intensities of ions from ɛ186–θ were summed and expressed as a percentage of the total intensities of all ions in ESI mass spectra. 2.0 M NH4OAc at pH 8 (▴); 0.1 M NH4OAc at pH 8 (▪); 0.01 M NH4OAc at pH 8 (•).
The hydrophobicity of the ɛ186–θ interaction was probed further by treating the complex in 0.1 M NH4OAc at pH 8, with the organic solvents methanol, ethanol, 1-propanol, 1-butanol, isopropanol, acetone, and acetonitrile. Organic solvent was added to ɛ186–θ (2 μM) in 1.0 M increments, except for 1-propanol and 1-butanol, where the increments were 0.2 M. ESI mass spectra were obtained within 1 min of mixing. Dissociation of ɛ186–θ was observed with increasing 1-propanol concentrations over the range 1–2 M (Fig. 3 ▶), and similar results were obtained with the other organic solvents, except that the concentration range over which the complex dissociated varied. The total intensity of ions from ɛ186–θ was expressed as a percentage of the total intensity of all ions and plotted against organic solvent concentration as shown in Figure 4 ▶. For the series of primary alkanols, the concentrations at which 50% of the complex was dissociated, which we now define as the C50 values for the solvents, were 8.9 M, 4.8 M, 1.7 M, and 1.1 M for methanol, ethanol, 1-propanol, and 1-butanol, respectively. The interpretation of results obtained from mixtures containing 1-butanol was a little complicated because it was miscible with the buffer only to ~1.2 M. Nevertheless, most of the ɛ186–θ complex had already dissociated at concentrations below 1.2 M. The propensity of the complex to dissociate appeared to increase with decreasing dielectric constant (Weast and Astle 1982) of the primary alkanol. The corresponding values of C50 for isopropanol, acetone, and acetonitrile were 3.1 M, 4.2 M, and 4.3 M, respectively. Acetonitrile has the highest dielectric constant, and would be expected to be the least effective solvent in dissociating ɛ186–θ if the dielectric constant were the only factor involved. This is not the case, which suggests that other intrinsic physical and structural features of the solvents (e.g., the ability to H-bond) may also be important. It is appropriate to note the importance of maintenance of consistent desolvation gas pressure in these studies; it was kept constant in all the experiments described here (see Materials and Methods). It was observed that at low pressures, lower concentrations of organic solvent were required to dissociate the complex.
Figure 3.

Positive ion ESI mass spectra of ɛ186–θ acquired using concentrations of 1-propanol. (A) ɛ186–θ in 0.1 M NH4OAc at pH 8, 1.0 M in 1-propanol. (B) ɛ186–θ in 0.1 M NH4OAc at pH 8, 1.4 M in 1-propanol. (C) ɛ186–θ in 0.1 M NH4OAc at pH 8, 1.8 M 1-propanol. (D) ɛ186–θ in 0.1 M NH4OAc at pH 8, 2.0 M 1-propanol. Ions from: θ (•); ɛ186 (▪); ɛ186–θ (♦).
Figure 4.

Stability of ɛ186–θ in various alkanols as a function of alkanol concentration. The intensities of ions from ɛ186–θ were summed and expressed as a percentage of the total intensities of all ions in ESI mass spectra.
Use of data like these to assign overall electrostatic or hydrophobic character to noncovalent protein–protein interactions undoubtedly oversimplifies the complexity of these interactions, which usually involve (favorable or unfavorable) energetic contributions from both of these types of interactions in addition to other polar (inter- and intramolecular H-bonds and H-bonds with water) and van der Waals forces. There are also other factors that may complicate interpretation of the data in Figure 4 ▶. First, the response factors (ability of the protein to be detected in the mass spectrometer) of ɛ186–θ, ɛ186, and/or θ might vary with changing solvent. Apparent response factors depend on intrinsic properties of the molecules in the gas phase, and on the proportion of them available in solution to be ionized. For example, varying desolvation rates that occur for different solvent mixtures may differentially affect the ionization efficiencies of components of the mixture, while their aggregation in solution may impair their ionization. If, as an example, the response factor for ɛ186 were to decrease as the organic solvent concentration increased, then the extent of dissociation of the complex would be underestimated in Figure 4 ▶.
Apparent response factors would also depend on the solubility of ɛ186–θ, ɛ186, and/or θ in the mixed solvent systems. The solubility of the protein components might also affect interpretation of these data more directly. Our objective was in essence to evaluate the effect of the organic solvent mixtures on the stability of the ɛ186–θ complex, as would be measured by KD in equation 1. Indeed, if the equilibrium described by equation 1 were to be sufficiently rapid on the time scale of the experiment, it would be perturbed directly by removal (e.g., by precipitation) of any of the three protein components (equations 2–4) regardless of any direct effect of solvent on KD.
 |
(1) |
 |
(2) |
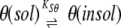 |
(3) |
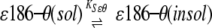 |
(4) |
Before their use was largely superceded by chromatographic methods in the 1950s, organic solvents (and ethanol in particular) had a history of use in biochemistry for fractionation of mixtures of proteins, and it would not be surprising if the solvents used here were to selectively precipitate one or other of the protein components of our mixtures without necessarily causing it to denature. Furthermore, ɛ186 is known to be particularly prone to denaturation and consequent aggregation (Hamdan et al. 2000; DeRose et al. 2002), but is stabilized by interaction with θ (Hamdan et al. 2002a; discussed further below).
Effects of organic solvents on the response factors for ɛ186 and θ were therefore examined by comparing ion counts in ESI mass spectra under all of the solvent conditions and at the same concentration (2 μM) used to examine effects of the primary alkanols on ɛ186–θ (Fig. 4 ▶). The ion currents for θ in 1.0, 3.0, 5.0, and 10.0 M ethanol (in 0.1 M NH4OAc at pH 8) were 250, 432, 455, and 495, respectively. All the ESI mass spectra were of high quality with high signal-to-noise ratios. Over the range 3–10 M ethanol, the ion counts for θ did not vary sufficiently to affect the data shown in Figure 4 ▶. In contrast, when ESI mass spectra of ɛ186 were acquired in 1.0, 2.0, 3.0, 4.0, and 5.0 M ethanol, the ion currents were 683, 841, 580, 19.5, and 0, respectively. Thus, an increase in ion current for ɛ186 was observed as the ethanol concentration was increased to 2.0 M, but declined dramatically thereafter. Similar observations were made for methanol (maximum near 4.0 M) and 1-propanol (1.0 M). The increases at low alkanol concentrations are consistent with enhanced evaporation of solvent droplets facilitating the electrospray process (Kebarle and Peschke 2000). However, the decrease in ion counts for ɛ186 at higher organic solvent concentrations means that the propensity for ɛ186–θ to dissociate in these solvents may have been underestimated in the earlier experiments (Fig. 4 ▶).
The dramatic decline in ion current for ɛ186 as alkanol concentration was increased suggests that this subunit may be poorly soluble under these solvent conditions. This poor solubility could result from aggregation of the native form of the protein, or from its denaturation. The instability of ɛ186 at temperatures above 20°C and in some aqueous buffer systems has been noted in previous studies (Hamdan et al. 2000; DeRose et al. 2002); ɛ186–θ is considerably more stable under similar conditions (Hamdan et al. 2002b; Pintacuda et al. 2004). For example, when NMR experiments were carried out in 50 mM phosphate buffer (pH 7.0), 50 mM in NaCl at 20°C, ɛ186 (1 mM) precipitated over a period of several hours (DeRose et al. 2002), while ɛ186–θ (0.5 mM) was observed to be stable for days at 25°C in 20 mM Tris-HCl (pH 7.0), 100 mM NaCl (Pintacuda et al. 2004). DeRose et al. (2002) noted that the precipitation of ɛ186 in phosphate buffer was concentration dependent, and that the low concentrations at which it was stable were well below those required for NMR measurements. The present ESI-MS experiments require only low protein concentrations (2 μM) and can be carried out rapidly.
A decrease in response factor arising solely from differences in ionization efficiency of a component of the mixture under different solvent conditions is difficult to separate experimentally from a decrease in ion current that is the result of precipitation of a component of the mixture. The possibility that ɛ186–θ, ɛ186, and/or θ might be unstable with respect to precipitation under the conditions of our experiments was tested by monitoring light scattering (absorbance at 360 nm) of solutions of ɛ186–θ, ɛ, and θ in each of the solvent mixtures containing alkanols at concentrations just above or just below their C50 values, over the time period required for ESI-MS analysis (≤10 min). The concentrations used were 8.0 and 10.0 M for methanol, 4.0 and 5.0 M for ethanol, and 1.6 and 2.0 M for 1-propanol.
When θ (alone) was treated with the alkanols at these concentrations in 0.1 M NH4OAc at pH 8, no increase in A360 was observed over 10 min (data not shown). This stability of θ was also observed at higher concentrations of the alkanols (e.g., 10.0 M ethanol). In contrast, precipitation of ɛ186 occurred as shown by an increase in A360 within 5 min of treatment with either 1.6 or 2.0 M 1-propanol (Fig. 5 ▶). The increase in A360 observed with 1.6 M was slower than with 2.0 M 1-propanol. Similar results were reproducibly obtained with the other alkanols. In contrast, ɛ186–θ was stable under these solvent conditions. When samples of ɛ186 that had been treated in 0.1 M ammonium acetate (pH 8), 2.0 M in 1-propanol for 5 min were centrifuged and the supernatants were analyzed for protein (Bradford 1976), only 25% of the original protein in the sample was found to remain in the supernatant. Under these conditions, the ion current in an ESI mass spectrum from a similar solution was negligible, suggesting that any ɛ186 remaining in solution was also not readily ionizable in the source of the mass spectrometer.
Figure 5.

A360 of various solutions containing ɛ186–θ, ɛ186, or θ (2 μM). (A) ɛ186 in 2.0 M 1-propanol in 0.1 M NH4OAc at pH 8; (B) ɛ186 in 1.6 M 1-propanol in 0.1 M NH4OAc at pH 8; (C) θ in 2.0 M 1-propanol in 0.1 M NH4OAc at pH 8; (D) ɛ186–θ in 2.0 M 1-propanol in 0.1 M NH4OAc at pH 8; (E) ɛ186 in 0.1 M NH4OAc at pH 8.
The results are consistent with a change in the solubility of ɛ186 over a critical range of alkanol concentration, affecting its transferal to the gas phase. It also appears that formation of soluble aggregates of ɛ186 that are not readily ionized precedes precipitation. For the purpose of determining the effect of solvents on the equilibrium described by equation 1, it has not mattered to this point in this discussion whether ɛ186 denatures when it aggregates (i.e., rendering equation 2 irreversible).
However, closer inspection of the data reveals greater complexity. For example, the concentration of ethanol (in 0.1 M ammonium acetate at pH 8) required to dissociate half of the complex as judged by ESI-MS (i.e., the C50 value for ethanol) was 4.8 M (Fig. 4 ▶). The data in Figure 4 ▶ could only be generated because ions from ɛ186 were reproducibly observed in experiments where the complex was treated with up to 5.8 M ethanol. That is, at concentrations of alkanol higher than the C50, ions from both binding partners could still be detected. These concentrations are significantly higher than the concentrations at which ions from ɛ186 were no longer detectable in spectra of ɛ186 alone (e.g., in 4.0 M ethanol). Similar results were obtained for the other alkanols. The simplest explanation for this apparent contradiction is that the ɛ186–θ complex is actually much more stable in solution than is revealed by the ESI-MS experiments, even at concentrations of alkanols above their C50 values, but that the organic solvents weaken the inter-subunit interactions such that the complex more easily dissociates in the mass spectrometer.
The effects of alkanols on the stability of ɛ186 alone or in the complex with θ (ɛ186–θ) in solution were tested further by carrying out the following experiments: (1) ɛ186 (2 μM) was pretreated for <1 min with alkanols (methanol, ethanol, or 1-propanol, above and below their C50 values) followed by addition of a small aliquot of concentrated θ (to 2 μM, in NH4OAc). ESI mass spectra were acquired within 1 min of the addition of θ; (2) θ was similarly pretreated for <1 min with alkanols (above and below their C50 values), followed by addition of concentrated ɛ186 (in NH4OAc). ESI mass spectra were again acquired within 1 min; and (3) controls were set up where the experiments (1) and (2) were repeated, but the solvent was 0.1 M NH4OAc (pH 8), without alkanols.
Figure 6 ▶ shows the results of these experiments when 1-propanol (1.6 or 1.8 M) was used as solvent. The control spectrum of ɛ186–θ in 0.1 M NH4OAc at pH 8 (Fig. 6A ▶) showed abundant ions from ɛ186–θ as previously observed under these conditions. In the experiments where ɛ186 or θ were pretreated with 1.6 M 1-propanol (Fig. 6, B and C ▶, respectively), the most abundant ions from ɛ186–θ were also observed, although they were in lower proportions when ɛ186 had been pretreated (Fig. 6B ▶). These results are consistent with previous experiments in which ɛ186–θ was treated with 1-propanol (Fig. 3 ▶). The complex formed readily and ions from ɛ186 were detectable. At this concentration and over this short time (1 min), a significant percentage of free ɛ186 remained soluble during pretreatment.
Figure 6.

Stabilization of ɛ186 by θ as judged by ESI-MS. (A) ɛ186–θ in 0.1 M NH4OAc at pH 8. (B) ɛ186 treated with 1.6 M 1-propanol (in 0.1 M NH4OAc at pH 8) with θ added after 1 min. (C) θ treated with 1.6 M 1-propanol (in 0.1 M NH4OAc at pH 8) with ɛ186 added after 1 min. (D) ɛ186 treated with 1.8 M 1-propanol (in 0.1 M NH4OAc at pH 8) with θ added after 1 min. (E) θ treated with 1.8 M 1-propanol (in 0.1 M NH4OAc at pH 8) with ɛ186 added after 1 min. Ions from: θ (•); ɛ186 (▪); ɛ186–θ (♦).
When 1.8 M 1-propanol (slightly above the C50 value of 1.7 M) was used, dramatic differences were observed. In fact, when ɛ186 had been pretreated with 1.8 M 1-propanol (Fig. 6D ▶), there were no ions from the complex or from free ɛ186, although ions from θ were observed, as expected from earlier data that showed θ to be stable under these conditions. This observation is consistent with aggregation of ɛ186 within 1 min in 1-propanol solutions at a concentration above its C50. This renders ɛ186 incapable of forming a complex with θ. This contrasts with the spectrum shown in Figure 3C ▶, where the ɛ186–θ complex prepared in ammonium acetate was treated later with 1.8 M 1-propanol. In this spectrum, ions from free ɛ186 and from the complex (in lower abundance than when lower concentrations of 1-propanol were used) were detected. The presence of θ in a complex with ɛ186 thus protects ɛ186 from aggregation at concentrations of 1-propanol above the C50 value.
In contrast, when θ was pretreated with 1.8 M 1-propanol (Fig. 6E ▶), ions from ɛ186–θ, θ, and to a lesser extent ɛ186, were all present, and the spectrum was similar to that obtained when the complex was formed in 0.1 M ammonium acetate at pH 8 (Fig. 6A ▶). The difference between these spectra is that there was an excess of ions from θ over ɛ186 in Figure 6E ▶, consistent with the aggregation of some of the ɛ186 decreasing its response factor and preventing its binding to θ. These data, however, show that under these conditions, formation of the ɛ186–θ complex occurs essentially quantitatively on a time scale considerably more rapid than aggregation of ɛ186 (i.e., much shorter than 1 min), even in solutions containing 1-propanol at a concentration above the C50 value. When ɛ186–θ complex was preformed and treated with 1.8 M 1-propanol (Fig. 3C ▶), ions from ɛ186–θ complex were present in addition to ions from both free subunits. The presence of ions from ɛ186 suggests that dissociation of the complex occurred after desolvation (removal of 1-propanol). Thus, the apparent instability of the complex, when analyzed directly in 1-propanol at these concentrations (Fig. 3 ▶), cannot be reflecting the situation in solution, but must be due to the influence of the organic solvent on the stability of the complex in the mass spectrometer.
Analogous results (data not shown) were obtained for methanol (8.0 and 10.0 M) and for ethanol (4.0 and 5.0 M), and all of these experiments were reproduced several times. Moreover, the same results were obtained when the times of pretreatment of ɛ186 and θ were extended from 1 min to 3 h.
The reaction mixtures prepared in 1.8 M 1-propanol were allowed to stand at room temperature for 9 h after acquisition of the spectra shown in Figure 6, D and E ▶, and new ESI mass spectra were recorded (Fig. 7 ▶). Figure 7A ▶ shows the spectrum of ɛ186–θ after 9 h in 0.1 M NH4OAc (control). Under these conditions the complex remains intact (cf. Fig. 6A ▶). In the mixture in which ɛ186 had been pretreated for 1 min with 1.8 M 1-propanol, the ESI mass spectrum after 9 h (Fig. 7B ▶) was essentially the same as the spectrum obtained immediately (Fig. 6D ▶). That is, no ions were detected from ɛ186–θ, but ions from θ were present, indicating that θ was stable after 9 h under these conditions. It is clear, therefore, that aggregation of ɛ186 in 1.8 M 1-propanol cannot be reversed by having free θ in solution.
Figure 7.

Stabilization of ɛ186 by θ as judged by ESI-MS monitored after 9 h. (A) ɛ186–θ in 0.1 M NH4OAc at pH 8. (B) ɛ186 treated with 1.8 M 1-propanol (in 0.1 M NH4OAc at pH 8) with θ added after 1 min. (C) θ treated with 1.8 M 1-propanol (in 0.1 M NH4OAc at pH 8) with ɛ186 added after 1 min. Ions from: θ (•); ɛ186 (▪); ɛ186–θ (♦).
In the mixture where θ had been pretreated with 1-propanol followed by addition of ɛ186, the spectrum obtained immediately showed an amount of complex comparable to that observed in the control (Fig. 6E ▶). However, the ESI mass spectrum acquired after 9 h (Fig. 7C ▶) no longer showed any ions from ɛ186 or the complex. ESI mass spectra acquired at various time points up to 9 h showed progressive loss of ions derived from the complex, and none from ɛ186 (not shown). Thus, even in 1.8 M 1-propanol, ɛ186–θ forms essentially quantitatively within a minute of adding ɛ186 to θ (each at 2 μM), and dissociates with a half-life on the order of hours. The ɛ186 produced by dissociation presumably aggregates within minutes. These data therefore indicate that KD, the dissociation constant of the complex (equation 1), must be no higher than 10−8 M under these conditions (determined as the ratio of estimated rate constants). Analogous results were obtained using 5.0 M ethanol as solvent, except that a significant amount of ɛ186–θ remained after 12 h, showing that the complex is more easily dissociated in 1.8 M 1-propanol than 5.0 M ethanol.
These experiments describe a novel use of ESI-MS to study the equilibrium between two proteins that are believed to associate largely through nonpolar interactions. Consistently with this, and in contrast with our earlier results with protein–DNA complexes that are largely held together by polar and electrostatic interactions (Kapur et al. 2002), the ɛ186–θ complex was found to resist dissociation in solutions containing up to 9 M ammonium acetate. We then examined the effects of organic solvent mixtures on ɛ186–θ and the individual protein subunits. The instability of ɛ186 (toward aggregation) was revealed by the dramatic diminution of ion current over narrow concentration ranges of organic solvents. On the other hand, although (or perhaps because) θ is known to be a poorly structured protein (Keniry et al. 2000), it proved to be remarkably resilient under these conditions. It was also shown to protect ɛ186 from the effects of these solvents, and the ɛ186–θ complex was shown to form rapidly and to dissociate very slowly in solutions containing high concentrations of alkanols at room temperature.
Our initial hope was that ESI mass spectrometry could provide a “snapshot” of the equilibrium composition of solutions containing ɛ186, θ and ɛ186–θ in the solvent mixtures. Although the complex appeared to dissociate over narrow concentration ranges of alkanols (Fig. 4 ▶) and other organic solvents (not shown), this dissociation was shown to occur in the mass spectrometer rather than in the analyte solution. Although further work is required to establish how this occurs, it appears to be related to the polarity of the solvent in a consistent way; solvents with lower dielectric constants promote dissociation of the complex at lower concentrations.
Despite this, the particular situation here where one of the binding partners (ɛ186) is unstable toward removal from the solvent mixtures by aggregation while the other (θ) and the complex (ɛ186–θ) appear to be almost indefinitely stable (toward aggregation) has afforded us a rare opportunity to use ESI-MS to study the time course of dissociation of the protein–protein complex. Further such studies should allow us to demonstrate clearly the effects of the organic solvents on the kinetics of dissociation of the complex under various conditions of ionic strength and solvent polarity.
Materials and methods
Proteins
The θ and ɛ186 subunits of DNA polymerase III were overproduced in E. coli and purified as described previously (Keniry et al. 2000; Hamdan et al. 2002a). These protein samples and the isolated ɛ186–θ complex had previously been characterized by mass spectrometry, giving masses in good agreement with the calculated values of 8848.0 (θ), 20,586.9 (ɛ186), and 29,434.9 (ɛ186–θ) (Hamdan et al. 2002a). Concentrations of θ and ɛ186 were determined by measurement of absorbance of solutions at 280 nm, using calculated values of ɛ280 of 8250 and 6400 M−1 cm1, respectively (Gill and von Hippel 1989).
For preparation of the ɛ186–θ complex, θ and ɛ186 were dialyzed separately against several changes of 10 mM NH4OAc (pH 6.8) at 4°C. Appropriate volumes of θ (50 μM) and ɛ186 (70 μM) subunits were mixed together in a 1:1 molar ratio at 0°C to give a final concentration of ɛ186–θ of 25 μM, in 0.01 M NH4OAc (pH 6.8). To obtain the optimum ESI mass spectrum, the complex was diluted giving a final concentration of 2 μM in 0.1 M NH4OAc (pH 8). In most experiments, the solvent used for this final dilution step was varied; details are given below and elsewhere in the text.
Electrospray ionization mass spectrometry
ESI mass spectra were acquired using a Qtof2 mass spectrometer (Micromass) equipped with a Z-spray electrospray ionization source. This spectrometer has an m/z range of 10,000. Samples were injected directly into the source using a Harvard Model 22 syringe pump at flow rates between 10 and 20 μL/min. The best conditions for obtaining mass spectra of the ɛ186–θ complex were: capillary, 2.5 kV; cone, 30 V; source block temperature, 60°C; desolvation temperature, 150°C; aperture, 13; transport, 2. The desolvation gas pressure was set at 400 Lh−1, as judged by the gauge on the front of the mass spectrometer. Spectra were acquired in positive ion mode over an m/z range 500–5000. Typically 30 to 35 scans were summed to give representative spectra. The data were calibrated against a standard CsI solution (750 μM) over the same m/z range. The ESI spectra shown in this work and those used for measurements of intensities of ions were subjected to background subtraction, and were smoothed using 2 × 10 (channels) m/z window and the Savitzky-Golay algorithm.
Effects of solvents on ESI mass spectra of ɛ186, θ and the ɛ186–θ complex
The ɛ186–θ complex (25 μM) was diluted to 2 μM with the appropriate solvent (containing NH4OAc at pH 8), in the presence or absence of organic solvents, methanol, ethanol, 1-propanol, 1-butanol, acetonitrile, and isopropanol prior to ESI mass analysis. In all experiments, the final concentration of NH4OAc was 0.1 M. Samples were prepared immediately before ESI-MS analysis. Typically, 2 μL of NH4OAc (5 M at pH 8), appropriate volumes of water and organic solvent were mixed, followed by a small volume of protein (1–3 μL), giving a final volume of 50–100 μL. The maximum concentration of 1-butanol that was miscible with the aqueous phase was between 1.0 and 1.2 M. ESI mass spectra were acquired after 5 min of treatment of the ɛ186–θ complex with these solvents at 23°C. ESI mass spectra of θ and ɛ186 (alone, at 2 μM) were acquired in all of the solvent mixtures used to analyze the complex.
In some experiments θ and ɛ186 were pretreated separately at 2 μM with alkanols in 0.1 M NH4OAc at pH 8 (100 μL), followed by addition of a small volume (typically 1–3 μL, to 2 μM) of the other subunit (in 0.01 M NH4OAc at pH 6.8) and analysis by ESI-MS.
Light-scattering measurements
The stabilities of ɛ186, θ and ɛ186–θ with respect to aggregation in solution were judged by monitoring the absorbance at 360 nm of solutions of these proteins (2 μM) in 8 or 10 M methanol, 4 or 5 M ethanol, or 1.6 or 2.0 M 1-propanol, all in 0.1 M NH4OAc at pH 8. Proteins were added to various solvents at t = 0, and A360 was monitored over 10 min at 23°C using a Varian Cary 500 spectrophotometer. In some experiments where the effect of alkanols on ɛ186 was investigated, a sample of the mixture was taken after 5 min, centrifuged in an Eppendorf benchtop microcentrifuge at 16,000g, and the supernatant taken for protein analysis by the method of Bradford (1976); values of A590 were compared to a standard curve constructed using ɛ186 over the concentration range 0.2–2 μM. The presence of alkanols did not significantly affect values of A590.
Acknowledgments
Acknowledgments
We thank the Australian Research Council for research grant support.
Abbreviations
CID, collision-induced dissociation
ɛ186, the N-terminal domain (residues 2–186) of the ɛ subunit of E. coli DNA polymerase III
ESI-MS, electrospray ionization mass spectrometry
NH4OAc, ammonium acetate
NMR, nuclear magnetic resonance
SPR, surface plasmon resonance
Article published online ahead of print. Article and publication date are at http://www.proteinscience.org/cgi/doi/10.1110/ps.04889604.
References
- Benesch, J.L.P., Sobott, F., and Robinson, C.V. 2003. Thermal dissociation of multimeric protein complexes by using nanoelectrospray mass spectrometry. Anal. Chem. 75 2208–2214. [DOI] [PubMed] [Google Scholar]
- Bligh, S.W.A., Haley, T., and Lowe, P.N. 2003. Measurement of dissociation constants of inhibitors binding to Src domain protein by non-covalent electrospray ionization mass spectrometry. J. Mol. Recog. 16 139–147. [DOI] [PubMed] [Google Scholar]
- Bradford, M.M. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72 248–254. [DOI] [PubMed] [Google Scholar]
- Burkitt, W.I., Derrick, P.J., Lafitte, D., and Bronstein, I. 2003. Protein–ligand and protein–protein interactions studied by electrospray ionization mass spectrometry. Biochem. Soc. Trans. 31 985–989. [DOI] [PubMed] [Google Scholar]
- de Brouwer, A.P.M., Versluis, C., Westerman, J., Roelofsen, B., Heck, A.J.R., and Wirtz, K.W.A. 2002. Determination of the stability of the noncovalent lipid transfer protein–lipid complex by electrospray time-of-flight mass spectrometry. Biochemistry 41 8013–8018. [DOI] [PubMed] [Google Scholar]
- DeRose, E.F., Li, D., Darden, T., Harvey, S., Perrino, F.W., Schaaper, R.M., and London, R.E. 2002. Model for the catalytic domain of the proofreading ɛ subunit of Escherichia coli DNA polymerase III based on NMR structural data. Biochemistry 49 94–110. [DOI] [PubMed] [Google Scholar]
- DeRose, E.F., Darden, T., Harvey, S., Gabel, S., Perrino, F.W., Schaaper, R.M., and London, R.E. 2003. Elucidation of the ɛ–θ interface of Escherichia coli DNA polymerase III by NMR spectroscopy. Biochemistry 42 3635–3644. [DOI] [PubMed] [Google Scholar]
- Gill, S.C. and von Hippel, P.H. 1989. Calculation of protein extinction coefficients from amino acid sequence data. Anal. Biochem. 182 319–326. [DOI] [PubMed] [Google Scholar]
- Griffiths, W.J., Jonsson, A.P., Liu, S., Rai, D.K., and Wang, Y. 2001. Electrospray and tandem mass spectrometry in biochemistry. Biochem. J. 355 545–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta, R., Kapur, A., Beck, J.L., and Sheil, M.M. 2001. Positive ion electrospray ionization mass spectrometry of double-stranded DNA/drug complexes. Rapid Commun. Mass Spectrom. 15 2472–2480. [DOI] [PubMed] [Google Scholar]
- Hamdan, S., Brown, S.E., Thompson, P.R., Yang, J.Y., Carr, P.D., Ollis, D.L., Otting, G., and Dixon, N.E. 2000. Preliminary X-ray crystallographic and NMR studies on the exonuclease domain of the ɛ subunit of Escherichia coli DNA polymerase III. J. Struct. Biol. 131 164–169. [DOI] [PubMed] [Google Scholar]
- Hamdan, S., Bulloch, E.M., Thompson, P.R., Beck, J.L., Yang, J.Y., Crowther, J.A., Lilley, P.E., Carr, P.D., Ollis, D.L., Brown, S.E., et al. 2002a. Hydrolysis of the 5′-p-nitrophenyl ester of TMP by the proofreading exonuclease (ɛ) subunit of Escherichia coli DNA polymerase III. Biochemistry 41 5266–5275. [DOI] [PubMed] [Google Scholar]
- Hamdan, S., Carr, P.D., Brown, S.E., Ollis, D.L., and Dixon, N.E. 2002b. Structural basis for proofreading during replication of the Escherichia coli chromosome. Structure 10 535–546. [DOI] [PubMed] [Google Scholar]
- Jorgensen, T.J.D., Roepstorff, P., and Heck, A.J.R. 1998. Direct determination of solution binding constants for noncovalent complexes between bacterial cell wall peptide analogues and vancomycin group antibiotics by electrospray ionization mass spectrometry. Anal. Chem. 70 4427–4432. [Google Scholar]
- Jurchen, J.C. and Williams, E.R. 2003. Origin of asymmetric charge partitioning in the dissociation of gas-phase protein homodimers. J. Am. Chem. Soc. 125 2817–2826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamada, K., Horiuchi, T., Ohsumi, K., Shimamoto, N., and Morikawa, K. 1996. Structure of a replication-terminator protein complexed with DNA. Nature 383 598–603. [DOI] [PubMed] [Google Scholar]
- Kapur, A., Beck, J.L., Brown, S.E., Dixon, N.E., and Sheil, M.M. 2002. Use of electrospray ionisation mass spectrometry to study binding interactions between a replication terminator protein and DNA. Protein. Sci. 11 147–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kebarle, P. and Peschke, M. 2000. On the mechanisms by which charged droplets produced by electrospray lead to gas phase ions. Anal. Chim. Acta 406 11–35. [Google Scholar]
- Kelman, Z. and O’Donnell, M. 1995. DNA polymerase III holoenzyme: Structure and function of a chromosomal replicating machine. Annu. Rev. Biochem. 64 171–200. [DOI] [PubMed] [Google Scholar]
- Keniry, M.A., Berthon, H.A., Yang, J.Y., Miles, C.S., and Dixon, N.E. 2000. NMR solution structure of the θ subunit of DNA polymerase III from Escherichia coli. Protein Sci. 9 721–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunkel, T.A. and Bebenek, K. 2000. DNA replication fidelity. Annu. Rev. Biochem. 69 497–529. [DOI] [PubMed] [Google Scholar]
- Loo, J.A. 1997. Studying noncovalent protein complexes by electrospray ionization mass spectrometry. Mass Spec. Rev. 16 1–23. [DOI] [PubMed] [Google Scholar]
- McHenry, C.S. 2003. Chromosomal replicases as asymmetric dimers: Studies of subunit arrangement and functional consequences. Mol. Microbiol. 49 1157–1165. [DOI] [PubMed] [Google Scholar]
- Neylon, C., Brown, S.E., Kralicek, A.V., Miles, C.S., Love, C.A., and Dixon, N.E. 2000. Interaction of the Escherichia coli replication terminator protein (Tus) with DNA: A model derived from DNA-binding studies of mutant proteins by surface plasmon resonance. Biochemistry 39 11989–11999. [DOI] [PubMed] [Google Scholar]
- Nousiainen, M., Vainiotalo, P., Feng, X.D., and Derrick, P.J. 2001. Calmodulin-RS20-Ca-4 complex in the gas phase: Electrospray ionisation and Fourier transform ion cyclotron resonance. Eur. J. Mass Spectrom. 7 393–398. [Google Scholar]
- Perrino, F.W., Harvey, S., and McNeill, S.M. 1999. Two functional domains of the ɛ subunit of DNA polymerase III. Biochemistry 38 16001–16009. [DOI] [PubMed] [Google Scholar]
- Pintacuda, G., Keniry, M.A., Huber, T., Park, A.Y., Dixon, N.E., and Otting, G. 2004. Fast structure-based assignment of 15N-HSQC spectra of paramagnetic proteins. J. Am. Chem. Soc. 126 2963–2970. [DOI] [PubMed] [Google Scholar]
- Potier, N., Donald, L.J., Chernushevich, I., Ayed, A., Ens, W., Arrowsmith, C.H., Standing, K.G., and Duckworth, H.W. 1998. Study of a noncovalent trp repressor:DNA operator complex by electrospray ionization time-of-flight mass spectrometry. Protein Sci. 7 1388–1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanglier, S., Leize, E., Van Dorsselaer, A., and Zal, F. 2003. Comparative ESI-MS study of similar to 2.2 MDa native hemocyanins from deep-sea and shore crabs: From protein oligomeric state to biotope. J. Am. Soc. Mass Spectrom. 14 419–429. [DOI] [PubMed] [Google Scholar]
- Schnier, P.D., Klassen, J.S., Strittmatter, E.F., and Williams, E.R. 1998. Activation energies for dissociation of double strand oligonucleotide anions: Evidence for Watson-Crick base pairing in vacuo. J. Am. Chem. Soc. 120 9605–9613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobott, F., Benesch, J.L.P., Vierling, E., and Robinson, C.V. 2002. Subunit exchange of multimeric protein complexes. Real-time monitoring of subunit exchange between small heat shock proteins by using electrospray mass spectrometry. J. Biol. Chem. 277 38921–38929. [DOI] [PubMed] [Google Scholar]
- Studwell-Vaughan, P.S. and O’Donnell, M. 1993. DNA polymerase III accessory proteins. V. θ encoded by holE. J. Biol. Chem. 268 11785–11791. [PubMed] [Google Scholar]
- Taft-Benz, S.A. and Schaaper, R.M. 1999. The C-terminal domain of DnaQ contains the polymerase binding site. J. Bacteriol. 181 2963–2965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veenstra, T.D. 1999. Electrospray ionization mass spectrometry in the study of biomolecular non-covalent interactions. Biophys. Chem. 79 63–79. [DOI] [PubMed] [Google Scholar]
- Wang, H.J. and Agnes, G.R. 1999. Kinetically labile equilibrium shifts induced by the electrospray process. Anal. Chem. 71 4166–4172. [DOI] [PubMed] [Google Scholar]
- Weast, R.C. and Astle, M.J., eds. 1982–1983. Dielectric constants. In The CRC handbook of chemistry and physics, 63rd ed., pp. E-55. CRC Press, Inc., Boca Raton, FL.