

Abstract
Synthesis of carbamoyl phosphate by carbamoyl phosphate synthetase (CPS) requires the coordinated utilization of two molecules of ATP per reaction cycle on duplicated nucleotide-binding sites (N and C). To clarify the contributions of sites N and C to the overall reaction, we carried out site-directed mutagenesis aimed at changing the substrate specificity of either of the two sites from ATP to GTP. Mutant design was based in part on an analysis of the nucleotide-binding sites of succinyl-CoA synthetases, which share membership in the ATP-grasp family with CPS and occur as GTP- and ATP-specific isoforms. We constructed and analyzed Escherichia coli CPS single mutations A144Q, D207A, D207N, S209A, I211S, P690Q, D753A, D753N, and F755A, as well as combinations thereof. All of the mutants retained ATP specificity, arguing for a lack of plasticity of the ATP sites of CPS with respect to nucleotide recognition. GTP-specific ATP-grasp proteins appear to accommodate this substrate by a displacement of the base relative to the ATP-bound state, an interaction that is precluded by the architecture of the potassium-binding loop in CPS. Analysis of the ATP-dependent kinetic parameters revealed that mutation of several residues conserved in ATP-grasp proteins and CPSs had surprisingly small effects, whereas constructs containing either A144Q or P690Q exerted the strongest effects on ATP utilization. We propose that these mutations affect proper movement of the lids covering the active sites of CPS, and interfere with access of substrate.
Keywords: ATP-grasp, carbamoyl phosphate synthetase, ATP, GTP, arginine
Carbamoyl phosphate (CP) is a metabolite of central importance to life. It is a precursor in both pyrimidine and arginine biosynthesis, and, in ureotelic vertebrates, serves as the first reservoir for excess nitrogen in ammonia detoxification via the urea cycle (Powers-Lee and Meister 1988). The metabolic importance of CP is matched by the elegance of its structure, being composed of one moiety, each derived from three of life’s most important inorganic molecules: ammonia, bicarbonate, and phosphate. Given the central role of CP, it is not surprising that carbamoyl phosphate synthetases (CPSs) are found in most living organisms.
The small size of the CP molecule belies the complexities involved in its synthesis. Carbamoyl phosphate synthetases are large enzymes with a molecular mass of ~160 kD, are subject to regulation by a variety of different allosteric effectors, depending on the metabolic context, and can occur as part of even larger multienzyme complexes in which CP synthesis is coupled to subsequent steps in the biosynthetic pathway (Anderson 1995). Additionally, although some organisms possess only one CPS, in other cases, multiple CPSs provide CP for different metabolic needs and have distinct subcellular localizations and tissue distributions as well as different requirements for the nitrogen-donating substrate. In those CPSs that derive the amino moiety of CP from glutamine, catalysis involves three active sites that have been shown to be separated by considerable distances in the enzyme from Escherichia coli (Thoden et al. 1997, 1999a). The individual activities at these sites are synchronized through communication of the substrate-loaded state between sites in such a way that significant activity is observed only in the presence of all substrates (Meister 1989; Miles and Raushel 2000).
Synthesis of CP requires the coordinated utilization of two molecules of ATP per reaction cycle, as well as one molecule each of bicarbonate and ammonia (free or derived from glutamine through reaction on the glutamine amidotransferase domain of CPS) to form one molecule of CP, two molecules of ADP, and one molecule of Pi (Meister 1989). The ATP molecules react at two domains (termed N for the one closest to the amino terminus and C for the one closest to the carboxyl terminus) that share sequence identity and appear to have resulted from an ancestral gene duplication event (Nyunoya and Lusty 1983). The X-ray structure of E. coli CPS demonstrated that the domains are superimposable, with an rmsd of 1.1 Å for 255 equivalent α carbons (Thoden et al. 1997), and that both are members of the ATP-grasp fold (Fan et al. 1995; Artymiuk et al. 1996; Galperin and Koonin 1997).
The presence in CPS of duplicated ATP sites complicates the analysis of experiments investigating its mechanism, in particular, the interplay of the reactions occurring at both sites. For example, the dual ATP sites of CPS make it difficult to test distinguishing features of a nucleotide switch mechanism that we proposed for CPS (Kothe et al. 1997). In both this mechanism and the sequential one, carboxy phosphate and ADP are formed at ATPN from bicarbonate and ATP, and nucleophilic ammonia reacts with the activated carbonyl group to yield a tetrahedral intermediate. In the sequential mechanism, the intermediate collapses to carbamate with phosphate as the leaving group, and at ATPC, the carbamate reacts with ATP to yield ADP and CP. In the switch mechanism, the intermediate collapses directly to CP on domain N, with water as the leaving group, and with domain C acting as an ATP-driven molecular switch that allows the energetically unfavorable reaction to proceed on domain N. This scheme is analogous to mechanisms proposed for nicotinate phosphoribosyltransferase (Vinitsky and Grubmeyer 1993) and ATP sulfurylase (Wei and Leyh 1998).
To allow for a more detailed investigation of the interplay between the two nucleotide-binding sites, as well as to be able to clearly distinguish between the two proposed mechanisms for CPS, we aimed to change the specificity of either one of the two sites of the E. coli enzyme from ATP to GTP. To achieve this goal, we mutated corresponding residues in the adenine-binding pockets of the two ATP-grasp folds that have been implicated in nucleotide recognition. In addition to achieving changed substrate specificity, we also expected to gain insight into the individual contributions of the mutated residues to ATP utilization in CPS.
Results
Mutant design
A number of changes in the nucleotide specificity of proteins have been reported in the literature, including those from cGMP to cAMP (Turko et al. 1998), cAMP to cGMP (Shabb et al. 1990), GTP to ATP (Sunahara et al. 1998), and ATP to GTP (Wang and Kemp 1999), with mutations targeted to residues whose side chains interact with, or are in the immediate vicinity of the nucleotide base. In the ATP-grasp fold, ATP is bound between two α+β subdomains, with one subdomain forming a lid over the active site (Artymiuk et al. 1996; Thoden et al. 1999a). Whereas sequence similarities between ATP-grasp members are relatively low, a number of residues involved in ATP binding and catalysis are conserved at structurally equivalent positions (Fan et al. 1995; Artymiuk et al. 1996). Both sides of the adenine make nonpolar contacts with the protein, whereas a residue segment connecting the two ATP-grasp subdomains (residues 207–211 and 753–757 in CPS domains N and C, respectively; Fig. 1 ▶) interacts with the edge of the six-membered ring of adenine. Two well-conserved hydrogen bonds are made between residues of this segment and the bound adenine, one between the backbone carbonyl oxygen of the second residue (E208/H754 in domains N/C in CPS) and the N6 of adenine, and the other between the backbone amide of L210/L756 and the N1 of adenine. One residue of this segment (D207/D753 in CPS) is generally positioned to allow a hydrogen bond with the N6 of adenine. This hydrogen bond is observed in most ATP-grasp protein structures and in the crystal structure of CPS for D753, but not for D207. However, D207 is positioned (distance between side-chain carboxyl and adenine N of 4.2 Å instead of 3.05 Å in domain C) so that only a slight movement of the side chain would bring it into hydrogen-bonding range. The residue at position 207/753 represents an obvious candidate for mutagenesis, as it can be argued that a change at this position from aspartate to asparagine should favor binding of GTP over ATP via positive interaction of the asparagine side chain with the C6 oxo group of GTP. However, this residue represents the only obvious target for mutagenesis, as the other interactions of the protein with the adenine moiety of ATP are either nonspecific or mediated through main-chain atoms.
Figure 1.

Escherichia coli CPS ATP-binding sites. Selected residues in the vicinity of the bound nucleotide are shown. Hydrogen bonds made with the six-membered ring of adenine are represented by broken lines. Residues involved in these bonds are Glu 208 (main chain O) and Leu 210 (main chain N) in domain N and Asp 753 (side chain O), His 754 (main chain O), and Leu 756 (main chain N) in domain C. Created from PDB file 1BXR (Thoden et al. 1999b) using Swiss Pdb-Viewer (Guex and Peitsch 1997) and rendered with POV-Ray (www.povray.org).
Whereas the binding-site architecture of the CPS ATP sites seems to limit the choices available for rational mutagenesis with the aim of changing nucleotide specificity, there is a precedent for the ability to use and discriminate between ATP and GTP within the constraints of the ATP-grasp fold. Succinyl-CoA synthetases (SCSs) all contain the ATP-grasp fold, yet either exclusively use ATP or GTP, or have dual substrate specificity (Johnson et al. 1998). This allows insight into the mechanism of nucleotide recognition in ATP-grasp enzymes. The three-dimensional structures have been solved for the dual specificity E. coli SCS in the presence of ADP (Joyce et al. 2000) and for the GTP-specific pig heart enzyme in the absence of nucleotide (Fraser et al. 2000). In an analysis of E. coli SCS (Fraser et al. 1999), the authors identify three residues that they suggest might be responsible for nucleotide recognition in SCSs. Comparison of the solved structures of CPS, SCSs, and other ATP-grasp proteins allows the identification of structurally equivalent residues in the nucleotide-binding sites. Table 1 lists residues that are at equivalent positions of the CPS and SCS nucleotide-binding sites, including those that are implicated in nucleotide discrimination in SCS. The residue equivalent to D207/D753 in CPS is a glutamate (E99) in E. coli SCS and forms the side-chain hydrogen bond with the N6 of adenine that is also observed in other ATP-grasp enzymes. In the ATP-specific SCSs, the equivalent residue is a cysteine, whereas it is substituted by alanine in the GTP-specific SCSs. The second residue is at the position corresponding to A144/P690 in CPS. This residue is positioned directly above the C6 of the base and is either alanine or proline (P20 in E. coli SCS) in those SCSs that are capable of binding ATP. In the GTP-specific SCSs, the equivalent residue is a glutamine. Together, substitutions at these two positions should result in an environment that can interact favorably with the C6 oxo group of GTP in analogy to the interaction that could be achieved with an aspartate-to-asparagine substitution at position 207/753 as outlined above. In addition to differences at these two positions, those SCSs that can interact with GTP have an alanine (A101 and A108 in E. coli and pig heart SCS, respectively) at a position that in ATP-grasp enzymes is usually occupied by a bulky hydrophobic residue, which is positioned near the C2 of the base in such a way that it could sterically block the C2 amino moiety of GTP. The equivalent residue is F755 in domain C of CPS, arginine in ATP-specific SCSs, and usually tryptophan or tyrosine in other members of the ATP-grasp fold. An exception is domain N of CPS, in which the equivalent residue is S209, a small polar residue that could be expected to interact favorably with the C2 amino group of GTP. In this domain, the function analogous to F755 could potentially be fulfilled by I211, whose position with respect to the adenine ring might sterically impede GTP binding.
Table 1.
Structurally equivalent residues in the nucleotide-binding pockets of ATP-grasp enzymes
| CPS | SCS (β subunit) | DD ligase | |||
| Domain N ATP | Domain C ATP | Pigeon ATP | E. coli ATP/GTP | Pig heart GTP | E. coli ATP |
| A144 | P690 | P22 | P20 | Q20 | A112 |
| D207 | D753 | C108 | E99 | A106 | E180 |
| E208 | H754 | E109 | A100 | E107 | K181 |
| S209 | F755 | R110 | A101 | A108 | W182 |
| L210 | L756 | R111 | T102 | L109 | L183 |
| I211 | D757 | Y112 | D103 | D110 | S184 |
The indicated PDB files were used to determine structurally equivalent residues for both domains of CPS (1BXR), E. coli SCS (1CQI), pig heart SCS (1EUC), and D-alanine:D-alanine ligase (2DLN). Because the structure of the ATP-specific pigeon SCS has not been determined, residues for this protein are listed on the basis of a sequence alignment of SCSs (Johnson et al. 1998). The nucleotide specificity for each enzyme is also listed. D-alanine:D-alanine ligase is included as a typical representative of the ATP-grasp fold.
On the basis of the analysis of the CPS and SCS nucleotide-binding sites, we anticipated that mutations of residues A144, D207, S209, and I211 in domain N of CPS or residues P690, D753, and F755 in domain C could allow GTP to interact with the CPS active sites, even in the continued presence of the conserved backbone interactions with ATP that have been described above. We constructed the following mutants: A144Q, D207A, S209A, A144Q/D207A, A144Q/D207A/S209A (ADS), I211S, and D207N in domain N, and P690Q, D753A, F755A, P690Q/D753A/F755A (PDF), and D753N in domain C. We then characterized these constructs to (1) test for the ability to utilize GTP, and (2) determine the effect of these mutations on ATP utilization by CPS.
CP formation assays
In the first experiment, we monitored CP synthesis by coupling CPS activity to that of ornithine transcarbamoylase and measuring the resulting citrulline. All CPS mutants displayed significant ATP-dependent CP synthesis activity (Table 2). This indicates that none of the mutations eliminated the ability of CPS to utilize ATP. Four mutants had activities of only 10% of that for wild-type CPS. These were ADS and PDF (those constructs in which all three mutations suggested by comparison of the SCS-binding sites have been combined in either domain N or C of CPS), as well as both mutations at position 753 in domain C (D753A and D753N). The remaining mutants showed only modest effects on the overall CPS activity, with values ranging from 30% to 100% of wild-type activity. The fact that the mutants were able to synthesize CP with glutamine as the nitrogen donor indicates that the mutations did not introduce large-scale structural changes in the protein, as this activity requires the coordinated utilization of substrates at three active sites that are separated by considerable distances in the folded protein. Nonspecific structural changes would likely interfere with proper coordination and communication between the different active sites.
Table 2.
Glutamine-dependent CP synthesis activities for wild-type and mutant CPSs
| CP formation (μmoles • min−1 • mg−1) | ||
| ATP | ATP + GTP | |
| WT | 0.64 | 0.81 |
| A144Q | 0.81 | 0.85 |
| D207A | 0.24 | 0.35 |
| S209A | 0.32 | 0.30 |
| ADS | 0.06* | 0.07* |
| D207N | 0.26 | 0.38 |
| A144Q/D207A | 0.37 | 0.40* |
| I211S | 0.57 | 0.56 |
| P690Q | 0.71 | 0.78 |
| D753A | 0.06* | 0.08 |
| F755A | 0.58 | 0.62 |
| 0.08 | 0.07 | |
| D753N | 0.08 | 0.08 |
CP formation was measured by coupling to the reaction of ornithine transcarbamoylase in the presence of ornithine and determining the resulting citrulline. Assay mixtures included 50 mM HEPES, 100 mM KCl, 20 mM MgSO4, 40 mM NaHCO3, 10 mM glutamine, 1 mM DTT, 5 u/mL ornithine transcarbamoylase, 10 mM ornithine, and 5 mM each indicated nucleotide, final pH 7.6. CPS (5–10 μg) was incubated at 37°C for 5–60 min, depending on the activity of the particular mutant. Citrulline was determined by reaction with diacetyl monoxime (Guthohrlein and Knappe 1968). SEM for all values was less than 1%, except * = less than 5%.
To test whether the mutants were able to utilize GTP, we also performed the coupled CP-formation assay in the presence of both ATP and GTP (Table 2). The rationale was that a mutant that successfully uses GTP in one domain will show an increased activity in the presence of both nucleotides compared with the activity observed with ATP alone. However, the additional presence of GTP resulted in little change in activity for any of the mutants.
We also performed a CP formation assay, in which enzyme was incubated in the presence of all substrates and either radiolabeled ATP or GTP, with subsequent separation of the reaction products by paper chromatography in a solvent system that is capable of separating nucleoside triphosphate, CP, and Pi (Wood 1961). This assay allows direct detection of nucleotide usage through visualization of the labeled products CP and Pi, as well as determination of the stoichiometry of product formation by comparison of the relative amounts of CP and Pi.
To detect GTP utilization, the wild-type and mutant CPSs were incubated in reactions containing ATP (to react on the wild-type domain) and [γ-32P]GTP. We carried out various permutations of this experiment, with protein concentration from 5 μM to 10 nM, incubation times from 10 min to 2 h, varying ATP:GTP ratios (5:5 mM, 0.5:5 mM, 0.1:10 mM, 0.05:10 mM, 0.001:10 mM, 0.02 nM:10 mM), and different exposure times to either quantitate abundant products or try and detect faint signals. Under no condition did we observe any utilization of GTP by wild-type or mutant CPSs. Incubation in the presence of labeled GTP did not result in any detectable signal over the background Pi level observed in the minus CPS control, confirming the inability of these mutants to utilize GTP in an appreciable manner.
We also tested product formation from ATP alone. In the presence of labeled ATP, production of labeled CP and Pi generally mirrored the results of Table 2. Approximately equal signals were detected for CP and Pi for wild-type and the domain N mutants A144Q, D207A, D207N, and I211S. This is in keeping with productive utilization of ATP on both domains in the overall reaction. The constructs ADS, P690Q, D753N, and PDF produced an excess of Pi over CP, indicating a partial uncoupling of the reactions on the two ATP sites with a resulting stoichiometry of more than two ATP consumed per CP formed.
Inhibition screen for GTP binding
Because it appeared that none of the mutant CPSs could utilize GTP, we determined whether GTP or its analog ITP could at least bind to the active site, causing inhibition of the glutamine-dependent ATPase activity (Table 3). Only modest changes in activity were seen for most mutants, with the strongest decrease in activity in the presence of GTP seen for ADS (from 0.24 to 0.14 μmoles • min−1 • mg−1) and PDF (from 0.75 to 0.55 μmoles • min−1 • mg−1). Similar results were seen in the presence of ITP. It is possible that this reduced activity is caused by binding of GTP and ITP to the mutated active sites, especially as the mutants involved combine more than one mutation, which would be more likely to adequately change the environment of the sites. However, all of the changes were relatively minor, and with the exception of PDF, the activities in the absence of GTP and ITP were already quite low. Thus, the finding of this possible GTP inhibition did not provide additional insight into the design of mutants that would utilize GTP.
Table 3.
Inhibition of the glutamine-dependent ATPase activity of CPS by GTP and ITP
| Specific activity (μmoles • min−1 • mg−1) | Relative activity | ||||
| ATP | +GTP | +ITP | +GTP | +ITP | |
| WT | 1.53 | 1.64 | 1.82 | 1.07 | 1.19 |
| A144Q | 2.06 | 2.31 | 2.19 | 1.12 | 1.06 |
| D207A | 0.42 | 0.46 | 0.48 | 1.08 | 1.15 |
| S209A | 0.79 | 0.89 | 0.85 | 1.14 | 1.09 |
| ADS | 0.24 | 0.14 | 0.17 | 0.60 | 0.74 |
| D207N | 0.44 | 0.38 | 0.41 | 0.91 | 1.12 |
| A144Q/D207A | 0.48 | 0.31 | nd | 0.65 | nd |
| I211S | 1.49 | 1.50 | 1.68 | 1.01 | 0.82 |
| P690Q | 2.54 | 3.09 | 2.96 | 1.22 | 1.17 |
| D753A | 0.45 | 0.46 | 0.49 | 1.03 | 1.10 |
| F755A | 1.56 | 1.49 | 1.73 | 0.96 | 1.11 |
| 0.75 | 0.55 | 0.63 | 0.74 | 0.84 | |
| D753N | 1.42 | 1.43 | 1.56 | 1.01 | 1.10 |
Glutamine-dependent ATPase assays were performed with ATP levels at ~ 10-fold Km for the particular construct (5, 0.5, or 0.05 mM) and either 5 mM GTP or ITP. Absolute rates observed and fold change relative to reactions omitting GTP or ITP are given. Blank reactions for this assay consisted of samples without ATP but containing CPS and 5 mM GTP or ITP. ITP inhibition for A144Q/D207A was not determined (nd). SEM for all values is <6%.
ATP-dependent kinetic parameters
Next, we analyzed the effects of the mutations on ATP interaction by determining kinetic parameters for (1) glutamine-dependent ATPase activity, which requires utilization of both sites, (2) bicarbonate-dependent ATPase activity, which monitors ATPN, and (3) CP-dependent ATP synthesis, which occurs on ATPC (Table 4). The mutations mostly affected the Km rather than the kcat of the monitored reactions, a finding that is consistent with the fact that the mutations in this study target interactions with the adenine ring, rather than parts of the catalytic machinery of CPS.
Table 4.
ATP-dependent kinetic parameters for wild-type and mutant CPS
| Glutamine-dependent ATPase |
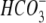 -dependent ATPase -dependent ATPase |
CP-dependent ATP synthesis | |||||||
| Km (mM) | kcat (s−1) | kcat/Km (mM−1 s−1) | Km (mM) | kcat (s−1) | kcat/Km (mM−1 s−1) | Km (mM) | kcat (s−1) | kcat/Km (mM−1 s−1) | |
| WT | 0.029 ± 0.002 | 4.04 ± 0.1 | 137.5 | 0.005 ± 3.10 • 10−4 | 0.09 ± 2.9 • 10−3 | 17.9 | 0.006 ± 0.001 | 0.36 ± 0.01 | 65.3 |
| A144Q | 0.285 ± 0.056 | 3.94 ± 0.51 | 13.9 | 0.034 ± 0.007 | 0.10 ± 0.01 | 2.9 | 0.005 ± 3.9 • 10−5 | 0.28 ± 8.0 • 10−4 | 56.4 |
| D207A | 0.015 ± 0.002 | 1.51 ± 0.11 | 100.9 | 0.006 ± 0.001 | 0.11 ± 0.01 | 18.9 | 0.006 ± 0.001 | 0.26 ± 0.02 | 44.6 |
| S209A | 0.031 ± 0.004 | 2.43 ± 0.13 | 78.0 | 0.016 ± 0.001 | 0.35 ± 0.01 | 22.7 | 0.011 ± 0.001 | 0.59 ± 0.01 | 51.4 |
| ADS | 0.571 ± 0.083 | 0.66 ± 0.04 | 1.1 | 0.319 ± 0.012 | 0.33 ± 0.01 | 1.0 | 0.008 ± 0.001 | 0.28 ± 0.01 | 36.3 |
| D207N | 0.020 ± 0.018 | 1.65 ± 0.02 | 84.2 | 0.009 ± 0.003 | 0.06 ± 0.01 | 7.2 | 0.004 ± 0.002 | 0.14 ± 0.01 | 36.6 |
| A144Q/D207A | 0.154 ± 0.001 | 1.70 ± 0.08 | 11.0 | 0.021 ± 0.002 | 0.12 ± 0.01 | 5.8 | 0.011 ± 4.0 • 10−4 | 0.37 ± 0.04 | 33.9 |
| I211S | 0.072 ± 0.009 | 4.77 ± 0.26 | 66.4 | 0.020 ± 0.002 | 0.29 ± 0.01 | 14.4 | 0.011 ± 0.002 | 0.48 ± 0.02 | 44.0 |
| P690Q | 0.383 ± 0.061 | 9.01 ± 0.79 | 23.5 | 0.009 ± 0.002 | 0.09 ± 0.01 | 10.4 | 0.039 ± 0.005 | 0.28 ± 0.01 | 7.1 |
| D753A | 0.064 ± 0.010 | 1.71 ± 0.13 | 26.8 | 0.007 ± 4.0 • 10−4 | 0.14 ± 2.9 • 10−3 | 19.4 | 0.002 ± 2.0 • 10−4 | 0.07 ± 2.4 • 10−3 | 41.2 |
| F755A | 0.135 ± 0.011 | 5.18 ± 0.17 | 38.3 | 0.004 ± 0.001 | 0.07 ± 2.4 • 10−3 | 14.8 | 0.031 ± 0.004 | 0.28 ± 0.01 | 9.0 |
| 0.199 ± 0.015 | 2.90 ± 0.11 | 14.6 | 0.022 ± 0.005 | 0.34 ± 0.03 | 15.6 | 0.027 ± 0.004 | 0.06 ± 3.5 • 10−3 | 2.3 | |
| D753N | 0.121 ± 0.026 | 5.60 ± 0.59 | 46.2 | 0.008 ± 0.001 | 0.13 ± 0.01 | 16.9 | 0.005 ± 3.0 • 10−4 | 0.15 ± 2.4 • 10−3 | 31.1 |
ATPase reactions contained 50 mM HEPES, 100 mM KCl, 20 mM MgSO4, 40 mM NaHCO3, 10 mM ornithine, 1 mM DTT, 10 mM phosphoenolpyruvate, 0.2 mM NADH, 18 u/mL pyruvate kinase, and 24 u/mL lactate dehydrogenase in the presence or absence of 10 mM glutamine. ATP synthesis reactions included 50 mM HEPES, 100 mM KCl, 20 mM MgSO4, 10 mM CP, 10 mM ornithine, 1 mM NADP, 20 mM glucose, 10 u/mL hexokinase, and 5 u/mL glucose-6-phosphate dehydrogenase. All reactions were performed with 5 or 50 μg CPS in 500 μL final volume at 25°C, final pH 7.6. Standard error of the kinetic parameters was determined from nonlinear regression curve fitting (GraFit, version 5.01) and was within ±6.1%.
Mutations at positions that are conserved in the ATP-grasp fold
As outlined above, residues D207 and S209 in domain N and the corresponding D753 and F755 in domain C of CPS are at positions that have been identified as part of a conserved fingerprint of residues in ATP-grasp proteins, and their mutation might therefore be expected to have strong effects on ATP utilization. Of these residues, only S209 does not conform to the observed consensus at its position (large hydrophobic residue), and we have targeted I211 as a possible functional substitute. All of the single mutations at these positions had surprisingly small effects on ATP utilization (Table 4).
Residues D207 and D753 are positioned to allow side-chain interactions with the N6 of the bound ATP. This interaction is generally conserved in ATP-grasp enzymes, but is not observed for D207 in domain N of CPS. Mutation of these residues to either alanine or asparagine had only modest effects on ATP utilization, arguing against a strong contribution of the aspartate side chain to nucleotide binding. It should be noted that D207A and D753A were subjected to previous study in which also only modest effects were observed (Javid-Majd et al. 1996; Stapleton et al. 1996).
The residue pair S209/F755, representing the second conserved position examined in our study, also showed only very small effects on ATP utilization. Mutation of either residue to alanine resulted in only a modest increase in ATP KM, an effect that was slightly stronger in the F755A mutation, in which the wild-type residue conforms to the consensus observed in ATP-grasp proteins. The mutant I211S, in which we examined the role of this residue as a potential functional analog to F755 in domain N, also exhibited behavior similar to wild-type CPS.
Constructs containing mutations at positions 144/690
The strongest effects were displayed by constructs containing the glutamine mutations at positions 144 in domain N or the equivalent 690 in domain C (Table 4). The A144Q and P690Q single mutants both showed an ~10-fold increase in ATP Km in the overall reaction and in the respective partial reaction. The A144Q mutant is also present in the ADS construct, the only other mutant with large Km changes. These findings confirm the important role of a small, uncharged residue at this position, and are consistent with a steric and/or electrostatic clash of the glutamine side chain with the N6 of adenine.
Combination of A144Q with the D207A mutation led to a slight lowering of Km compared with the A144Q mutant. The removal of the aspartate side chain that would be in close proximity to the newly introduced bulk of the glutamine could reduce the steric clash with ATP by allowing the glutamine side chain to adopt a different conformation compared with that in the A144Q mutant. Introduction of the S209A mutation into the A144Q/D207 double mutant led to a large increase in ATP Km, especially in the bicarbonate-dependent reaction. The serine-to-alanine change is a size neutral substitution that eliminates the hydroxyl group and, therefore, the polar character of the serine side chain. Because the kinetic parameters for the S209A single mutant did not differ significantly from those of wild type, the large change in ATP Km between A144Q/D207A and ADS, two mutants that differ only by one oxygen atom, is intriguing. Serine 209 is quite well conserved in domain N of CPSs (in 55 of 61 compared sequences; Cammarano et al. 2002), suggesting an important function that is distinct from that of the corresponding residue in domain C and most other ATP-grasp enzymes in which this position is conserved as a bulky hydrophobic side chain that is involved in nonpolar interactions with the adenine ring. Obviously, this nonpolar function cannot be fulfilled by S209, but it is unclear why this residue is so conserved in domain N, especially as the isolated mutation of this residue to alanine had only modest effects.
Despite the similar effect of the A144Q and P690Q mutations, the three combined SCS-based mutations in constructs ADS and PDF differed significantly, with PDF exhibiting a much smaller effect than its domain N counterpart. In the overall reaction, the Km increase for the PDF mutant was less than half of that of the ADS construct, and this was even more pronounced when comparing the effect on the respective partial reactions, in which there was an ~10-fold difference in the Km increase for ADS (bicarbonate-dependent reaction) and PDF (ATP synthesis reaction). Also, as opposed to the Km increase that was observed in ADS relative to A144Q, combination of all three mutations in domain C resulted in a decrease of ATP Km relative to P690Q. This differential effect of the ADS and PDF mutants might be a manifestation of the fact that fewer constraints are imposed on domain C compared with domain N, or it might be due to the difference in side-chain character at positions S209 and F755 in the two domains.
Discussion
Structural basis for inability of CPS to utilize GTP
The fact that all of our CPS mutants retained specificity for ATP demonstrates that the active sites of CPS are very resistant to changes with respect to nucleotide base recognition. Although this might not come as a surprise, given the nature of the ATP-grasp fold with its heavy reliance on backbone interactions for interaction with the adenine base, it is nonetheless confounding, as SCSs are able to utilize both ATP and GTP either exclusively or with dual specificity. This demonstrates a flexibility within the ATP-grasp fold of SCSs that is in stark contrast to the rigidity that is encountered in the case of CPS. Unfortunately, the structure of the GTP-specific SCS was solved in the absence of nucleotide (Fraser et al. 2000), leaving the details of how an ATP-grasp fold can effectively interact with GTP open to speculation. A precedent for a protein with dual ATP/GTP specificity with known structures for both substrate complexes exists, however, in protein kinase CK2 (Niefind et al. 1999), a member of the serine/threonine kinase family. This family belongs to the protein kinase-like fold (SCOP classification), and has been shown recently to possess structural similarities to enzymes of the ATP-grasp fold (Kobayashi and Go 1997; Denessiouk and Johnson 2000) and to display some analogous residue interactions with the bound nucleotide, including residues corresponding to D207-L210 and D753-L756 in domain N and C of CPS, respectively. In CK2, the adenine ring makes interactions with the protein in a manner analogous to that observed in CPS (Fig. 2A ▶), including the conserved backbone-mediated hydrogen bonds, in this case, between the carbonyl of E114 and amide of V116 and the N6 and N1 of adenine, respectively. The difference in hydrogen-bonding character at these positions in guanine is satisfied in the GTP-bound state of CK2 by displacement of the base in such a way that the O6 of guanine superimposes with the N1 of adenine and can therefore make the analogous interaction with the backbone amide of V116 (Fig. 2B,C ▶). At the position of the N6 in the ATP-bound state of CK2, there is a water molecule in the GTP-bound state that is hydrogen bonded to both the E114 carbonyl and the O6 of guanine. The N1 of guanine is hydrogen bonded to the backbone carbonyl of V116. Somewhat surprisingly, no specific interactions are made with the C2 amino group of guanine. As a result of the displacement relative to one another, the bases of both ATP and GTP make two direct hydrogen bonds with the protein, a fact that is reflected in similar Km values for both substrates in protein kinase CK2 (Niefind et al. 1999).
Figure 2.

Nucleotide binding in protein kinase CK2, SCS, and CPS. (A) ATP-bound state of CK2 (PDB file 1DAW; Niefind et al. 1999). Only the backbone atoms are shown for residues 114–117, whereas backbone and side-chain atoms are shown for Asn 118. Water molecules are represented as red spheres. Hydrogen bonds are shown as broken lines. (B) GTP-bound state of CK2 (PDB file 1DAY; Niefind et al. 1999). (C) Superposition of ATP- and GTP-bound states of CK2 with the GTP shown semitransparent. Note the displacement of GTP relative to ATP and differences in water structure that allow analogous hydrogen bonding for both substrates. (D) CPS segment interacting with N3 of adenine, and SCS counterpart. Cartoon representations of CPS residues 228–250 (in domain N, green), 773–795 (in domain C, orange), and the corresponding segment in E. coli SCS (121–144, blue) are superimposed. CPS residues 240–241 and 785–786 (shown in stick representation in Fig. 1 ▶) are colored red. These residues make van der Waals contacts with the N3 of the bound nucleotide and could prevent binding of GTP in a manner analogous to that in protein kinase CK2. A potassium ion that is present in both domains N and C of CPS is shown as a pink sphere. PDB files 1BXR (Thoden et al. 1999b) and 1CQI (Joyce et al. 2000) were used, respectively, for the CPS and SCS structures. All panels were created with Swiss Pdb-Viewer (Guex and Peitsch 1997) and rendered with POV-Ray.
The example of protein kinase CK2 demonstrates an elegant solution to the problem of using the same binding-site architecture to bind two substrates with different hydrogen-bonding requirements, and doing so through interactions with the protein backbone, rather than the more flexible situation in which side chains are used to interact with the substrate. A similar ratcheting model has been proposed for GTP interaction in the dual-specificity E. coli SCS (Fraser et al. 2000) and might also be necessary in CPS to satisfy the distinct hydrogen-bonding requirements of GTP. Analysis of the CPS nucleotide-binding sites reveals the presence of a loop (containing M240 and G241 in domain N and A785 and G786 in domain C; Fig. 2D ▶) that is positioned underneath the N3 of the bound adenine, and could prevent a similar dislocation of GTP relative to ATP, thereby not allowing CPS to accommodate this alternative substrate. The equivalent sequence in SCS forms a helix that is positioned in such a way as to not interact with the bound nucleotide (Fig. 2D ▶). The N3 of the bound ADP in the E. coli SCS is exposed to solvent, allowing for movement of the nucleotide in a manner similar to that observed for CK2 if GTP were to bind in the active site. Residues in the loop in both domains N and C of CPS are involved intimately in coordinating a potassium ion that is known to be essential for activity (Lusty 1978). It seems unlikely that this segment can adopt the radically different conformation seen in the SCS structure and still allow for the potassium ion to bind. In addition, M240 in domain N makes extensive contacts with the hydrophobic side of the adenine base (Fig. 1 ▶), and disruption of this interaction is unlikely to be tolerated. Thus, the occurrence of multiple backbone interactions with the adenine rings, combined with steric interference of the K+-loop, appears to preclude GTP utilization by CPS.
Effects of mutations on ATP-dependent CPS activity
Whereas the mutations did not allow alternative nucleotide usage, they did provide insight into the detailed architecture of the two ATP sites and the extent to which the targeted residues contribute to ATP recognition by CPS.
The first interesting result was that mutation of several residues generally conserved in ATP-grasp proteins did not result in significant changes in kinetic parameters, as would be expected given their suggested involvement in nucleotide base recognition. The first residue of this category is D207 in domain N and the corresponding D753 in domain C. It is noteworthy that a hydrogen bond between this residue and the N6 of the bound adenine generally occurs in ATP-grasp proteins but is not observed for D207 in domain N of CPS. It is not clear whether the observed difference in hydrogen bonding of the glutamate side chains at positions 207 and 753 represents a functional difference between the two ATP-binding sites in CPS, or whether the different conformation is an artifact of the crystallization and D207 has a different conformation in solution or during the reaction cycle that allows it to hydrogen bond to the N6 of adenine.
A possible explanation for the small effect of the asparagine mutations can be obtained by examining the environment of D207 and D753 in more detail. Both side chains are hydrogen bonded to an arginine side chain (R169 and R715, respectively), which also interacts with the α-phosphate of the bound nucleotide. This hydrogen bond between the two residues involves the same carbonyl oxygen of D207 and D753 that is also bonded to the N6 of the ATP in domain C. If the asparagine in the D207N and D753N mutants makes the same interaction with the arginine residue, its side chain will be positioned for productive bonding to ATP, whereas the part of the side chain that has hydrogen bond-donating potential (which we hoped would interact favorably with GTP) is pointed away from the nucleotide. In this orientation, the asparagine side chain would look very similar to the wild-type aspartate from the perspective of the bound nucleotide and not be expected to display radically different kinetics. More surprising is the fact that the effect of the alanine mutations at these positions did not differ from those of the asparagine substitutions. Because in this case the functionality of the side chain is completely removed, this finding suggests that residues D207 and D753 in CPS are not contributing significantly to ATP recognition and/or binding.
Small effects were also observed for mutations at the second position conserved in ATP-grasp proteins, S209 and F755. Whereas F755 conforms to the consensus observed in ATP-grasp proteins, S209 does not, but is strongly conserved in domain N of CPSs. An indication for the important role of S209, despite the marginal effect of the serine-to-alanine single mutant, can be seen in the dramatic ATP Km increase observed in the ADS triple mutant compared with the A144Q/D207A double mutant. The side chain of S209 is involved in two hydrogen bonds to the backbone amides of neighboring L210 and I211, thereby stabilizing the conformation of this residue segment that is connecting the two subdomains of the ATP-grasp fold. It is possible that this stabilizing effect of S209 becomes more critical in the A144Q/D207A double mutant, in which the environment of this segment is disturbed, leading to the strong effect observed when the serine side chain is removed in this context. The corresponding segment in domain C seems to be stabilized by backbone hydrogen bonds from F755 to D753 and D757. The involvement of backbone hydrogen bonds in this case might explain the lack of a strong effect in the PDF triple mutant.
In contrast to the surprisingly small effects of mutations at the conserved ATP-grasp positions 207/209 and 753/755, we observed strong effects on the ATP Km in mutants at position A144 in domain N and the corresponding P690 in domain C. In addition to the direct steric clash of the introduced glutamine side chain with the bound ATP as reasoned above, the large effect of the A144Q and P690Q mutations compared with the surprisingly small effects of mutations of the conserved ATP-grasp residues can also be rationalized by the localization of all of the targeted residues in the hinge region connecting the two subdomains of the ATP-grasp fold. This positions them ideally to interfere with proper movement of the lid over the ATP site, and could therefore limit access of the nucleotide to the active site. The rigid-body movement of the lid (residues 141–210 in domain N and 687–756 in domain C) has been demonstrated by X-ray crystallography for domain C (Thoden et al. 1999b), and has also been described for the corresponding domain in other ATP-grasp proteins (Artymiuk et al. 1996). The domain N lid is in a closed conformation for all solved structures of CPS, but must open to admit substrates and release products. Introduction of the rather bulky glutamine side chain in place of the small alanine and proline residues at positions 144 and 690 would be expected to have a larger negative effect than the other substitutions in which the size of the side chain was either maintained (D207N, S209A, D753N) or reduced (D207A, I211S, D753A, F755A). The relatively mild effect of PDF compared with ADS could be explained by the fact that combined removal of the bulky D753 and F755 side chains counteracts the effect of the P690Q mutation in the hinge of domain C.
In summary, we show here that the active sites of CPS are very resistant toward changes in nucleotide specificity, most likely owing to the presence of the K+-binding loop that prevents binding of GTP in a manner analogous to that observed in protein kinase CK2, and that might also operate in GTP-specific SCSs. We further show that several residues that are conserved in ATP-grasp proteins in general, or CPSs in particular, show surprisingly small effects when mutated, whereas another pair of residues that has so far received little attention seems more important for ATP utilization by CPS. We propose that the effect of mutations at positions 144 and 690 is primarily due to their position in the hinge region connecting the two subdomains of the ATP-grasp fold, thereby interfering with proper movement of the lid and access of substrate to the active sites.
Materials and methods
Strains, plasmids, and DNA methods
XL1-Blue and Top10 E. coli were used for DNA manipulations. The CPS-deficient E. coli strain L673 (Guillou et al. 1989) was used as an expression host for purification. The E. coli CPS expression plasmid pUCAB, encoding both carA and carB under the control of the trc promoter, was created from pUCABI (Tuchman et al. 1997) by excision of a BamHI–KpnI fragment containing the argI gene as described previously (Saeed-Kothe and Powers-Lee 2002). Site-directed mutagenesis was performed using the PCR-based primer-extension method (Higuchi 1990) or either the QuikChange or Chameleon kits from Stratagene, followed by subcloning of the sequence-verified mutagenesis cassette. Sequencing was done at the DNA sequencing facility of the University of Maine. All other DNA methods were essentially as described by Sambrook et al. (1989).
Protein purification and activity determination
Wild-type and mutant E. coli CPSs were prepared as described previously (Saeed-Kothe and Powers-Lee 2002) with purity of >95% indicated by SDS-PAGE. Yield and chromatographic behavior for all mutants was very similar to that of wild-type CPS, indicating proper folding and assembly of the mutant enzymes and the absence of large-scale structural changes. Protein concentrations were determined either by the Bradford dye-binding assay (Bradford 1976), or by measuring the absorbance at 280 nm (0.685 for 1 mg/mL CPS [Rubino et al. 1986], corresponding to 6.3 μM).
CP synthesis was measured as citrulline formation by coupling CP formation to the reaction of ornithine transcarbamoylase in the presence of ornithine (Guthohrlein and Knappe 1968). ADP formation was followed at 340 nm by coupling to NADH oxidation with pyruvate kinase and lactate dehydrogenase (Mareya and Raushel 1994). ATP synthesis was followed by using a coupled system of hexokinase and glucose-6-phosphate dehydrogenase and following NAD reduction at A340 (Mareya and Raushel 1994). Kinetic data were collected on a Beckman DU 640 spectrophotometer and were fit by nonlinear regression (GraFit 5.0 software; Leatherbarrow 2001) to the equation v = Vmax S/(Km+S), in which v is the initial velocity, Vmax is the maximal velocity, S is the substrate concentration, and Km is the Michaelis-Menten constant.
Paper chromatography using Whatman 3MM chromatography paper and the GW3 solvent system (Wood 1961) was used to separate γ-32P-labeled nucleotide, CP, and Pi. The GW3 system consists of (per 100 mL) the following: 32 mL n-butanol, 16 mL n-propanol, 20 mL acetone, 20 mL 80% formic acid, 12 mL 30% trichloroacetic acid, and 0.3 g EDTA. The reactions included 10 nM CPS, 50 mM HEPES, 100 mM KCl, 20 mM MgSO4, 40 mM NaHCO3, 10 mM glutamine, 1 mM DTT, and either 0.5 mM [γ-32P]ATP (when assaying ATP utilization), or 0.5 mM ATP and 5 mM [γ-32P]GTP (when assaying GTP utilization). [γ-32P]ATP and [γ-32P]GTP (specific activity 6000 Ci/mmole, radiochemical concentration 10 mCi/mL) were from Perkin Elmer Life Sciences, Inc. The specific radioactivity in the assay was 0.2 and 0.02 μCi/nmole for ATP and GTP, respectively. The assay volume was 50 μL and the final pH was 7.6. After 120 min incubation, 2 μL of each reaction was spotted 2 cm from the bottom of the paper, developed for 1–1.5 h at room temperature, and dried for 15 min before exposure on Fuji medical X-ray film. The identity of the CP signal was confirmed by its absence in control reactions with either glutamine omitted or with ornithine and ornithine transcarbamoylase added to convert CP to citrulline and Pi. Quantification of the CP and Pi signals was done using the gel analysis command of the program ImageJ, developed by Wayne Rasband at the NIH (http://rsb.info.nih.gov/ij/).
Acknowledgments
This work was supported by National Institutes of Health Grant DK54423, and by GAANN fellowship support for M.K. from the U.S. Department of Education.
We thank Mendel Tuchman for the plasmid pUCABI and Amna Saeed-Kothe for insightful discussion.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Abbreviations
ADS, CPS mutant A144Q/D207A/S209A
CP, carbamoyl phosphate
CPS, carbamoyl phosphate synthetase
PDF, CPS mutant P690Q/D753A/F755A
SCS, succinyl-coenzyme A synthetase
Article published online ahead of print. Article and publication date are at http://www.proteinscience.org/cgi/doi/10.1110/ps.03416804.
References
- Anderson, P.M. 1995. Molecular aspects of carbamoyl phosphate synthesis. In Nitrogen metabolism and excretion (eds. P.J. Walsh et al.), pp. 33–49. CRC Press, Boca Raton, FL.
- Artymiuk, P.J., Poirrette, A.R., Rice, D.W., and Willett, P. 1996. Biotin carboxylase comes into the fold. Nat. Struct. Biol. 3 128–132. [DOI] [PubMed] [Google Scholar]
- Bradford, M.M. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72 248–254. [DOI] [PubMed] [Google Scholar]
- Cammarano, P., Gribaldo, S., and Johann, A. 2002. Updating carbamoylphosphate synthase (CPS) phylogenies: Occurrence and phylogenetic identity of archaeal CPS genes. J. Mol. Evol. 55 153–160. [DOI] [PubMed] [Google Scholar]
- Denessiouk, K.A. and Johnson, M.S. 2000. When fold is not important: A common structural framework for adenine and AMP binding in 12 unrelated protein families. Proteins 38 310–326. [PubMed] [Google Scholar]
- Fan, C., Moews, P.C., Shi, Y., Walsh, C.T., and Knox, J.R. 1995. A common fold for peptide synthetases cleaving ATP to ADP: Glutathione synthetase and D-alanine:D-alanine ligase of Escherichia coli. Proc. Natl. Acad. Sci. 92 1172–1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser, M.E., James, M.N., Bridger, W.A., and Wolodko, W.T. 1999. A detailed structural description of Escherichia coli succinyl-CoA synthetase. J. Mol. Biol. 285 1633–1653. [DOI] [PubMed] [Google Scholar]
- ———. 2000. Phosphorylated and dephosphorylated structures of pig heart, GTP-specific succinyl-CoA synthetase. J. Mol. Biol. 299 1325–1339. [DOI] [PubMed] [Google Scholar]
- Galperin, M.Y. and Koonin, E.V. 1997. A diverse superfamily of enzymes with ATP-dependent carboxylate- amine/thiol ligase activity. Protein Sci. 6 2639–2643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guex, N. and Peitsch, M.C. 1997. SWISS-MODEL and the Swiss-PdbViewer: An environment for comparative protein modeling. Electrophoresis 18 2714–2723. [DOI] [PubMed] [Google Scholar]
- Guillou, F., Rubino, S.D., Markovitz, R.S., Kinney, D.M., and Lusty, C.J. 1989. Escherichia coli carbamoyl-phosphate synthetase: Domains of glutaminase and synthetase subunit interaction. Proc. Natl. Acad. Sci. 86 8304–8308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guthohrlein, G. and Knappe, J. 1968. Modified determination of citrulline. Anal. Biochem. 26 188–191. [DOI] [PubMed] [Google Scholar]
- Higuchi, R. 1990. Recombinant PCR. In PCR protocols: A guide to methods and applications. (eds. M.A. Innis et al.), pp. 177–183. Academic Press, San Diego, CA.
- Javid-Majd, F., Stapleton, M.A., Harmon, M.F., Hanks, B.A., Mullins, L.S., and Raushel, F.M. 1996. Comparison of the functional differences for the homologous residues within the carboxy phosphate and carbamate domains of carbamoyl phosphate synthetase. Biochemistry 35 14362–14369. [DOI] [PubMed] [Google Scholar]
- Johnson, J.D., Mehus, J.G., Tews, K., Milavetz, B.I., and Lambeth, D.O. 1998. Genetic evidence for the expression of ATP- and GTP-specific succinyl-CoA synthetases in multicellular eucaryotes. J. Biol. Chem. 273 27580–27586. [DOI] [PubMed] [Google Scholar]
- Joyce, M.A., Fraser, M.E., James, M.N., Bridger, W.A., and Wolodko, W.T. 2000. ADP-binding site of Escherichia coli succinyl-CoA synthetase revealed by x-ray crystallography. Biochemistry 39 17–25. [DOI] [PubMed] [Google Scholar]
- Kobayashi, N. and Go, N. 1997. A method to search for similar protein local structures at ligand binding sites and its application to adenine recognition. Eur. Biophys. J. 26 135–144. [DOI] [PubMed] [Google Scholar]
- Kothe, M., Eroglu, B., Mazza, H., Samudera, H., and Powers-Lee, S.G. 1997. Novel mechanism for carbamoyl-phosphate synthetase: A nucleotide switch for functionally equivalent domains. Proc. Natl. Acad. Sci. 94 12348–12353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leatherbarrow, R.J. 2001. GraFit Version 5. Erithacus Software, Horley, U.K.
- Lusty, C.J. 1978. Carbamyl phosphate synthetase. Bicarbonate-dependent hydrolysis of ATP and potassium activation. J. Biol. Chem. 253 4270–4278. [PubMed] [Google Scholar]
- Mareya, S.M. and Raushel, F.M. 1994. A molecular wedge for triggering the amidotransferase activity of carbamoyl phosphate synthetase. Biochemistry 33 2945–2950. [DOI] [PubMed] [Google Scholar]
- Meister, A. 1989. Mechanism and regulation of the glutamine-dependent carbamyl phosphate synthetase of Escherichia coli. Adv. Enzymol. Relat. Areas Mol. Biol. 62 315–374. [DOI] [PubMed] [Google Scholar]
- Miles, B.W. and Raushel, F.M. 2000. Synchronization of the three reaction centers within carbamoyl phosphate synthetase. Biochemistry 39 5051–5056. [DOI] [PubMed] [Google Scholar]
- Niefind, K., Putter, M., Guerra, B., Issinger, O.G., and Schomburg, D. 1999. GTP plus water mimic ATP in the active site of protein kinase CK2. Nat. Struct. Biol. 6 1100–1103. [DOI] [PubMed] [Google Scholar]
- Nyunoya, H. and Lusty, C.J. 1983. The carB gene of Escherichia coli: A duplicated gene coding for the large subunit of carbamoyl-phosphate synthetase. Proc. Natl. Acad. Sci.. 80 4629–4633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powers-Lee, S.G. and Meister, A. 1988. Urea synthesis and ammonia metabolism. In The liver: Biology and pathology (eds. I.M. Arias et al.), pp. 317–329. Raven Press, NY.
- Rubino, S.D., Nyunoya, H., and Lusty, C.J. 1986. Catalytic domains of carbamyl phosphate synthetase. Glutamine- hydrolyzing site of Escherichia coli carbamyl phosphate synthetase. J. Biol. Chem. 261 11320–11327. [PubMed] [Google Scholar]
- Saeed-Kothe, A. and Powers-Lee, S.G. 2002. Specificity determining residues in ammonia- and glutamine-dependent carbamoyl phosphate synthetases. J. Biol. Chem. 277 7231–7238. [DOI] [PubMed] [Google Scholar]
- Sambrook, J., Fritsch, E.F., and Maniatis, T. 1989. Molecular cloning: A laboratory manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- Shabb, J.B., Ng, L., and Corbin, J.D. 1990. One amino acid change produces a high affinity cGMP-binding site in cAMP-dependent protein kinase. J. Biol. Chem. 265 16031–16034. [PubMed] [Google Scholar]
- Stapleton, M.A., Javid-Majd, F., Harmon, M.F., Hanks, B.A., Grahmann, J.L., Mullins, L.S., and Raushel, F.M. 1996. Role of conserved residues within the carboxy phosphate domain of carbamoyl phosphate synthetase. Biochemistry 35 14352–14361. [DOI] [PubMed] [Google Scholar]
- Sunahara, R.K., Beuve, A., Tesmer, J.J., Sprang, S.R., Garbers, D.L., and Gilman, A.G. 1998. Exchange of substrate and inhibitor specificities between adenylyl and guanylyl cyclases. J. Biol. Chem. 273 16332–16338. [DOI] [PubMed] [Google Scholar]
- Thoden, J.B., Holden, H.M., Wesenberg, G., Raushel, F.M., and Rayment, I. 1997. Structure of carbamoyl phosphate synthetase: A journey of 96 Å from substrate to product. Biochemistry 36 6305–6316. [DOI] [PubMed] [Google Scholar]
- Thoden, J.B., Raushel, F.M., Benning, M.M., Rayment, I., and Holden, H.M. 1999a. The structure of carbamoyl phosphate synthetase determined to 2.1 Å resolution. Acta Crystallogr., Sect. D: Biol. Crystallogr. 55 8–24. [DOI] [PubMed] [Google Scholar]
- Thoden, J.B., Wesenberg, G., Raushel, F.M., Holden, H.M., Wang, W., Kappock, T.J., Stubbe, J., and Ealick, S.E. 1999b. Carbamoyl phosphate synthetase: Closure of the B-domain as a result of nucleotide binding. Biochemistry 38 2347–2357. [DOI] [PubMed] [Google Scholar]
- Tuchman, M., Rajagopal, B.S., McCann, M.T., and Malamy, M.H. 1997. Enhanced production of arginine and urea by genetically engineered Escherichia coli K-12 strains. Appl. Environ. Microbiol. 63 33–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turko, I.V., Francis, S.H., and Corbin, J.D. 1998. Hydropathic analysis and mutagenesis of the catalytic domain of the cGMP-binding cGMP-specific phosphodiesterase (PDE5). cGMP versus cAMP substrate selectivity. Biochemistry 37 4200–4205. [DOI] [PubMed] [Google Scholar]
- Vinitsky, A. and Grubmeyer, C. 1993. A new paradigm for biochemical energy coupling. Salmonella typhimurium nicotinate phosphoribosyltransferase. J. Biol. Chem. 268 26004–26010. [PubMed] [Google Scholar]
- Wang, X. and Kemp, R.G. 1999. Identification of residues of Escherichia coli phosphofructokinase that contribute to nucleotide binding and specificity. Biochemistry 38 4313–4318. [DOI] [PubMed] [Google Scholar]
- Wei, J. and Leyh, T.S. 1998. Conformational change rate-limits GTP hydrolysis: The mechanism of the ATP sulfurylase-GTPase. Biochemistry 37 17163–17169. [DOI] [PubMed] [Google Scholar]
- Wood, T. 1961. A procedure for the analysis of acid-soluble phosphorous compounds and related substances in muscle and other tissues. J. Chromatogr. 6 142–154. [Google Scholar]