

Abstract
Corynebacterium 2,5-Diketo-D-gluconic acid reductase (2,5-DKGR) catalyzes the reduction of 2,5-diketo-D-gluconic acid (2,5-DKG) to 2-Keto-L-gulonic acid (2-KLG). 2-KLG is an immediate precursor to L-ascorbic acid (vitamin C), and 2,5-DKGR is, therefore, an important enzyme in a novel industrial method for the production of vitamin C. 2,5-DKGR, as with most other members of the aldo-keto reductase (AKR) superfamily, exhibits a preference for NADPH compared to NADH as a cofactor in the stereo-specific reduction of substrate. The application of 2,5-DKGR in the industrial production of vitamin C would be greatly enhanced if NADH could be efficiently utilized as a cofactor. A mutant form of 2,5-DKGR has previously been identified that exhibits two orders of magnitude higher activity with NADH in comparison to the wild-type enzyme, while retaining a high level of activity with NADPH. We report here an X-ray crystal structure of the holo form of this mutant in complex with NADH cofactor, as well as thermodynamic stability data. By comparing the results to our previously reported X-ray structure of the holo form of wild-type 2,5-DKGR in complex with NADPH, the structural basis of the differential NAD(P)H selectivity of wild-type and mutant 2,5-DKGR enzymes has been identified.
Keywords: aldo keto reductase; vitamin C; enzyme engineering; 2,5-diketo-D-gluconic acid reductase; ascorbic acid
Between 10 and 40 million years ago, the common ancestor of the great apes suffered a catastrophic mutation in the gene for L-gulonolactone oxidase—the last enzyme in the biosynthetic pathway for L-ascorbate. Since that event, L-ascorbate has been an essential dietary nutrient (vitamin C) for all great apes, including man. Application as an antioxidant and preservative in the food and beverage industry, in addition to dietary supplements, has resulted in a current worldwide consumption of vitamin C in excess of 50,000 tons per year (Banta et al. 2002b), making vitamin C one of the most important specialty chemicals in the world.
Vitamin C is currently produced from D-glucose using a modified Reichstein–Grussner synthesis, devised by the Nobel Prize winning chemist Thaddeus Reichstein (Reichstein and Grussner 1934), and involving five chemical steps and a single fermentation step. The penultimate intermediate in this process, 2-keto-L-gulonic acid (2-KLG), is a relatively stable compound that can be stored for later conversion (via acid or base cyclization) to L-ascorbate. A long-standing goal has been the development of a new method for the production of vitamin C that replaces the chemical steps with direct fermentation of D-glucose to 2-KLG. In the early 1980s Sonoyama and coworkers identified a strain of Erwinia herbicola capable of fermenting D-glucose to 2,5-diketo-D-gluconate (2,5-DKG; an intermediate that is not part of the Reichstein–Grussner synthesis), and a strain of Corynebacterium able to subsequently ferment 2,5-DKG to 2-KLG (Sonoyama et al. 1982). The two fermentation steps were combined in tandem to produce 2-KLG (and thus L-ascorbate) from D-glucose with effective elimination of intermediate chemical steps. The fermentation of 2,5-DKG to 2-KLG by the Corynebacterium was identified as the product of a single catalytic step involving the NADPH-dependent enzyme 2,5-diketo-D-gluconate reductase (2,5-DKGR). Two distinct forms of 2,5-DKGR, types “A” and “B,” were characterized (Sonoyama and Kobayashi 1987) and identified as members of the aldo-keto reductase (AKR) family that share a characteristic TIM barrel tertiary structure and non–Rossman-fold nucleotide-binding domain. Subsequently, Anderson and coworkers introduced the gene for 2,5-DKGR “A” (2,5-DKGRA) into Erwinia herbicola to develop a genetically engineered strain capable of direct fermentation of 2-KLG from D-glucose (Anderson et al. 1985). A further simplification of the fermentation of 2-KLG from D-glucose has been developed by utilizing permeabilized Pantoea citrea cells (that can convert D-glucose to 2,5-DKG via membrane-bound dehydrogenases) in a bioreactor scheme utilizing exogenously added 2,5-DKGR enzyme, NADP+, and a soluble NADP+-dependent glucose dehydrogenase (Boston and Swanson 2003). In this method, the NADP+ is recycled by the action of the two exogenously added enzymes.
Being a commodity item of an industrial process, the cost associated with production of vitamin C is of paramount importance. Although the above-described methodologies can produce vitamin C, their ability to compete in cost with the existing method remains to be proven. The typical cell concentration of NADH is approximately threefold higher than NADPH (Lundquist and Olivera 1971). Thus, in fermentation-based production methods, NADH would be the more abundant cofactor. NADH is also commercially less expensive than NADPH (by approximately an order of magnitude) and is more stable than NADPH. Thus, NADH is also preferred for exogenously added cofactor in the bioreactor scheme of Boston and Swanson. However, the dissociation constant for NADH with 2,5-DKGRA is 2.6 × 10−3M (Banta et al. 2002b) whereas it is approximately 10 × 10−6M with NADPH (Miller et al. 1987; Sonoyama and Kobayashi 1987; Powers 1996). In light of the above considerations, Anderson and coworkers have argued that a significant enhancement could be achieved if the 2,5-DKGR enzyme could be engineered to function using NADH as a cofactor instead of NADPH (Banta et al. 2002b). Recent work involving a series of directed and combinatorial mutagenesis experiments has identified a mutant form of the enzyme (K232G, R238H; we will refer to mutations using the single letter code, thus K232G refers to a Lys 232 → Gly point mutation) with an almost threefold reduction in the dissociation constant for NADH in addition to a sevenfold increase in overall kcat using NADH as cofactor (Powers 1996; Banta and Anderson 2002; Banta et al. 2002b,c). This double mutant has been combined with additional substrate binding site mutations (F22Y, A272G) resulting in a quadruple mutant exhibiting the lowest dissociation constant (6.6 × 10−4M) for NADH identified to date (Banta et al. 2002c). Furthermore, mathematical modeling of 2-KLG production suggests that the use of the F22Y/K232G/R238H/A272G 2,5-DKGRA mutant with NAD(H), in combination with a small amount of NADP(H), could provide a significant cost benefit for in vitro enzymatic production of 2-KLG (Banta et al. 2002a).
The Corynebacterium from which 2,5-DKGRA and 2,5-DKGRB have been isolated is a soil-dwelling bacterium with a growth optimum of approximately 22°C. Irreversible thermal denaturation studies, monitoring unfolding by circular dichroism (CD), indicate a melting transition midpoint of approximately 38°C for 2,5-DKGRA and 32°C for 2,5-DKGRB (Powers 1996). Thus, although the stability of either form is appropriate for the normal growth temperature, it is problematic for the higher fermentation temperatures more typical of enteric bacteria (37°C). At such higher temperatures, an increase in the apparent kcat value could, therefore, also be achieved by thermal stabilization of the enzyme. Furthermore, the low thermal stability limits the ability to freely mutagenize the protein, as several potentially interesting activity site mutations have proven to be too destabilized to isolate (Powers 1996).
We report here the 2.0 Å X-ray structure determination, and isothermal equilibrium denaturation characterization, of a quadruple mutant (F22Y/K232G/R238H/A272G) of Cory- nebacterium 2,5-DKGRA in complex with NADH. In conjunction with our previously reported X-ray structure of wild-type 2,5-DKGRA in complex with NADPH, we are able to elucidate the details of the structural basis of the improved dissociation constant with NADH cofactor. The results show how this mutation is able to enhance binding to NADH, while retaining to a large extent the ability to bind NADPH. Furthermore, the thermodynamic data show that this quadruple mutant is also stabilized in relationship to the wild-type protein; thus, this mutant form is ideally suited for function at higher temperatures associated with industrial fermentation or bioreactor processes, or as a background for further mutagenic studies.
Results
Crystal data, processing, and refinement statistics for the F22Y/K232G/R238H/A272G 2,5-DKGRA mutant in complex with NADH are listed in Table 1. The space group was identified as C2 with one molecule per asymmetric unit. This is a different space group in comparison to the wild-type apo enzyme (no cofactor; space group, P212121; PDB accession number 1HW6) and wild-type holo enzyme (with NADPH; space group, P21; PDB accession number 1A80) X-ray structures (Khurana et al. 1998; Sanli and Blaber 2001). The crystals of the mutant 2,5-DKGRA were somewhat radiation sensitive, and the image plate and CCD data sets were therefore combined to achieve maximum completeness while retaining an acceptable value for Rmerge (Table 1). The Wilson temperature factor for the mutant data set is essentially identical to the P21 crystal form of the cofactor-bound form of wild-type 2,5-DKGRA. Final model building and refinement resulted in excellent stereochemistry and crystallographic residual (Table 1). Analysis of the Ramachandran plot distribution indicates that 90.3% of the residue positions are located in the most favored regions, with no residues in disallowed regions. Unambiguous density within the active site indicated that the NADH cofactor added to the protein solution during crystallization was bound within the active site. We note that although the cofactor was added to the crystallization solution as the reduced form, the crystallographic data cannot distinguish between oxidized and reduced forms. Thus, the bound cofactor may be present as the oxidized (NAD+) form.
Table 1.
Crystal, data collection, and refinement statistics for F22Y/K232G/R238H/A272G 2,5-DKGRA in complex with NADH
| Crystal data | |
| Space group | C2 |
| Cell constants | a = 111.95 Å, b = 55.24 Å, c = 51.54 Å, β = 111.7° |
| Molecules/asymmetric unit | 1 |
| Matthews coefficient (Vm) Å3/D | 2.2 |
| Resolution range (Å) | 27.0–2.0 |
| Data collection and processing | |
| Total/unique reflections | 242,073/18,364 |
| Completion (%; 27.0–2.0 Å) | 92.9 |
| Completion (%; 2.05–2.0 Å) | 83.3 |
| I/σ (overall) | 16.6 |
| I/σ (2.05–2.0 Å) | 4.2 |
| Rmerge (%) | 11.4 |
| Wilson temperature factor (Å2) | 15.0 |
| Refinement | |
| Rcryst (%; 27.0–2.0 Å) | 21.4 |
| Rfree (%; 27.0–2.0 Å) | 26.1 |
| R.m.s. bond length deviation (Å) | 0.008 |
| R.m.s. bond angle deviation (°) | 1.5 |
| R.m.s. B factor deviation (σ)a | 2.1 |
a Library of Tronrud (Tronrud 1996).
The overall structure of the mutant 2,5-DKGRA follows closely that of the wild-type 2,5-DKGRA/NADPH holo structure (Khurana et al. 1998), and the main chain atoms of both structures overlay with a root mean square (r.m.s.) deviation of 0.43 Å. However, if the first five residues are excluded from the overlay, the r.m.s. deviation improves to 0.38 Å. Thus, the mutations have been accommodated with minimal overall structural perturbation. Similarly, the NADH cofactor in the mutant structure overlays the NADPH cofactor from the holo wild-type structure (sans the 2′ phosphate moiety) with an r.m.s. deviation of 0.35 Å. Thus, the NAD cofactor is bound within the mutant structure with minimal structural perturbation.
A detailed description of the interactions between NADPH cofactor in the holo wild-type structure has previously been described (Khurana et al. 1998). Briefly, the interactions comprise a total of 16 hydrogen bonds, 2 ionic bonds, and 1 aromatic stacking interaction with the nicotinamide ring. Of these interactions, those directly interacting with the 2′ phosphate of the NADPH cofactor include Lys 232 Nζ, Val 234 N, Ser 233 Oγ, and Arg 238 Nη1. The Lys 232 Nζ interaction is eliminated with the K232G mutation, and the Arg238 Nη1 interaction is similarly eliminated with the R238H mutation. However, the structure of the mutant 2,5-DKGRA with bound NADH shows that the introduced His side chain at position 238 introduces a novel base-stacking interaction with the adenine ring of the NADH cofactor (Fig. 1 ▶). The Ser side chain at position 233 has not been subject to mutation; however, in response to the lack of a 2′ phosphate moiety in the bound NADH, this side chain has adopted an alternative rotamer that positions the Oγ away from the cofactor binding site (Fig. 1 ▶). Finally, the main chain amide of position 234 remains available for hydrogen bonding with a cofactor 2′ phosphate, and the main chain conformation at this position is essentially unchanged in the mutant 2,5-DKGRA (Fig. 1 ▶).
Figure 1.

Relaxed stereo diagram of the overlay of F22Y/K232G/R238H/A272G 2,5-DKGRA (dark gray) with wild type (1A80; light gray). The view is toward the adenine of the bound NADPH/NADH cofactor and provides structural details of the region involving the K232G and R238H mutations. The hydrogen bonding interactions of the 2′ adenosine phosphate group (of the NADPH bound in the wild-type structure) are indicated.
Mutation F22Y is located within the substrate-binding site, and is approximately 13 Å distal to the location of the NADPH 2′ phosphate position (Khurana et al. 1998). Similarly, the A272G mutation is approximately 21 Å distal. The introduced Tyr side chain at position 22 adopts an identical rotamer to the wild-type Phe residue (Fig. 2 ▶). The A272G mutation is accommodated with little, if any, structural perturbation (Fig. 2 ▶).
Figure 2.

Relaxed stereo diagram of the overlay of F22Y/K232G/R238H/A272G 2,5-DKGRA (dark gray) with wild type (1A80; light gray). The view is oriented toward the substrate-binding pocket (Khurana et al. 1998, 2000) and provides structural details of the region involving the F22Y and A272G mutations. The orientation of the NAD(P)H cofactor is identical to Figure 1 ▶.
The isothermal equilibrium denaturation data for both wild-type and mutant 2,5-DKGRA exhibited excellent agreement with the two-state denaturation model. The ΔG of unfolding for wild-type 2,5-DKGRA is 33.5 kJ/mole under conditions of 0 M denaturant (ΔG0; Table 2). The ΔG of unfolding for the mutant 2,5-DKGRA is 36.4 kJ/mole in 0 M denaturant. The isothermal equilibrium denaturation m values are slightly different for the two proteins, with the mutant exhibiting moderately reduced cooperativity. In such cases, a comparison of the extrapolated ΔG0 values is less accurate than a comparison at the midpoint of denaturation (i.e., ΔG0 = 0; Pace and Scholtz 1997). The mutation increases the midpoint of denaturation by 0.16 M GuHCl (Fig. 3 ▶), corresponding to an increase in stability (ΔΔG) of 5.2 kJ/mole (Table 2).
Table 2.
Thermodynamic parameters for wild-type and F22Y/K232G/R238H/A272G 2,5-DKGRA determined from isothermal (298K) equilibrium denaturation monitored using CD spectroscopy
| Protein | ΔG0 (kJ/mole) | m-value (kJ/mole M) | Cm (M) | ΔΔGa (kJ/mole) |
| Wild type | 33.5 ± 0.4 | 32.8 ± 0.3 | 1.02 ± 0.01 | — |
| F22Y/K232G/R238H/A272G | 36.4 ± 0.3 | 31.9 ± 0.5 | 1.18 ± 0.01 | −5.2 |
a ΔΔG = (CmWT − Cmmutant) * (mWT + mmutant)/2 (Pace and Scholtz 1997). A negative value for ΔΔG indicates a more stable mutant. All errors are listed as standard error of multiple data sets.
Figure 3.

Fraction of the unfolded state as a function of GuHCl denaturant, as determined from isothermal equilibrium denaturation data monitored by CD, for wild-type 2,5-DKGRA (filled circles) and the F22Y/K232G/R238H/A272G 2,5-DKGRA mutant (open circles). The solid line shows the fit to the two-state model in each case.
Discussion
Corynebacterium expresses two distinct variants of 2,5-DKGR, form “A” and form “B,” that are the products of distinctly different genes (Sonoyama and Kobayashi 1987). The two forms share 38% amino acid identity and different biophysical and enzymatic properties. With regard to biophysical properties, as previously mentioned, 2,5-DKGRA exhibits a higher melting temperature than 2,5-DKGRB. With regard to enzymatic properties, 2,5-DKGRA follows Michaelis–Menten kinetics, with a Km of 31 mM and kcat of 8.34 sec−1, with 2,5-DKG as substrate (Miller et al. 1987; Powers 1996). Conversely, 2,5-DKGRB exhibits a Km of 2.26 mM and kcat 18.8 sec−1 with 2,5-DKG as substrate, but exhibits pronounced substrate inhibition at concentrations higher than 10 mM (Powers 1996). Thus, 2,5-DKGRA is more stable than 2,5-DKGRB, and is not inhibited by high substrate concentrations, but is generally less active. In an attempt to incorporate the favorable activity properties of 2,5-DKGRB into the more stable 2,5-DKGRA, Powers (1996) constructed a series of chimeric and scanning mutations involving both forms. These studies identified a crucial role for a Tyr side chain at position 22 (F22Y mutation in 2,5-DKGRA) in both reducing Km (to ~10 mM) and increasing kcat (by ~50%). Additionally, a Gly mutation at position 272 (A272G in 2,5-DKGRA) resulted in a twofold increase in kcat, but also an increase in Km (to ~43mM). The 2,5-DKGRA double mutant (F22Y/A272G) exhibited additive effects upon the kinetic properties of the individual mutations, and surpassed the activity of 2,5-DKGRB at substrate concentrations greater than 17.5 mM (although it did exhibit some substrate inhibition; Powers 1996).
Molecular modeling studies of substrate binding (utilizing the holo wild-type 2,5-DKGRA X-ray structure; 1A80), suggested that the introduced Tyr residue at position 22 would be adopted with minimal perturbation of the structure (Khurana et al. 2000). Furthermore, it was proposed that if the introduced Tyr residue adopted the same rotamer as the wild-type Phe residue, the side chain hydroxyl would be positioned to hydrogen bond with the 2,5-DKG substrate C1 carboxyl group (Khurana et al. 2000). The structure of the F22Y/K232G/R238H/A272G 2,5-DKGRA mutant reported here shows that the introduced Tyr side chain at position 22 is adopted with minimal perturbation of the structure and assumes the same rotamer as the wild-type Phe residue (Fig. 2 ▶). Thus, the hypothesized basis for the improved kinetic constants with this mutation (primarily a reduction in Km) is supported by the present structural data.
No similar hypothesis for the basis of the improved catalytic effects of the A272G mutation in 2,5-DKGRA have been proposed. However, its location within the carboxyl-terminal region, and the role of this region in forming a lip of the substrate binding pocket, suggest that an Ala → Gly mutation could affect either access of substrate to the binding pocket or the shape of the pocket (Powers 1996). A structural determination of the apo 2,5-DKGRA indicated that cofactor binding is associated with substantial structural changes, including ordering of the carboxyl terminal region involving residues 264–278 (Sanli and Blaber 2001). The structural details of the F22Y/K232G/R238H/A272G 2,5-DKGRA mutant reported here show that the A272G mutation is not associated with significant structural changes of the local region (Fig. 2 ▶). Thus, the A272G mutation most likely improves the kinetic properties by either promoting access of substrate or possibly increasing the kinetics associated with binding pocket formation upon cofactor binding or, conversely, cofactor release. Further studies will be required to resolve this question.
The Lys residue at position 232 is known to be one of four residue positions in 2,5-DKGRA that interact directly with the 2′ phosphate group of the bound NADPH cofactor (Khurana et al. 1998). A study of various mutations at this position, involving chemically and structurally different amino acids, and screening for activity with NADH cofactor, indicated that a Gly substitution resulted in a slight reduction in cofactor Ka and a slight increase in overall kcat (albeit with a significant reduction in activity using NADPH cofactor; Banta et al. 2002b). The structural details of the F22Y/K232G/R238H/A272G 2,5-DKGRA mutant reported here show that the Gly mutation at position 232 results in little, if any, structural perturbation (Fig. 1 ▶). The primary effect of this mutation appears to be the elimination of a potential electrostatic interaction between the Lys Nζ and NADPH. With the exception of possible increased access to the cofactor-binding site, there appears to be little selective advantage for NADH binding afforded by this mutation.
The Arg side chain at position 238 is known to form a direct electrostatic interaction with the 2′ phosphate of bound NADPH (Khurana et al. 1998), and mutation by His potentially abolishes this interaction. A modeling study of a His mutation at this position has been reported and proposes a gauche+ rotamer (χ1 = −60°) with a χ2 value of ±90° (Banta and Anderson 2002). This set of conformations represents a commonly observed rotamer distribution (~42%) of His side chains, and, in the 2,5-DKGRA structure, results in an edge-to-face ring interaction between the introduced His side chain and the adenine ring of the bound cofactor. The His/adenine ring interaction has been suggested as a possible basis for improved NADH binding, and is supported by the observation that aromatic residues substituted at this position generally result in increased NADH-mediated catalysis by 2,5-DKGRA (although only the His mutation retains substantial activity with NADPH as cofactor; Banta and Anderson 2002). The structural details of the F22Y/K232G/R238H/A272G 2,5-DKGRA mutant reported here show that the introduced His side chain adopts a gauche+ rotamer, but with a χ2 value of +161° (Fig. 1 ▶). This rotamer is less frequently observed (~7%), and, in the 2,5-DKGRA structure, allows a face-to-face stacking interaction with the adenine ring of the NADH cofactor. This His rotamer subsequently positions the side chain Nδ1 atom at the former Arg Cδ location (Fig. 1 ▶). In the holo wild-type 2,5-DKGRA structure, the cofactor AOP1 oxygen (i.e., one of the 2′ phosphate oxygens) to Arg 238 Cδ distance is a close 3.1 Å (Khurana et al. 1998). Thus, the introduced His side chain, in adopting a face-to-face stacking interaction, also positions its side chain Nδ1 atom in ideal juxtaposition to form a novel charged hydrogen bond interaction with the 2′ phosphate of a bound NADPH cofactor.
The observed ring stacking interaction between the introduced His side chain at position 238 and the NADH adenine ring should also provide a similar stabilizing interaction with the NADPH cofactor. Thus, the combination of the K232G and R238H mutations, and their effects upon NADH or NADPH cofactor binding, can be summarized as follows: NADPH binding has lost two electrostatic interactions involving the 2′ phosphate (with Lys 232 Nζ and Arg 238 Nη1) but has gained an electrostatic interaction with His 238 Nδ1; furthermore, it has gained a stacking interaction between the adenine ring and His 238 side chain. NADH binding has gained a stacking interaction between the adenine ring and His 238 side chain. Thus, the kinetic studies of Banta (Banta et al. 2002c), indicating that this mutation has improved enzymatic activity with NADH and that it retains a unique ability to function to a significant extent with NADPH, are explained by the X-ray structural data presented here.
The atoms of the bound NADH cofactor in the F22Y/K232G/R238H/A272G 2,5-DKGRA mutant structure have an average thermal factor of 50 Å2, whereas the atoms of the NADPH cofactor bound in the wild-type structure have an average thermal factor of 20 Å2. As previously mentioned, the Wilson B factor value is essentially identical between the two data sets, and the average main chain B factors for the mutant are 18.3 Å2 in comparison to 16.3 Å2 for the wild-type apo structure. Although the average thermal factor value of the NADH cofactor is higher in the mutant structure, certain atoms of the cofactor exhibit thermal factors characteristic of the NADPH cofactor bound in the wild-type structure. In particular, the adenine base, the pyrophosphate AO1 oxygen, the NO2* oxygen of the nicotinamide ribose, as well as the NO7 oxygen and NN7 nitrogen of the nicotinamide, all exhibit thermal factors similar to the NADPH cofactor bound within the wild-type structure (Fig. 4 ▶). Conversely, the greatest increases in thermal factors are observed within the adenine ribose moiety (Fig. 4 ▶). With the exception of the 2′ phosphate, this ribose group hydrogen bonds exclusively with solvent (Khurana et al. 1998), and the increase in thermal factors is likely a direct consequence of the elimination of the 2′ phosphate and the associated interactions with enzyme. The increase in the ribose thermal factors propagates along the length of the cofactor, with the exception of the apparent “anchor points” listed above (Fig. 4 ▶). The introduction of the Arg 238 → His mutation has, to a large degree, resulted in the retention of wild-type thermal factors for the cofactor adenine (despite the loss of the adjacent phosphate moiety; Fig. 4 ▶). In the wild-type structure, the cofactor AO1 oxygen participates in a charged hydrogen bond interaction with the main chain amide of Lys 232 (Khurana et al. 1998), and the stereochemistry of this interaction is maintained despite the Gly mutation at this position. The NO1 and NO2 oxygens, located within the second phosphate of the central pyrophosphate, also hydrogen bond primarily with solvent and exhibit an increase in thermal factors (Fig. 4 ▶). The NO2* oxygen within the nicotinamide ribose is involved in a charged hydrogen bond interaction with the side chain of Asp 45. This interaction is maintained in the mutant structure. The nicotinamide NO7 oxygen and NN7 nitrogen form interactions with Asn 140, Ser 139, and Gln 161, and these are conserved in the mutant structure, as is a ring-stacking interaction with the side chain of Trp 187. Finally, the adenine ring exhibits thermal factors similar to the NADPH bound in the wild-type structure and hydrogen bonds through its AN6 and AN7 nitrogens to the side chains of Asn 242 and Glu 241 (both conserved in the mutant structure).
Figure 4.

Relaxed stereo diagram of the NADPH cofactor bound within wild-type 2,5-DKRA (Khurana et al. 1998; left) and the NADH cofactor bound within the F22Y/K232G/R238H/A272G 2,5-DKGRA mutant structure reported here. The colors of the atoms indicate the relative values of the thermal factors (blue is low, red is high). Both structures are rendered on the same color scale. The orientation of the cofactors is the same as in Figures 1 ▶ and 2 ▶.
The 2′ phosphate appears to provide key interactions that contribute to the ordered positioning of the majority of atoms comprising the cofactor. Penning and coworkers have demonstrated that in 3-α-hydroxysteroid dehydrogenase the “anchoring” of the 2′ phosphate of NADPH, by an Arg residue equivalent to Arg 238, is necessary for pre–steady-state kinetic transients to be observed upon NADPH binding (Ratnam et al. 1999). The results of the thermal factor analysis reported here suggest that anchoring of the phosphate is key for positional ordering throughout the length of the cofactor (mediated through the adenine ribose), and that it is not enough to simply maintain interactions with the adenine moiety. Thus, in the absence of the 2′ phosphate, secondary binding interactions may need to be introduced throughout the length of the cofactor for optimum utilization of NADH as cofactor.
Xylose reductase (XR) is a member of a related family of aldo-keto reductases that is able to utilize both NADH and NADPH as a cofactor in the reduction of the open-chain form of D-Xylose to the corresponding polyol xylitol (Lee 1998). The crystal structures of XR in complex with either NADPH or NADH have recently been reported (Kavanagh et al. 2002, 2003). The ability of XR to efficiently bind NADH cofactor is due to conformational changes of two local loops that permit novel hydrogen bonding interactions with the 2′ and 3′ hydroxyl groups of the cofactor adenosine ribose. These interactions involve side chains Glu 227 and Asn 276 (numbering scheme of XR), with Glu 227 identified as the most significant interaction (Kavanagh et al. 2003). Glu 227 in XR is located within a short α-helix that has no corresponding structural element in 2,5-DKGRA. The residue in 2,5-DKGRA that is structurally equivalent to Asn 276 in XR is Val 234, which has no potential to participate in hydrogen bonding interactions. Furthermore, the ribose group in the NADH cofactor bound to XR adopts a 3′-endo conformation, and not the 2′-endo conformation seen in the NADH bound to 2,5-DKGRA (or in NADPH cofactor bound to XR and 2,5-DKGRA). Thus, the interactions that facilitate NADH binding in the F22Y/K232G/R238H/A272G 2,5-DKGRA mutant represent a uniquely different solution to the efficient utilization of NADH cofactor. However, both the F22Y/K232G/R238H/A272G 2,5-DKGRA mutant and XR structures highlight the importance of interactions with the adenine ribose.
The relatively low thermal stability of both 2,5-DGRA and B has two important implications in the industrial application for vitamin C production. The fermentation or bioreactor temperatures must be kept below the melting temperature of the enzymes involved. In the case of 2,5-DKGRA (the more stable of the two Corynebacterium enzymes) the reported melting temperature is 38°C. Thus, at a temperature typical of enteric bacterial fermentation, approximately half the population of enzyme molecules will be denatured and inactive. The presence of irreversible pathways from the denatured state (e.g., aggregation or proteolysis) will result in a continual and rapid decline in effective enzyme activity. Secondly, low thermal stability translates to a limited tolerance for any mutagenic changes that will further destabilize the enzyme. With regard to desired mutations, 2,5-DKG has never been identified as a metabolite in Corynebacterium; furthermore, the active site appears to be substantially larger than the 2,5-DKG substrate (Khurana et al. 2000). Thus, although 2,5-DKGRA and B can stereospecifically reduce 2,5-DKG to 2-KLG, 2,5-DKG may not represent the natural substrate within the cell. Coupled with the relatively high Km for 2,5-DKG, these facts suggest that the active site might be mutated to improve binding or catalysis with 2,5-DKG (Powers 1996; Khurana et al. 2000). However, if such mutations destabilize the protein further, the effective enzymatic activity could be substantially reduced, or the protein may not fold at all. In this regard, the data in Table 2 present some rather good news. The thermodynamic results show that the F22Y/K232G/R238H/A272G 2,5-DKGRA mutant is also more stable and can, therefore, be expected to exhibit greater effective activity at elevated temperatures in comparison to wild type. The added stability also identifies this mutant as an appropriate background for future mutagenesis studies.
Materials and methods
Crystallization of F22Y/K232G/R238H/A272G 2,5-DKGRA in complex with NADH
The construction, expression, and purification of F22Y/K232G/R238H/A272G 2,5-DKGRA have previously been described (Powers 1996; Banta et al. 2002c). The purified protein was concentrated to 10 mg/mL in 25 mM Tris-HCl (pH 7.5) for crystallization, with the addition of NADH cofactor to a concentration of 1 mM. Crystallization trials were set up using the hanging drop vapor diffusion method (Jancarik and Kim 1991) with Hampton Crystal Screen I and II kit solutions (Hampton Research) at room temperature. Hanging drops contained a 1:1 mixture of protein and crystallization buffer (5 μL each) and vapor diffused against 1 mL reservoirs of crystallization buffer. Thin, plate-shaped crystals grew from 1.5 M lithium sulfate and 0.1 M Na HEPES (pH 7.5) within 7–10 d.
Data collection and processing
Two crystals (each approximately 0.2 × 0.3 × 0.05 mm) were mounted on a cryo loop (Hampton Research) and X-ray diffraction data were collected at low temperature (103K). No additional cryoprotectant was added to the crystallization buffer for protection against ice formation. Data collection was performed for the first crystal using a Rigaku RU-H2R rotating anode X-ray source (40 kV, 100 mA, graphite monochromatic CuKα radiation; Rigaku MSC) equipped with Osmic Blue confocal mirrors (MarUSA) and an R-Axis IIc imaging plate system. Data collection for the second crystal was accomplished using a similar X-ray source equipped with Osmic Purple confocal mirrors and a MarCCD 165 detector. Both crystals diffracted to approximately 1.9 Å resolution, and indexing suggested a monoclinic C2 unit cell with dimensions a = 111.95 Å, b = 55.24 Å, c = 51.54 Å, and unique angle β = 111.7°. The diffraction data from each crystal were combined to achieve maximum redundancy, completeness, and signal-to-noise ratio for a 2.0 Å data set. Data were processed using the DENZO software program (Otwinowski 1993; Otwinowski and Minor 1997).
Molecular replacement
Based on a molecular mass of approximately 29,000 D (Sonoyama and Kobayashi 1987) one molecule in the asymmetric unit yielded a Matthews coefficient (Vm) of 2.2 Å3/D, suggesting that a single molecule represented the contents of the asymmetric unit (Matthews 1968). Molecular replacement was attempted using the refined 2.1 Å crystal structure of 2,5-DKGRA in complex with NADPH (Khurana et al. 1998) as a search model (PDB accession number 1A80; space group P21). The NADPH cofactor and solvent were omitted from the search model. Rotation and translation function searches were attempted using the MRCHK software package (Zhang and Matthews 1994). A rotation function search, using data from 7.0Å to 2.3 Å, resulted in a top solution with a signal of 5.8σ (next nearest peak 1.8σ). The correctly rotated model was used for a subsequent translation function search in space group C2. This search resulted in a top peak of 5.4σ, with the next highest peak at 4.3σ. The applied rotation and translation solutions resulted in an initial value for Rcryst of 49.9%, using all data from 27.0 Å to 3.5 Å. Rigid body refinement of the model resulted in a slight improvement of Rcryst to 48.8%. At this point, 10% of the data were separated for calculation of Rfree (Brunger 1992).
Refinement
The structure was refined using the TNT least-squares refinement package (Tronrud et al. 1987; Tronrud 1992, 1996). An initial round of individual atomic position refinement, using data from 40.0 Å to 3.0 Å improved Rcryst to 37.2% (Rfree to 39.0%) while maintaining excellent stereochemistry. Inspection of the 2Fo-Fc electron density map for the entire structure indicated that several residue positions were in the incorrect rotamer and required rebuilding. These residues of the model were, therefore, deleted prior to subsequent rounds of refinement. Manual model building was performed using 2Fo-Fc, and Fo-Fc omit maps and the molecular graphics program O (Jones et al. 1991). Individual atomic coordinate refinement was performed in stages using data out to 2.5 Å resolution, at which point atomic thermal factors were simultaneously refined using a correlated thermal factor restraint library (Tronrud 1996). With the inclusion of all data to 2.0 Å, solvent molecules were added to the structure if they exhibited well-defined density in both the 2Fo-Fc and Fo-Fc electron density maps, appropriate hydrogen bonding stereochemistry with neighboring groups, and refined thermal factors of 60 Å2 or better. At this point in the refinement the density for the NADH cofactor was unambiguous and the NADH molecule was added to the structure. All coordinate and structure factor files have been deposited in the protein data bank (1M9H).
Isothermal equilibrium denaturation
Purified mutant protein was dialyzed against 10 mM sodium phosphate (pH 7.0) and concentrated to approximately 1 mM. Thermally induced denaturation of 2,5-DKGRA results in irreversible aggregation (Powers 1996), and accurate thermodynamic parameters cannot be determined. For this reason, isothermal equilibrium denaturation was utilized. Isothermal equilibrium denaturation was quantitated using CD spectroscopy and guanidine hydrochloride (GuHCl) as a denaturant. GuHCl was prepared as a 7.9 M stock solution (determined by refractive index) in 10 mM phosphate buffer (pH 7.0). Protein samples (at a final concentration of 10 μM) were allowed to equilibrate with GuHCl (0 M–2 M with 0.1-M increments) by overnight incubation at 4°C. CD measurements were carried out with an AVIV 202 circular dichroism spectrometer (AVIV), fitted with a thermoelectric cuvette holder, and interfaced with a model CTF-33 refrigerated recirculator bath (Thermo NESLAB) maintained at 298K. Isothermal CD spectra were acquired by scanning from 260 nm down to 200 nm in 1-nm increments with 1 nm bandwidth. Duplicate scans were recorded for each protein sample; triplicate experiments were collected and averaged. Buffer traces were subsequently subtracted from protein scans. The general purpose nonlinear least squares fitting program DataFit (Oakdale Engineering) was used to fit the CD signal versus GuHCl concentration to a six-parameter two-state model (Eftink 1994):
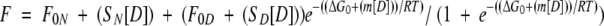 |
(1) |
where the denaturant concentration is given by [D], the native state (0 M denaturant) CD signal intercept and slope are F0N and SN, respectively, the denatured state CD signal intercept and slope are FOD and SD, respectively, and the free energy of unfolding function intercept and slope are ΔG0 and m, respectively. ΔG0 and m describe the linear function of the unfolding free energy versus denaturant concentration under isothermal equilibrium conditions. The effect of mutation upon the stability of the protein (ΔΔG) was calculated using the method of Pace and Sholtz (1997):
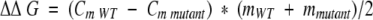 |
(2) |
where Cm WT and Cm mutant are the denaturant concentrations for the midpoint of the isothermal denaturation (ΔG = 0) for wild-type and mutant 2,5-DKGRA, respectively, and mWT and mmutant refer to the m values relating ΔG as a function of denaturant concentration for the wild-type and mutant protein, respectively. In this case, a negative value for ΔΔG indicates the mutant is stabilized in comparison to the wild type.
Acknowledgments
S.B. was supported by a N.I.H. Predoctoral Training Grant in Biotechnology (5T32GM08339). This work was supported by an American Heart Association Established Investigator Grant (0040235N) to M.B.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Abbreviations
and symbols: 2,5-DKG, 2,5-diketo-D-gluconic acid
2,5-DKGR, 2,5-diketo-D-gluconic acid reductase
2-KLG, 2-keto-L-gulonic acid
AKR, aldo keto reductase
NADH, nicotinamide adenine dinucleotide (reduced form)
NADPH, nicotinamide adenine dinucleotide phosphate (reduced form)
Tris-HCl, tris hydroxymethylaminoethane hydrochloride
HEPES, N-2-Hydroxyethylpiperazine-N′-2-ethanesulfonic acid
CD, circular dichroism
GuHCl, guanidinium hydrochloride
r.m.s., root mean square
XR, xylose reductase
Article published online ahead of print. Article and publication date are at http://www.proteinscience.org/cgi/doi/10.1110/ps.03450704.
References
- Anderson, S., Marks, C.B., Lazarus, R., Miller, J., Stafford, K., Seymour, J., Light, D., Rastetter, W., and Estell, D. 1985. Production of 2-keto-L-gulonate, an intermediate in L-ascorbate synthesis, by a genetically modified Erwinia herbicola. Science 230 144–149. [DOI] [PubMed] [Google Scholar]
- Banta, S. and Anderson, S. 2002. Verification of a novel NADH-binding motif: Combinatorial mutagenesis of three amino acids in the cofactor-binding pocket of Corynebacterium 2,5-diketo-D-gluconic acid reductase. J. Mol. Evol. 55 623–631. [DOI] [PubMed] [Google Scholar]
- Banta, S., Boston, M., Jarnagin, A., and Anderson, S. 2002a. Mathematical modeling of in vitro enzymatic production of 2-keto-L-gulonic acid using NAD(H) or NADP(H) as cofactors. Metab. Eng. 4 273–284. [DOI] [PubMed] [Google Scholar]
- Banta, S., Swanson, B.A., Wu, S., Jarnagin, A., and Anderson, S. 2002b. Alteration of the specificity of the cofactor-binding pocket of Corynebacterium 2,5-diketo-D-gluconic acid reductase A. Protein Eng. 15 131–140. [DOI] [PubMed] [Google Scholar]
- ———. 2002c. Optimizing an artificial metabolic pathway: Engineering the cofactor specificity of Corynebacterium 2,5-diketo-D-gluconic acid reductase for use in vitamin C biosynthesis. Biochemistry 41 6226–6236. [DOI] [PubMed] [Google Scholar]
- Boston, M.G. and Swanson, B.A. 2003. Method for producing ascorbic acid intermediates. U.S. Patent 6,599,722.
- Brunger, A.T. 1992. Free R value: A novel statistical quantity for assessing the accuracy of crystal structures. Nature 355 472–475. [DOI] [PubMed] [Google Scholar]
- Eftink, M.R. 1994. The use of fluorescence methods to monitor unfolding transitions in proteins. Biophys. J. 66 482–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jancarik, J. and Kim, S.-H. 1991. Sparse matrix sampling: A screening method for crystallization of proteins. J. Appl. Crystallogr. 24 409–411. [Google Scholar]
- Jones, T.A., Zou, J.Y., Cowan, S.W., and Kjeldgaard, M. 1991. Improved methods for the building of protein models in electron density maps and the location of errors in these models. Acta Crystallogr. A 47 110–119. [DOI] [PubMed] [Google Scholar]
- Kavanagh, K.L., Klimacek, M., Nidetzky, B., and Wilson, D.K. 2002. The structure of apo and holo forms of xylose reductase, a dimeric aldo-keto reductase from Candida tenuis. Biochemistry 41 8785–8795. [DOI] [PubMed] [Google Scholar]
- ———. 2003. Structure of xylose reductase bound to NAD+ and the basis for single and dual co-substrate specificity in family 2 aldo-keto reductases. Biochem. J. 373 319–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khurana, S., Powers, D.B., Anderson, S., and Blaber, M. 1998. Crystal structure of 2,5-diketo-D-gluconic acid reductase A complexed with NADPH at 2.1Å resolution. Proc. Natl. Acad. Sci. USA 95 6768–6773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khurana, S., Sanli, G., Powers, D.B., Anderson, S., and Blaber, M. 2000. Molecular modeling of substrate binding in wild-type and mutant Corynebacteria 2,5-diketo-D-gluconate reductases. Proteins Struct. Funct. Genet. 39 68–75. [DOI] [PubMed] [Google Scholar]
- Lee, H. 1998. The structure and function of yeast xylose (aldose) reductases. Yeast 14 977–984. [DOI] [PubMed] [Google Scholar]
- Lundquist, R. and Olivera, B.M. 1971. Pyridine nucleotide metabolism in Escherichia coli. J. Biol. Chem. 246 1107–1116. [PubMed] [Google Scholar]
- Matthews, B.W. 1968. Solvent content of protein crystals. J. Mol. Biol. 33 491–497. [DOI] [PubMed] [Google Scholar]
- Miller, J.V., Estell, D.A., and Lazarus, R.A. 1987. Purification and characterization of 2,5-Diketo-D-gluconate reductase from Corynebacterium Sp. J. Biol. Chem. 262 9016–9020. [PubMed] [Google Scholar]
- Otwinowski, Z. 1993. Oscillation data reduction program. In Proceedings of the CCP4 Study Weekend: Data collection and processing (eds. L. Sawyer et al.), pp. 56–62. SERC Daresbury Laboratory, Cheshire, England.
- Otwinowski, Z. and Minor, W. 1997. Processing of x-ray diffraction data collected in oscillation mode. Methods in Enzymol. 276 307–326. [DOI] [PubMed] [Google Scholar]
- Pace, C.N. and Scholtz, J.M. 1997. Measuring the conformational stability of a protein. In Protein structure: A practical approach (ed. T.E. Creighton), pp. 299–321. Oxford University Press, Oxford, UK.
- Powers, D.P. 1996. “Structure/function studies of 2,5-diketo-D-gluconic acid reductases.” Ph.D. thesis, University of Medicine and Dentistry of New Jersey.
- Ratnam, K., Ma, H., and Penning, T.M. 1999. The arginine 276 anchor for NADP(H) dictates fluorescence kinetic transients in 3 α-hydroxysteroid dehydrogenase, a representative aldo-keto reductase. Biochemistry 38 7856–7864. [DOI] [PubMed] [Google Scholar]
- Reichstein, T. and Grussner, A. 1934. Eine ergiebige synthese der L-ascorbinsaure (C-Vitamin). Helvetica Chimica Acta 17 311–328. [Google Scholar]
- Sanli, G. and Blaber, M. 2001. Structural assembly of the active site in an aldo-keto reductase by NADPH cofactor. J. Mol. Biol. 309 1209–1218. [DOI] [PubMed] [Google Scholar]
- Sonoyama, T. and Kobayashi, K. 1987. Purification and properties of two 2,5-diketo-D-gluconate reductases from a mutant strain derived from Corynebacterium sp. J. Ferment. Technol. 65 311–317. [Google Scholar]
- Sonoyama, T., Tani, H., Matsuda, K., Kageyama, B., Tanimoto, M., Kobayashi, K., Yagi, S., Kyotani, H., and Mitsushima, K. 1982. Production of 2-keto-L-gulonic acid from D-glucose by two-stage fermentation. Appl. Environ. Microbiol. 43 1064–1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tronrud, D.E. 1992. Conjugate-direction minimization: An improved method for the refinement of macromolecules. Acta Crystallogr. A 48 912–916. [DOI] [PubMed] [Google Scholar]
- ———1996. Knowledge-based B-factor restraints for the refinement of proteins. J. Appl. Crystallogr. 29 100–104. [Google Scholar]
- Tronrud, D.E., Ten Eyck, L.F., and Matthews, B.W. 1987. An efficient general-purpose least-squares refinement program for macromolecular structures. Acta Crystallogr. A 43 489–501. [Google Scholar]
- Zhang, X.-J. and Matthews, B.W. 1994. Enhancement of the method of molecular replacement by incorporation of known structural information. Acta Crystallogr. D 50 675–686. [DOI] [PubMed] [Google Scholar]