

Abstract
The Ca2+ binding 70–80 loop of factor X (fX) contains one basic (Arg71) and three acidic (Glu74, Glu76, and Glu77) residues whose contributions to the zymogenic and enzymatic properties of the protein have not been evaluated. We prepared four Ala substitution mutants of fX (R71A, E74A, E76A, and E77A) and characterized their activation kinetics by the factor VIIa and factor IXa in both the absence and presence of cofactors. Factor VIIa exhibited normal activity toward E74A and E76A and less than a twofold impaired activity toward R71A and E77A in both the absence and presence of tissue factor. Similarly, factor IXa in the absence of factor VIIIa exhibited normal activity toward both E74A and E76A; however, its activity toward R71A and E77A was impaired approximately two- to threefold. In the presence of factor VIIIa, factor IX activated all mutants with approximately two- to fivefold impaired catalytic efficiency. In contrast to changes in their zymogenic properties, all mutant enzymes exhibited normal affinities for factor Va, and catalyzed the conversion of prothrombin to thrombin with normal catalytic efficiencies. However, further studies revealed that the affinity of mutant enzymes for interaction with metal ions Na+ and Ca2+ was impaired. These results suggest that although charged residues of the 70–80 loop play an insignificant role in fX recognition by the factor VIIa-tissue factor complex, they are critical for the substrate recognition by factor IXa in the intrinsic Xase complex. The results further suggest that mutant residues do not play a specific role in the catalytic function of fXa in the prothrombinase complex.
Keywords: Factor X, factor Xa, factor VIIa, factor IXa, extrinsic Xase, intrinsic Xase
Factor X (fX) is a vitamin K-dependent serine protease zymogen in plasma that upon activation to factor Xa (fXa) assembles into the prothrombinase complex (fXa, factor Va, negatively charged membrane surface, and Ca2+ ion) to convert prothrombin to thrombin in the final stage of the blood coagulation cascade (Furie and Furie 1988; Mann et al. 1990; Davie et al. 1991). FX can be activated by either one of the two physiological activators—the factor VIIa (fVIIa)– tissue factor (TF) complex (extrinsic Xase), or the factor IXa (IXa)–factor VIIIa (fVIIIa) complex (intrinsic Xase)—on the negatively charged membrane surfaces in the presence of Ca2+ (Jackson and Nemerson 1980; Edgington et al. 1991). Both activators specifically cleave a single peptide bond, Arg15–Ile16 (chymotrypsinogen numbering system has been used throughout the manuscript) on the activation peptide of fX to release a 52-residue peptide thereby generating an active enzyme. An efficient cleavage of this peptide bond by either one of the physiological Xase complexes occurs only in the presence of cofactors because neither fVIIa nor fIXa alone can activate fX at a significant rate. The exact mechanism by which cofactors improve the catalytic efficiency of these and other coagulation proteases is not well understood.
Structural data suggest that the catalytic domain of fXa, similar to those of other coagulation proteases, has several surface loops including 39, 60, 70, 90, and 148 loops that surround the substrate-binding pocket of the protease (Furie et al. 1982; Bode et al. 1992; Padmanabhan et al. 1993). Although these surface loops are conserved at similar three-dimensional locations on all coagulation proteases, the amino acid residues forming the loops are not conserved in all members of the family. It has become clear in recent years that these variant surface loops provide exosite-dependent recognition sites for substrates and/or cofactors thereby facilitating the specific assembly of the coagulation activation complexes on membrane surfaces (Chattopadhyay and Fair 1989; Anderson et al. 2000; Baugh et al. 2000). A well-studied example is the pivotal role that the basic residues of the 70–80 loop play in determining the substrate, cofactor, and inhibitor specificity of both prothrombin/thrombin and protein C/activated protein C in the procoagulant and the anticoagulant pathways, respectively (Bode et al. 1992; Mather et al. 1996; Pineda et al. 2002; Chen et al. 2003). Unlike prothrombin and protein C, this loop in fX has a reversed polarity, because it contains only one basic (Arg71), but five acidic residues (Asp70, Glu74, Glu76, Glu77, and Glu80). Similar to trypsin, and with the exception of prothrombin, an acidic residue at both positions 70 and 80 has been conserved in all vitamin K-dependent coagulation proteases (Bode and Schwager 1975). Structural, mutagenesis, and antibody binding data have indicated that acidic residues of the 70 and 80 sites are involved in Ca2+ coordination in all of these proteases (Bode and Schwager 1975; Padmanabhan et al. 1993; Persson et al. 1993; Rezaie and Esmon 1994). In the case of fXa, we previously showed that the substitution of Asp70 with a Lys results in a functionally active mutant that loses its ability to interact with Ca2+, but retains its ability to activate prothrombin (Rezaie and Esmon 1994). The role of other charged residues of the 70–80 loop to the zymogenic and enzymatic properties of fX/fXa has not been examined.
To address this question, we substituted Arg71, and the three acidic residues Glu74, Glu76, and Glu77 of fX, with Ala in separate constructs and expressed the mutant zymogens in mammalian cells. Following purification to homogeneity, these mutants were characterized with respect to their ability to function as zymogens for the two physiological activators fVIIa and fIXa, and then as enzymes in the prothrombinase complex to activate prothrombin to thrombin. It was found that the activation of E74A and E76A by fVIIa was unaffected, but the activation of R71A and E77A was slightly impaired (<twofold) in both the absence and presence of TF. Similarly, with fIXa as the activating enzyme, the activation of E74A and E76A was normal in the absence of fVIIIa, but impaired approximately twofold in the presence of the cofactor. Unlike the latter two residues, the fIXa activation of R71A and E77A was impaired approximately two- to fivefold in both the absence and presence of fVIIIa. These results suggest that none of the residues under study interact with TF in the extrinsic Xase complex. Moreover, although all four residues of fX are required for its proper recognition by the intrinsic Xase complex, only Glu74 and Glu76 have an approximately twofold effect on the cofactor-dependent recognition of the substrate by the activation complex. Nevertheless, cofactor-independent interaction of fX residues Arg71 and Glu74 with fIXa is required for effective activation of the substrate. Results of plasma-based clotting assays are in agreement with results obtained in the purified system. In contrast to observed impairments in the zymogenic properties, all mutant enzymes exhibited normal activity toward prothrombin in both the absence and presence factor Va (fVa), suggesting that these residues do not play an apparent role in the catalytic function of fXa in the prothrombinase complex. However, further studies revealed that both the Ca2+ and Na+ binding properties of the mutant enzymes had been impaired. These results confirm our previous observation that the conformation of the Ca2+-binding 70–80 loop of fXa is allosterically linked to the Na+-binding loop of the protease (Rezaie and He 2000).
Results
Expression and purification of recombinant proteins
The wild-type and mutant fX zymogens were expressed in a novel expression/purification vector system as described (Manithody et al. 2002). This vector system replaces the first 12 N-terminal residues of the activation peptide of fX with the epitope for the Ca2+-dependent monoclonal antibody, HPC4, for the purification purposes. Wild-type and mutant proteins were separated from the cell culture supernatants by a combination of immunoaffinity and ion exchange chromatography using the monoclonal antibody HPC4 and a Mono Q column as described (Manithody et al. 2002). Both plasma and recombinant fX yielded a Gla content of ~10 mole Gla/mole of protein (10.2 and 10.7 for the plasma-derived fX and recombinant fX, respectively). The theoretical value for the Gla content of fX is 11 mole Gla/mole of protein. Consistent with a full γ-carboxylation, all recombinant fXa derivatives also exhibited indistinguishable prothrombinase activity on PC/PS vesicles (see below). SDS-PAGE analysis of fX derivatives suggested that the recombinant proteins have been purified to homogeneity, and that they all migrate with similar molecular masses as the plasma-derived fX (Fig. 1 ▶). All derivatives could be converted to their active forms by RVV-X as determined by an amidolytic activity assay using Spectrozyme FXa (SpFXa). Following complete activation, the concentration of fXa derivatives were determined by an amidolytic activity assay and active-site titration with known concentrations of human antithrombin.
Figure 1.

SDS-PAGE analysis of the recombinant fX derivatives expressed in HEK293 cells. Under nonreducing conditions: (Lane 1) Plasma-derived fX, (lane 2) wild-type recombinant fX, (lane 3) R71A, (lane 4) E74A, (lane 5) E76A, (lane 6) E77A, and (lane 7) molecular mass standards in kDa.
Activation by the extrinsic Xase complex
The zymogenic properties of fX mutants were evaluated in activation reactions by fVIIa in both the absence and presence of TF and Ca2+ on PC/PS vesicles (extrinsic Xase complex). FVIIa by itself exhibited normal activity toward both E74A and E76A, but its activity toward R71A and E77A was slightly affected (Fig. 2A ▶). Similarly, the concentration dependence of zymogen activation suggested that the catalytic efficiency of the extrinsic Xase complex toward the mutant is also minimally affected (Fig. 2B ▶; Table 1). A notable observation was that the kcat of the activation for both R71A and E77A were slightly impaired (Fig. 2B ▶). However, the activation of both mutants by fVIIa alone was also affected to a similar extent (Fig. 2A ▶). Thus, none of the residues of the 70–80 loop makes a significant contribution to fX recognition by fVIIa in the extrinsic Xase complex. The normal activation rates of mutants by both fVIIa and the extrinsic Xase complex suggest that the mutagenesis did not adversely affect the proper folding of the mutant zymogens.
Figure 2.
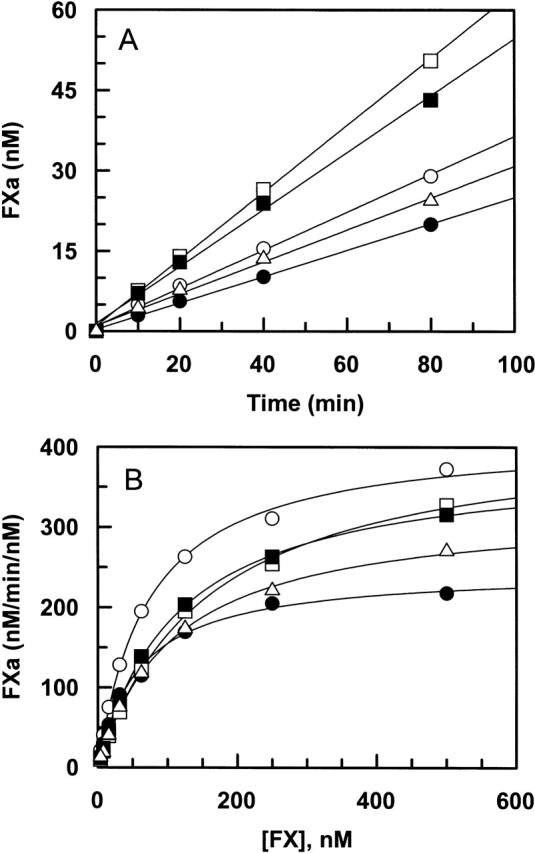
Activation of fX derivatives by fVIIa in the absence and presence of TF. (A) In the absence of TF, fVIIa (50 nM) was incubated with recombinant fX derivatives: wild type (open circles), R71A (filled circles), E74A (open squares), E76A (filled squares), and E77A (open triangles) (0.5 μM each) on PC/PS vesicles (50 μM) at room temperature in TBS/Ca2+. At different time intervals, small aliquots of the activation reactions were transferred to wells of a 96-well assay plate containing 20 mM EDTA, and the rate of fXa generation was measured from the cleavage rate of SpFXa, as described in Materials and Methods. (B) FVIIa (0.025 nM) in complex with the relipidated dcTF (2.5 nM) was incubated with different concentrations of fX derivatives: wild type (open circles), R71A (filled circles), E74A (open squares), E76A (filled squares), and E77A (open triangles) in TBS/Ca2+. After 1–2 min of incubation at room temperature, the reactions were terminated by addition of EDTA, and the initial rate of fXa generation was measured as described above. Solid lines in B are nonlinear regression fits of kinetic data to the Michaelis-Menten equation, and those in A are fits to a linear equation.
Table 1.
Kinetic constants for the activation of fX derivatives by extrinsic and intrinsic Xase complexes
| Extrinsic Xase complex | Intrinsic Xase complex | |||||
| Km (nM) | kcat (nM/min/nM) | kcat/Km | Km (nM) | kcat (nM/min/nM) | kcat/Km | |
| WT | 75.8 ± 2.8 | 457.4 ± 5.5 | 6.0 | 51.6 ± 4.6 | 234.9 ± 6.4 | 4.6 |
| R71A | 59.6 ± 5.3 | 246.9 ± 7.0 | 4.1 | 55.0 ± 3.6 | 54.7 ± 1.1 | 1.0 |
| E74A | 107.2 ± 5.7 | 451.6 ± 8.5 | 4.2 | 80.6 ± 4.1 | 109.1 ± 1.9 | 1.4 |
| E76A | 91.4 ± 2.4 | 428.0 ± 4.2 | 4.7 | 49.2 ± 2.7 | 148.4 ± 2.4 | 3.0 |
| E77A | 100.1 ± 4.9 | 358.5 ± 6.5 | 3.6 | 54.8 ± 4.4 | 147.9 ± 3.7 | 2.7 |
The kinetic constants were determined from the concentration dependence of the activation of fX derivatives by either the extrinsic or the intrinsic Xase complex in TBS/Ca2+ at room temperature as described under Materials and Methods. All values are the average of at least two to three measurements ± standard errors.
Activation by the intrinsic Xase complex
The zymogenic properties of fX mutants were also evaluated in activation studies by fIXa in both the absence and presence of fVIIIa and Ca2+ on PC/PS vesicles (intrinsic Xase complex). Similar to fVIIa, fIXa by itself exhibited normal activity toward both E74A and E76A mutants of fX (Fig. 3A ▶). However, relative to wild type, the activation of R71A and E77A by fIXa was impaired two- to threefold (Fig. 3A ▶). On the other hand, the activity of the intrinsic Xase complex toward all mutants was impaired at varying degrees (Fig. 3B ▶). The most impairment in the catalytic efficiency (approximately fivefold) was observed for the activation of the R71A mutant. The activation of all other mutants was impaired approximately twofold. The concentration dependence of zymogen activation by the intrinsic Xase complex revealed that the primary defect with all mutants is in the kcat of the activation reaction (Fig. 3B ▶; Table 1). These results suggest that all fX residues under study are important for the substrate activation by fIXa in the intrinsic Xase complex. However, a similar extent of impairment in the activation of Arg71 and Glu77 mutants by fIXa in both the absence and presence of fVIIIa suggests that neither residue interacts with the cofactor.
Figure 3.

Activation of fX derivatives by fIXa in the absence and presence of fVIIIa. (A) In the absence of fVIIIa, fIXa (50 nM) was incubated with recombinant fX derivatives: wild type (open circles), R71A (filled circles), E74A (open squares), E76A (filled squares), and E77A (open triangles) (0.5 μM each) on PC/PS vesicles (50 μM) at room temperature in TBS/Ca2+. At different time intervals, small aliquots of the activation reactions were stopped by 20 mM EDTA, and the rate of fXa generation was measured from the cleavage rate of SpFXa as described in Materials and Methods. (B) FIXa (0.025 nM) in complex with fVIIIa (25 nM) was incubated with different concentrations of fX derivatives: wild type (open circles), R71A (filled circles), E74A (open squares), E76A (filled squares), and E77A (open triangles) on PC/PS vesicles (50 μM) in TBS/Ca2+. After 1–2 min of incubation at room temperature, the reactions were terminated by addition of EDTA, and the initial rate of fXa generation was measured as described above. Solid lines in B are nonlinear regression fits of kinetic data to the Michaelis-Menten equation, and those in A are fits to a linear equation.
It is worth mentioning that during activation by the extrinsic Xase complex, identical activation parameters (both Km and kcat) were obtained for both the plasma-derived fX and recombinant fX in which the first 12 residues of the activation peptide were exchanged with the epitope for the HPC4 monoclonal antibody. However, with the intrinsic Xase complex as the activator, the activation of recombinant fX was impaired approximately fourfold (data not shown). To determine whether the mutations at the N terminus of the activation peptide of recombinant fX or other posttranslational modifications (i.e., differential glycosylation) are responsible for the differences in the activation rates between the two Xase complexes, another wild-type recombinant fX was prepared in which the activation peptide of the zymogen was not modified; thus, it was chemically identical to plasma-derived fX. This recombinant wild-type fX was purified to homogeneity on a fX monoclonal antibody as described (Le Bonniec et al. 1992). As expected, both forms of wild-type recombinant fX were activated normally by the extrinsic Xase complex; however, the activation rate of the unmodified recombinant fX by the intrinsic Xase complex was still impaired approximately twofold (data not shown). Thus, it appears that recombinant fX expressed in HEK293 cells is not efficiently recognized by the intrinsic Xase complex. Monitoring the initial rate of activation in both the absence and presence of fVIIIa revealed that the slower activation of recombinant fX by fIXa is independent of the cofactor (data not shown). Previous results have indicated that the activation peptide of fX plays a critical role in efficient substrate activation by the intrinsic (Duffy and Lollar 1992), but not by the extrinsic Xase complex (Baugh and Krishnaswamy 1996). Moreover, it is known that the carbohydrate moieties of fX are required for its optimal activation by both physiological activators (Sinha and Wolf 1993). Because the activation peptide of human fX has two N-linked and two O-linked glycosylation sites (Inoue and Morita 1993; Sinha and Wolf 1993), one possibility for the slower activation rate of recombinant fX by the intrinsic Xase complex may be that the carbohydrates on the activation peptide play a more critical role in the interaction of the substrate with fIXa and that the differential modification of either one of these glycosylation sites negatively affects this interaction. Further mutagenesis work is in progress to determine the validity of this hypothesis.
Clotting assays
Both PT and APTT assays were used to evaluate the clotting activities of recombinant zymogens. As expected, both plasma-derived fX and recombinant fX exhibited indistinguishable clotting activities in the PT assay. In this assay, the clotting activities of E74A, E76A, and E77A mutants were slightly impaired (Fig. 4A ▶). The extent of impairments in the PT assay correlated well with results obtained in the zymogen activation by the extrinsic Xase complex in the purified system (Fig. 2A ▶). The most impairment (approximately twofold) was observed for the clotting activity of the R71A mutant (Fig. 4A ▶, filled circles). By doubling the concentration of R71A in the PT assay, it was not possible to restore the defect in the clotting activity of this mutant, suggesting that the defect in the clotting activity is due to a defect in the kcat value. Inspection of the kinetic data shown in Table 1 is consistent with this hypothesis. Thus, Arg71 contributes approximately twofold to kcat of fX activation by fVIIa. This defect is not related to TF because the activation of the R71A mutant by fVIIa alone was impaired to nearly the same extent (Fig. 2A ▶). On the other hand, in the APTT assay, the clotting activities of all four mutants were significantly impaired (Fig. 4B ▶). Similar to results in the purified system (Fig. 3B ▶), the greatest impairment in the APTT assay was observed for R71A, followed by the R74A mutant. Similar to the PT assay, the extent of the impairments in the APTT assays correlated relatively well with the zymogen activation by the intrinsic Xase complex in the purified system (Fig. 3 ▶). Similarly, by increasing the concentration of the mutants it was not possible to restore the defects in the clotting activities, suggesting that these residues are important for the kcat of fX activation by the intrinsic Xase complex. This is consistent with results presented in Table 1.
Figure 4.
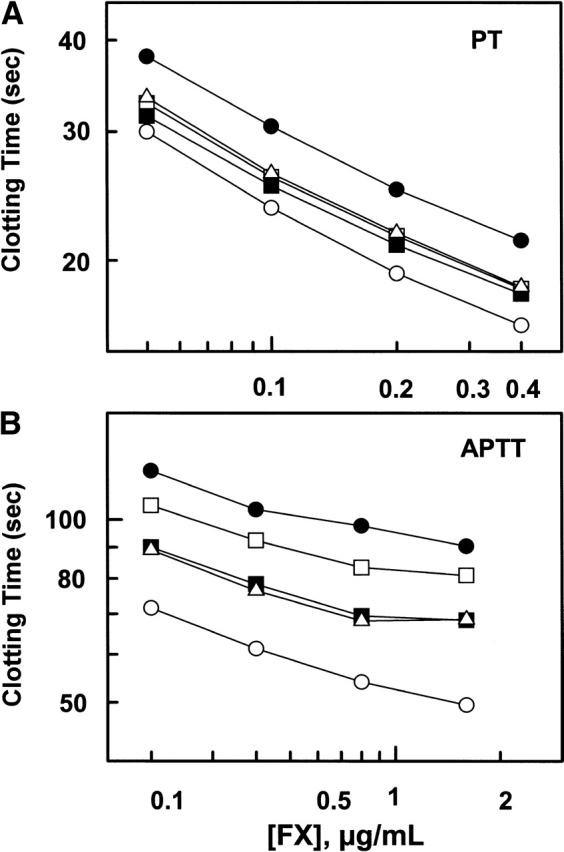
Clotting activities of fX derivatives in PT and APTT assays. (A) The clotting activities of fX derivatives wild type (open circles), R71A (filled circles), E74A (open squares), E76A (filled squares), and E77A (open triangles) at four different dilutions (0.05–0.4 μg/mL) were determined by a PT assay as described in Materials and Methods. (B) The clotting activities of the same fX derivatives (the same symbols) at four different dilutions (0.2–1.6 μg/mL) were determined by an APTT assay as described in Materials and Methods.
Catalytic properties of the FXa derivatives
To determine whether the residues of the 70–80 loop play a role in the catalytic function of fXa, the kinetic properties of mutants were studied with respect to their ability to: (1) hydrolyze SpFXa and bind to the S1 site-specific competitive inhibitor of serine proteases, p-aminobenzamidine (PAB), (2) bind monovalent and divalent cations, (3) activate prothrombin, and finally, (4) react with antithrombin.
Amidolytic activity and binding to PAB
Kinetic parameters for the hydrolysis of the chromogenic substrate SpFXa by fXa derivatives are presented in Table 2. Relative to wild-type fXa, a similar kcat for the substrate hydrolysis was observed for all mutants; however, the Km value for the R71A mutant was elevated approximately twofold. These results suggest that with the exception of R71A, the mutagenesis did not adversely affect the conformation of the S3–S1 binding pocket of fXa mutants. Similar to hydrolysis of the chromogenic substrate, the Ki values for the interaction of PAB were nearly normal for all mutants, with the exception of an approximately twofold impairment for the R71A mutant. These results suggest that the S1 binding pocket of the R71A mutant enzyme has been slightly affected (Table 2).
Table 2.
Kinetic constants for the cleavage of the chromogenic substrate Spectrozyme FXa by fXa derivatives
| Km (μM) | kcat (sec−1) | kcat/Km (μM−1 s−1) | Ki (PAB, μM) | Kd(app) (Na+, mM) | |
| WT | 83.0 ± 6.0 | 173.5 ± 3.5 | 2.1 ± 0.2 | 53.9 ± 2.3 | 13.2 ± 5.5 |
| R71A | 154.7 ± 14.3 | 188.7 ± 4.9 | 1.2 ± 0.1 | 110.6 ± 15.8 | 61.4 ± 20.1 |
| E74A | 102.3 ± 7.7 | 147.5 ± 3.3 | 1.4 ± 0.1 | 79.2 ± 4.1 | 24.7 ± 8.8 |
| E76A | 82.7 ± 4.3 | 149.0 ± 2.0 | 1.8 ± 0.1 | 63.2 ± 2.5 | 21.1 ± 8.3 |
| E77A | 110.0 ± 8.0 | 188.3 ± 3.6 | 1.7 ± 0.2 | 70.2 ± 2.5 | 26.2 ± 10.0 |
The kinetic constants were calculated from the cleavage rate of increasing concentrations of SpFXa (7.5–1000 μM) by each fXa derivative (0.5 nM) in TBS/Ca2+. The Ki values for p-aminobenzamidine (PAB) were measured from the decrease in initial rates of SpFXa hydrolysis produced by the competitive inhibitor, and the Kd(app) values for the interaction with Na+ was determined from the concentration dependence enhancement in the amidolytic activity of fXa derivatives in TBS/Ca2+ as described under Materials and Methods. Kinetic values are the average of three measurements ± standard errors.
Interaction with Na+ and Ca2+
We previously demonstrated that the conformation of the Ca2+ binding 70–80 loop of fXa is allosterically linked to the Na+ binding loop of the protease (Rezaie and He 2000). In addition, results of other recent studies have indicated that the S1 binding site of fXa is also thermodynamically linked to the Na+ binding site of the protease (Underwood et al. 2000; Camire 2002). To determine whether the mutagenesis of these residues influenced the affinity of mutants for interaction with the monovalent and divalent cations, the amidolytic activities of mutants were monitored in the presence of increasing concentrations of Na+ and Ca2+ as described (Rezaie and He 2000). In the presence of 5 mM Ca2+, the Na+ concentration dependence of the amidolytic activity of mutant enzymes revealed that the affinity of mutants for Na+ has been altered (Table 2). Interestingly, the greatest effect (approximtely fivefold impairment) was observed with the Na+ binding to the R71A mutant, and a less than twofold effect was observed for the other three mutants (Table 2). The affinity of mutants for interaction with Ca2+ was also evaluated. The amidolytic activity of wild-type fXa toward S2222 in TBS containing 0.1 M NaCl was enhanced ~30% to 40% with Kd(app) of ~178 μM. A similar extent of Ca2+-dependent enhancement in the amidolytic activities E74A and E76A mutants was observed with Kd(app) values of ~356 μM and ~690 μM, respectively. However, the enhancement (~70%) in the amidolytic activity of the R71A mutant exhibited a biphasic dependence on the concentration of Ca2+ yielding Kd(app) values of ~0.6 mM and ~2 mM (data not shown). The underlying basis for changes in the Ca2+ dependence of the R71A amidolytic activity may be due to a significant impairment in its affinity for Na+. Interestingly, no Kd(app) value for the E77A mutant could be calculated because Ca2+ did not influence the amidolytic activity of this mutant. These results suggest that the mutagenesis of charged residues of the 70–80 loop affects the Na+ and Ca2+ ion binding properties of fXa, confirming our initial observation that the two metal ion binding sites are allosterically linked (Rezaie and He 2000).
Prothrombin activation
The catalytic efficiency of fXa derivatives toward prothrombin was studied in both the absence and presence of fVa and Ca2+ on PC/PS vesicles (prothrombinase complex). Time course of the activation studies in the absence of the cofactor indicated that all fXa derivatives have similar activity toward prothrombin (Fig. 5A ▶). Similar to results obtained with the hydrolysis of SpFXa and interaction with PAB, the ability of R71A to activate prothrombin was slightly impaired relative to the wild-type enzyme (Fig. 5A ▶). The fVa concentration dependence of prothrombin activation on PC/PS vesicles suggested that all mutants interact with the cofactor with similar Kd(app) values of ~1.5–3 μM (Table 3). Detailed kinetic analysis further suggested that all fXa derivatives have a similar prothrombinase activity (kcat/Km) toward the activation of prothrombin, which suggests that the residues of the 70–80 loop under study do not interact with either the cofactor or the substrate in the activation complex (Fig. 5B ▶; Table 3).
Figure 5.
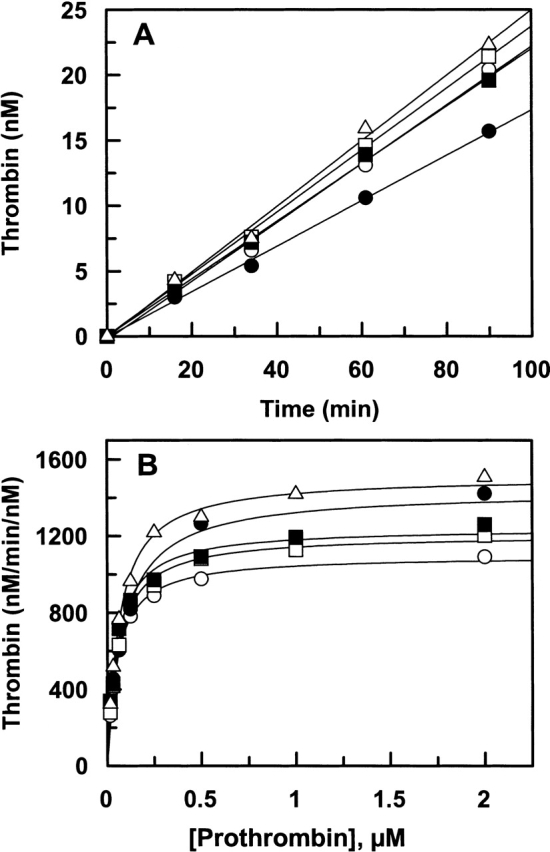
Prothrombin activation by recombinant fXa derivatives in the absence and presence of fVa. (A). In the absence of fVa, human prothrombin (1 μM) was incubated with each fXa derivatives: wild type (open circles), R71A (filled circles), E74A (open squares), E76A (filled squares), and E77A (open triangles) (10 nM each) on PC/PS vesicles (25 μM) at room temperature in TBS/Ca2+. At different time intervals, small aliquots of the activation reactions were transferred to wells of a 96-well assay plate containing 20 mM EDTA, and the rate of thrombin generation was measured from the cleavage rate of S2238 as described in Materials and Methods. (B) In the presence of human fVa (30 nM), fXa derivatives: wild type (open circles), R71A (filled circles), E74A (open squares), E76A (filled squares), and E77A (open triangles) (0.025 nM each) was incubated with different concentrations of prothrombin on PC/PS vesicles (25 μM) in TBS/Ca2+. After 0.5–2 min of incubation at room temperature, the reactions were terminated by addition of EDTA and the initial rate of thrombin generation was measured as described above. Solid lines in B are nonlinear regression fits of kinetic data to the Michaelis-Menten equation, and those in A are fits to a linear equation.
Table 3.
Kinetic constants for prothrombin activation by fXa derivatives assembled into the prothrombinase complex
| Km (nM) | kcat (nM/min/nM) | Kd(app) (fVa, nM) | |
| WT | 51.6 ± 2.5 | 1096.4 ± 13.4 | 2.1 ± 0.3 |
| R71A | 85.6 ± 14.7 | 1434.3 ± 70.1 | 1.5 ± 0.2 |
| E74A | 56.7 ± 3.4 | 1205.9 ± 16.8 | 1.7 ± 0.3 |
| E76A | 52.4 ± 5.5 | 1241.1 ± 29.3 | 2.9 ± 0.3 |
| E77A | 62.9 ± 3.9 | 1510.0 ± 22.3 | 3.3 ± 0.6 |
The kinetic constants were determined from the concentration dependence of prothrombin activation by fXa derivatives in the prothrombinase complex in TBS/Ca2+ at room temperature as described under Materials and Methods. All values are the average of at least two to three measurements ± standard errors.
Reaction with antithrombin
The reactivity of the fXa derivatives with antithrombin was evaluated in both the absence and presence of the pentasaccharide. Relative to the wild-type fXa, antithrombin inactivated E76A and E77A with a similar second-order association rate constant (k2) in both the absence and presence of the pentasaccharide (Table 4). However, an approximately twofold impairment in the k2 values was observed for the antithrombin inhibition of both R71A and E74A mutants, although the extent of the cofactor effect of the pentasaccharide was not affected with either one of the mutants (Table 4).
Table 4.
Second-order rate constants (M−1 sec−1) for the inhibition of fXa derivatives by antithrombin in the absence and presence of pentasaccharide
| −H5 (×103) | +H5 (×105) | +H5/−H5 | |
| WT | 2.7 ± 0.1 | 8.0 ± 0.1 | 296 ± 15 |
| R71A | 1.3 ± 0.1 | 4.4 ± 0.6 | 339 ± 72 |
| E74A | 1.3 ± 0.1 | 4.2 ± 0.6 | 323 ± 71 |
| E76A | 2.2 ± 0.1 | 5.7 ± 0.4 | 259 ± 30 |
| E77A | 1.9 ± 0.2 | 5.3 ± 0.4 | 279 ± 50 |
The second-order association rate constants were determined from the remaining activities of enzymes (1 nM) after incubation with antithrombin (250 nM in the absence and 25 nM in the presence of 500 nM pentasaccharide [H5]) in TBS/Ca2+ as described under Materials and Methods. All values are the average of at least two to three measurements ± standard errors.
Discussion
Recent results have indicated that variant residues on the exposed surface loops play pivotal roles in determining the substrate, cofactor, and inhibitor specificity of coagulation proteases (Baugh et al. 2000; Kolkman and Mertens 2000; Rezaie 2000; Pineda et al. 2002; Chen et al. 2003). One of these surface loops, the 70–80 loop, has been thoroughly studied for both prothrombin (Anderson et al. 2000; Chen et al. 2003) and protein C (Friedrich et al. 2001; Gale et al. 2002; Yang and Rezaie 2003), and contains several basic residues in both molecules. Previous mutagenesis of these residues in both prothrombin and protein C has been shown to severely impair the zymogenic and enzymatic properties of the mutant molecules. Unlike prothrombin and protein C, the 70–80 loop of fX has a reversed polarity because it contains only one positively charged (Arg71) but five negatively charged residues (Asp70, Glu74, Glu76, Glu77, and Glu80). With the exception of Asp70 and Glu80, which are involved in Ca2+ coordination (Padmanabhan et al. 1993; Rezaie and Esmon 1994), the contribution of other residues of this loop to structure and function of fX/fXa has not been studied. Thus, the current study was undertaken to address this question. The results obtained suggest that these residues are important for the efficient activation of fX by fIXa in the intrinsic Xase complex. Unlike prothrombin and protein C, however, in which the basic residues of the 70–80 loop were cofactor-dependent recognition sites for prothrombinase (Chen et al. 2003) and the thrombin–thrombomodulin complex (Yang and Rezaie 2003), respectively, the residues of this loop in fX do not appear to interact with fVIIIa in the intrinsic Xase complex. This was evidenced by the observation that the activation of both R71A and E77A mutants by fIXa was impaired to nearly the same extent in both the absence and presence of fVIIIa (Fig. 3 ▶). Although the activation of E74A and E76A was specifically impaired approximately twofold in the presence of fVIIIa, in light of allosteric linkage of this loop to other parts of the molecule and the modest effect of the mutation on the activation rates, a direct interaction for either one of these residues with the cofactor cannot be ascertained based on results of this study. Unlike the intrinsic Xase complex, results of both activation kinetic data and the clotting assays suggest that the mutant residues play an insignificant role (~1.5-fold) in fX recognition by the extrinsic Xase complex (Fig. 2 ▶; Table 1). These results are in agreement with a previous peptide inhibition study that investigated the ability of synthetic peptides derived from selected regions of fX to inhibit the substrate activation by the intrinsic and extrinsic Xase complexes (Chattopadhyay and Fair 1989). In that study, a peptide spanning all residues of the 70–80 loop exhibited no inhibitory effect on fX activation by the extrinsic Xase complex. However, an ~30% inhibitory effect for the same peptide was observed in fX activation by the intrinsic Xase complex.
Unlike the zymogenic properties, no significant defect in the proteolytic properties of the mutant enzymes was observed with respect to their ability to interact with fVa, or activate prothrombin when assembled into the prothrombinase complex. These results are also consistent with the peptide inhibition study mentioned above, in which a synthetic peptide containing the residues of the 70–80 loop, including those mutated in this study, had no inhibitory effect on the rate of prothrombin activation by the prothrombinase complex (Chattopadhyay et al. 1992). Thus, neither one of the mutant residues under study in this loop interacts with the cofactor fVa or the substrate prothrombin in the activation complex. With the exception of an approximately twofold elevated Km value for the hydrolysis of the tripeptidyl substrate, SpFXa, by the R71A mutant, all fXa mutants cleaved this substrate with similar catalytic efficiencies. The observation that the Ki for the interaction of the R71A mutant with the S1 site-specific inhibitor PAB was also elevated approximately twofold suggests that the S1 binding site of the mutant fXa has been affected by the mutagenesis. The Kd(app) for the interaction of R71A with both Na+ and Ca2+ ions were also impaired. These results are in agreement with results of our previous mutagenesis study in which we showed that the conformation of the 70–80 loop of fXa is allosterically linked to the Na+ binding loop of the protease (Rezaie and He 2000). In addition to the metal ion binding loops, it has been recently demonstrated that the S1 and the Na+ binding sites of fXa are also thermodynamically linked (Underwood et al. 2000). Because the ability of the R71A mutant to interact with Na+ was elevated approximately fivefold, it therefore follows that the mutagenesis of Arg71 may have affected the conformation of the S1 site of the mutant enzyme. Nevertheless, despite possible changes in the conformation of both the Ca2+ and Na+ binding loops, the R71A mutant of fXa in the prothrombinase complex activated prothrombin with a catalytic efficiency that was similar to that of the wild-type fXa. In a recent study, it was proposed that the Na+ binding site of fXa is also thermodynamically linked to the fVa binding helix of the protease (Camire 2002). Because the affinity of R71A for interaction with Na+ was impaired approximately fivefold, we expected that the mutant may also exhibit altered affinity for fVa. However, the fVa concentration dependence of prothrombin activation yielded essentially identical Kd(app) values for fVa interaction with both the wild-type and mutant enzymes. Thus, direct binding studies with this mutant will be required to further investigate this question. The observation that the mutagenesis of Arg71 had a more deleterious effect than the mutagenesis of the acidic residues suggests that Arg71 plays a critical role in the structure and function of fX/fXa.
With the exception of prothrombin, an Asp or a Glu has been conserved at both 70 and 80 positions of all vitamin K-dependent coagulation proteins as well as in trypsin. It is known that both acidic residues 70 and 80 in fXa and other coagulation proteases are involved in Ca2+ coordination (Padmanabhan et al. 1993; Persson et al. 1993; Rezaie and Esmon 1994). The observation that the chromogenic substrate activity of the E77A mutant was independent of Ca2+ suggests that the carboxylate group of Glu77 is also involved in Ca2+ coordination. The structural basis for this observation may be provided by examining the relative orientation of the side chains of these residues in the X-ray crystal structure of the protease domain of the CI-1031-inhibited fXa (Fig. 6 ▶; Adler et al. 2000). The side chains of all three residues Asp70, Glu77, and Glu80 are pointing inward, and thus capable of liganding a Ca2+ ion. On the other hand, side chains of both residues Glu74 and Glu76 are pointing outward toward the solvent, and not expected to be involved in interaction with Ca2+. Consistent with the structural data, the amidolytic activity of the latter two mutants was Ca2+-dependent. However, the observation that the neutralization of the charges of these residues lead to approximately two- to fourfold impairments in Kd(app) for interaction of mutants with Ca2+ suggests that both Glu74 and Glu76 contribute to the overall electronegativity of this loop to support the high affinity interaction of fXa with the metal ion. Although the E77A mutant lost its ability to bind Ca2+, it assembled into the prothrombinase complex and activated prothrombin with a similar kinetic efficiency as the wild-type fXa. Thus, the binding of Ca2+ to the 70–80 loop may not be required for the prothrombinase activity of fXa. The structural data also revealed that the side chain of Arg71 is not exposed to the solvent, but oriented toward a cavity in the vicinity of Asp24 (~3.7 Å) behind the 70–80 loop (Fig. 6 ▶; Adler et al. 2000). Assuming that this side-chain arrangement is also true for the Ca2+-stablized uninhibited fXa, Arg71 may not be available for interaction with the fXa ligands. However, this appears not to be the case with the zymogen fX, because its activation by both fIXa alone and the intrinsic Xase complex was impaired approximately three- to fivefold. Nevertheless, because there is no crystal structure for the zymogen fX, this possibility cannot be ruled out. If this is the case, then the impairment in the zymogenic properties of the R71A mutant during activation by the intrinsic Xase complex may not actually be caused by the loss of specific interactions of fIXa with the side chain of Arg71 as proposed above, but rather by an overall change in the conformation of the 70–80 loop in the mutant zymogen. A crystal structure of fX zymogen is required to answer this question with certainty.
Figure 6.

Crystal structure of the catalytic domain of fXa in complex with CI-1031. The side chain of acidic residues of the 70–80 loop of fXa are shown in red and the basic residue is shown in blue. The black sphere represents a Ca2+ ion placed arbitrarily in the middle of the loop. The autolysis loop of fXa from residues 143–155 is colored in purple. The Na+-binding loop from residues 218–225 is colored in black. The catalytic triad residues Asp102, His57, and Ser195 are shown in red, green, and blue, respectively. The coordinates (Protein Data Bank code 1FJS) of the C-terminal EGF and catalytic domains of fXa were used to prepare the figure (Adler et al. 2000).
Finally, a minimal change in the reactivity of E76A and E77A mutants of fXa with antithrombin, in either the absence or presence of the pentasaccharide, suggests that neither residue significantly contributes to the specificity of the fXa–antithrombin interaction. However, the reactivity of R71A and E74A with the serpin was impaired approximately twofold, independent of the pentasaccharide. We previously showed that the autolysis loop of fXa is critical for interaction of the protease with antithrombin (Manithody et al. 2002). Both Arg71 and Glu74 of the 70–80 loop are located adjacent to the autolysis loop of fXa (Fig. 6 ▶). Thus, it is possible that these two residues of the 70–80 loop also interact with the serpin. It should, however, be noted that the impairment in the reactivity of both mutants with antithrombin could also be due to an indirect effect stemming from possible alterations in the conformations of the Na+-binding and/or the autolysis loop of the protease. The latter possibility is strengthened by the observation that the carbonyl oxygen of Arg71 is in hydrogen bond contact with the main chain nitrogen atom of Leu155 at the C-terminal end of the autolysis loop (2.734 Å; Adler et al. 2000). Similarly, the main-chain nitrogen atom of Thr73 makes a hydrogen bond with the side-chain oxygen of Thr153 (2.762 Å). Thus, the mutagenesis of Arg71 and Glu74 could lead to alterations in the conformation of the autolysis loop, which in turn, could lead to its altered interaction with antithrombin. A conformational linkage between the autolysis loop and the 70–80 loop is supported by the observation that the Ca2+ occupancy of the 70–80 loop protects fXa from proteolytic cleavage of its own autolysis loop (Sabharwal et al. 1997).
Materials and methods
Mutagenesis and expression of recombinant proteins
The construction and expression of fX in a novel expression/purification vector system, in which the first 12 residues of the activation peptide of fX (residues 183–194 in fX numbering) on the heavy chain have been replaced with the sequence of a 12-residue epitope for the Ca2+-dependent monoclonal antibody, HPC4, was described previously (Manithody et al. 2002). The Arg71 → Ala (R71A), Glu74 → Ala (E74A), Glu76 → Ala (E76A), and Glu77 → Ala (E77A) mutants of fX were prepared by PCR mutagenesis methods in the same vector system as described (Manithody et al. 2002). After confirmation of the accuracy of mutagenesis by DNA sequencing, the constructs were transfected into HEK293 cells and the mutant proteins were isolated from 20-L cell culture supernatants by a combination of immunoaffinity and ion exchange chromatography using the HPC4 monoclonal antibody and an FPLC Mono Q column as described (Manithody et al. 2002). Recombinant tissue factor lacking the cytoplasmic domain (dcTF), expressed in bacteria (Neuenschwander et al. 1995), was generously provided by Dr. James Morrissey (University of Illinois at Urbana–Champaign).
Human plasma proteins including factors Va, VIIa, IXa, Xa, and X, and the factor X-activating enzyme from Russell’s viper venom (RVV-X) were purchased from Haematologic Technologies Inc. Phospholipid vesicles containing 80% phosphatidylcholine and 20% phosphatidylserine (PC/PS) were prepared as described (Smirnov and Esmon 1994). Human recombinant fVIIIa was a generous gift from Dr. Philip Fay (University of Rochester). Human recombinant antithrombin was prepared as described (Rezaie and Yang 2001). The active antithrombin-binding pentasaccharide fragment of heparin (fondaparinux sodium) was purchased from Quintiles Clinical Supplies. The chromogenic substrate Spectrozyme FXa (SpFXa) was purchased from American Diagnostica, and S2222 and S2238 were purchased from Kabi Pharmacia/Chromogenix. The prothrombin time (PT; Thrombomax with Ca2+) and activated partial thromboplastin time (APTT; Alexin) reagents were purchased from Sigma. Normal pooled plasma and fX deficient plasma from a genetically deficient patient were purchased from George King Bio-Medical, Inc.
γ-Carboxyglutamic acid (Gla) analysis
The Gla content of both plasma-derived and recombinant fX were determined following alkaline hydrolysis and derivatization with phenylisothiocyanate as described (Smalley and Preusch 1988).
Activation by the FIXa–VIIIa complex
The initial rate of activation of fX derivatives by fIXa was studied in both the absence and presence of fVIIIa on PC/PS vesicles in 0.1 M NaCl, 0.02 M Tris-HCl, pH 7.4 (TBS) containing 0.1 mg/mL BSA, 0.1% PEG 8000, and 5 mM Ca2+ (TBS/Ca2+). In the absence of the cofactor, the time course of the activation of fX derivatives (0.5 μM) by fIXa (50 nM) on PC/PS vesicles (50 μM) was monitored in TBS/Ca2+ at room temperature (~22–25°C). At different time points, a small aliquot of each reaction was transferred to TBS containing 20 mM EDTA and the concentration of fXa generated was determined from standard curves by an amidolytic activity assay from the hydrolysis of 200 μM SpFXa in TBS containing 0.1 mg/mL BSA and 0.1% PEG 8000. The rate of hydrolysis was measured at 405 nm at room temperature in a Vmax kinetic plate reader (Molecular Devices). In the presence of fVIIIa (25 nM), the concentration dependence of fX activation (7.8–1000 nM) by fIXa (25 pM) was monitored on PC/PS vesicles (50 μM) in TBS/Ca2+ at room temperature for 1–2 min. The activation reactions (0.03 mL volumes) were terminated by addition of 0.02 mL EDTA to obtain a final concentration of 20 mM. The rate of fXa generation was determined from standard curves using an amidolytic activity assay as described above. The Km and kcat values were calculated from the Michaelis-Menten equation.
Activation by the factor VIIa–TF complex
Activation of fX derivatives by fVIIa was monitored in both the absence and presence of TF on PC/PS vesicles in TBS/Ca2+. In the absence of the cofactor, the time course of the activation of fX derivatives (0.5 μM) by fVIIa (50 nM) on PC/PS vesicles (50 μM) was monitored in TBS/Ca2+ at room temperature. At different time points, a small aliquot of each reaction was transferred to TBS containing 20 mM EDTA and the concentration of fXa generated was determined from standard curves as described above. Kinetics of activation of fX derivatives were also studied by fVIIa in complex with dcTF incorporated into phospholipid vesicles containing 80% phosphatidylchloine (PC) and 20% phosphatidylserine (PS) as described (Neuenschwander et al. 1995). Briefly, dcTF (400 nM final) was incubated with PC/PS vesicles (1 mM final) in the presence of the detergent, CHAPS (3.75 mM final) in 0.1 M NaCl, 0.02 M Tris-HCl, pH 7.4 in 200 μL reaction volume for 2 h at 37°C. Relipidated dcTF was extensively dialyzed (12–14-kD cutoff membrane) against 5 × 4 liters of TBS at 4°C. The concentration of functional dcTF, exposed on the outside of PC/PS vesicles, was determined by an amidolytic activity assay using known concentrations of fVIIa as described (Neuenschwander and Morrissey 1994). This assay indicated that 60% (240 nM) of dcTF is exposed on the outside of vesicle surface. The initial rate of activation by factor VIIa (25 pM) in complex with relipidated dcTF (2.5 nM) was carried out as a function of increasing concentrations of the substrate (7.8–1000 nM) in TBS/Ca2+ as described (Neuenschwander and Morrissey 1994). The reactions were carried out at room temperature for 0.5–2 min. The activation reactions (0.03 mL volumes) were terminated by addition of 0.02 mL EDTA to obtain a final concentration of 20 mM. The concentration of fXa generated was determined from standard curves by a chromogenic substrate assay as described above. It was ensured that less than 15% of fX was activated at all substrate concentrations. The Km and kcat values for activation were calculated from the Michaelis-Menten equation.
Measurement of clotting activities
Clotting activities of fX derivatives were evaluated by both prothrombin time (PT) and activated partial thromboplastin time (APTT) assays using a STart 4 fibrinometer (Diagnostica/Stago) as described (Manithody et al. 2002, 2003). In the PT assay, 0.1 mL of the PT reagent (Thrombomax with Ca2+) was added to a mixture of 0.05 mL of the test sample (human plasma, plasma derived fX, and recombinant wild-type and mutant fX derivatives) and 0.05 mL of fX deficient plasma at 37°C. Activities of all samples were measured at four different dilutions ranging from 0.05 to 0.4 μg/mL fX (final concentrations). In the case of APTT assay, 0.05 mL of APTT reagent (Alexin) was incubated with a mixture of 0.05 mL fX deficient plasma and 0.05 mL of the test sample for 5 min before the initiation of clotting by the addition of 0.05 mL of 35 mM CaCl2 at 37°C. The clotting activities of samples were measured at four different dilutions ranging from 0.2 to 1.6 μg/mL fX (final concentrations). The clotting activities of plasma samples ranged from 17–32 sec for PT and 27–55 sec for APTT.
Cleavage of the chromogenic substrates by the factor Xa derivatives
For measuring the amidolytic activities, fX derivatives (1 μM each) were completely activated to fXa using 20 nM RVV-X in 100 μL reactions at 37°C for 1 h. Time-course analysis indicated that this time was sufficient for a complete activation of all fX derivatives. Then, the active-site concentrations of the fXa derivatives were determined by titrations with human antithrombin assuming a 1:1 stoichiometry as described (Rezaie 1998). The steady-state kinetics of hydrolysis of SpFXa (7.5–1000 μM) by the fXa derivatives (0.5 nM) were studied in TBS/Ca2+ as described above. The rate of hydrolysis was measured at 405 nm at room temperature in a Vmax kinetic plate reader as described (Manithody et al. 2002). The Km and kcat values for substrate hydrolysis were calculated from the Michaelis-Menten equation.
Dissociation constants (Kd(app)) for Na+ and Ca2+
The Kd(app) values for the interaction of fXa derivatives with Na+ and Ca2+ were determined from the effect of varying concentrations of the metal ions on the amidolytic activities of proteases toward the synthetic substrates SpFXa or S2222 as described (Rezaie and He 2000). The Ca2+ titration was carried out in TBS containing 0.1 M NaCl and 0.02 M Tris-HCl, pH, 7.4. For the Na+ titration (0–200 mM) experiments, Tris-HCl was used to adjust the ionic strength of buffer in all reactions as described (Rezaie and He 2000).
Prothrombinase assay
The initial rate of prothrombin activation by the wild-type and mutant fXa derivatives was determined in both the absence and presence of fVa at room temperature as described (Manithody et al. 2002). In the absence of the cofactor, prothrombin (1 μM) was incubated with each fXa derivative (10 nM) in TBS/Ca2+ on PC/PS vesicles (25 μM). The time course of prothrombin activation was monitored from the rate of thrombin generation by an amidolytic activity assay using 100 μM S2238. The concentrations of thrombin generated in the activation reactions were determined from a standard curve prepared from the cleavage rate of S2238 by known concentrations of recombinant thrombin under exactly the same conditions. The initial rate of prothrombin activation in the presence of human fVa (30 nM) was measured by incubating each fXa derivative (25 pM) with increasing concentration of prothrombin (15–1000 nM) in TBS/Ca2+ on PC/PS vesicles (25 μM). Following 0.5–2-min incubation at room temperature, EDTA was added to a final concentration of 20 mM, and the concentrations of thrombin generated were determined from a standard curve as described above. It was ensured that less than 15% of prothrombin was activated at all concentrations of the substrate.
Inactivation by antithrombin
The rate of inactivation of fXa derivatives by antithrombin in both the absence and presence of the pentasaccharide fragment of high affinity heparin was measured under pseudofirst-order rate conditions by a discontinuous assay as described (Manithody et al. 2002). Briefly, 1 nM fXa was incubated with 250 nM human antithrombin in the absence, or 25 nM antithrombin in the presence of 500 nM pentasaccharide in TBS/Ca2+. All reactions were carried out at room temperature in 50 μL volumes in 96-well polystyrene plates. After a period of time (20 min in the absence and 0.5 min in the presence of pentasaccharide), 50 μL of SpFXa (500 μM) in TBS was added to each well and the remaining enzyme activity was measured with a Vmax kinetics plate reader as described above. The second-order inactivation rate constants were obtained as described (Manithody et al. 2002).
Inhibition by p-aminobenzamidine
The affinity of p-aminobenzamidine (PAB) for interaction with the active-site pocket of the wild-type and mutant fXa derivatives was evaluated. In all cases, fXa (1 nM) was incubated with increasing concentrations of the inhibitor (0–320 μM) in the presence of different fixed concentrations of SpFXa (50–400 μM) in TBS/Ca2+. The enzyme activity was measured from the cleavage rate of the chromogenic substrate as described above and the Ki values were determined by global fitting of data to a competitive binding equation as described (Rezaie 2003).
Acknowledgments
We would like to thank Audrey Rezaie for her proofreading of the manuscript. The research discussed herein was supported by grants awarded by the National Heart, Lung, and Blood Institute of the National Institutes of Health (HL 62565 and HL 68571 to A.R.R.).
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Abbreviations
fX, factor X
fXa, activated fX
R71A, E74A, E76A, and E77A, fX derivatives in which Arg71, Glu74, Glu76, and Glu77 in the chymotrypsinogen numbering system (Bode et al. 1989) have been replaced with Ala
fVIIa, active factor VII
TF, tissue factor
dcTF, TF in which cytoplasmic domain of the cofactor has been deleted
fIXa, active factor IX
fVIIIa, active factor VIII
fVa, active factor V
PEG, polyethylene glycol
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.03406904.
References
- Adler, M., Davey, D.D., Phillips, G.B., Kim, S.H., Jancarik, J., Rumennik, G., Light, D.R., and Whitlow, M. 2000. Preparation, characterization, and the crystal structure of the inhibitor ZK-807834 (CI-1031) complexed with factor Xa. Biochemistry 39 12534–12542. [DOI] [PubMed] [Google Scholar]
- Anderson, P.J., Nesset, A., Dharmawardana, K.R., and Bock, P. 2000. Role of proexosite I in factor Va-dependent substrate interactions of prothrombin activation. J. Biol. Chem. 275 16435–16442. [DOI] [PubMed] [Google Scholar]
- Baugh, R.J. and Krishnaswamy, S. 1996. Role of the activation peptide domain in human factor X activation by the extrinsic Xase complex. J. Biol. Chem. 271 16126–16134. [DOI] [PubMed] [Google Scholar]
- Baugh, R.J., Dickinson, C.D., Ruf, W., and Krishnaswamy, S. 2000. Exosite Interactions determine the affinity of factor X for the extrinsic Xase complex. J. Biol. Chem. 275 28826–28833. [DOI] [PubMed] [Google Scholar]
- Bode, W. and Schwager, P. 1975. The refined crystal structure of bovine β-trypsin at 1.8 Å resolution. II. Crystallographic refinement, calcium binding site, benzamidine binding site and active site at pH 7.0. J. Mol. Biol. 98 693–717. [DOI] [PubMed] [Google Scholar]
- Bode, W., Mayr, I., Baumann, U., Huber, R., Stone, S.R., and Hofsteenge, J. 1989. The refined 1.9 Å crystal structure of human α-thrombin: Interaction with D-Phe-Pro-Arg chlorometheylketone and significance of the Tyr-Pro-Pro-Trp insertion segment. EMBO J. 8 3467–3475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bode, W., Turk, D., and Karshikov, A. 1992. The refined 1.9-Å X-ray crystal structure of D-Phe-Pro-Arg chloromethylketone-inhibited human α-thrombin: Structure analysis, overall structure, electrostatic properties, detailed active-site geometry, and structure–function relationships. Protein Sci. 1 426–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camire, R.M. 2002. Prothrombinase assembly and S1 site occupation restore the catalytic activity of FXa impaired by mutation at the sodium-binding site. J. Biol. Chem. 277 37863–37870. [DOI] [PubMed] [Google Scholar]
- Chattopadhyay, A. and Fair, D.S. 1989. Molecular recognition in the activation of human blood coagulation factor X. J. Biol. Chem. 264 11035–11043. [PubMed] [Google Scholar]
- Chattopadhyay, A., James, H.L., and Fair, D.S. 1992. Molecular recognition sites on factor Xa which participate in the prothrombinase complex. J. Biol. Chem. 267 12323–12329. [PubMed] [Google Scholar]
- Chen, L., Yang, L., and Rezaie, A.R. 2003. Proexosite-1 on prothrombin is a factor Va-dependent recognition site for the prothrombinase complex. J. Biol. Chem. 278 27564–27569. [DOI] [PubMed] [Google Scholar]
- Davie, E.W., Fujikawa, K., and Kisiel, W. 1991. The coagulation cascade: Initiation, maintenance and regulation. Biochemistry 30 10363–10370. [DOI] [PubMed] [Google Scholar]
- Duffy, E.J. and Lollar, P. 1992. Intrinsic pathway activation of factor X and its activation peptide-deficient derivative, factor Xdes-143–191. J. Biol. Chem. 267 7821–7827. [PubMed] [Google Scholar]
- Edgington, T.S., Mackman, N., Brand, K., and Ruf, W. 1991. The structural biology of expression and function of tissue factor. Thromb. Haemost. 66 67–79. [PubMed] [Google Scholar]
- Friedrich, U., Niccolaes, G.A.F., Villoutreix, B.O., and Dahlback, B. 2001. Secondary substrate-binding exosite in the serine protease domain of activated protein C important for cleavage at Arg-506 but not at Arg-306 in factor Va. J. Biol. Chem. 276 23105–23108. [DOI] [PubMed] [Google Scholar]
- Furie, B. and Furie, B.C. 1988. The molecular basis of blood coagulation. Cell 53 505–518. [DOI] [PubMed] [Google Scholar]
- Furie, B., Bing, D.H., Feldman, R.J., Robison, D.J., Burnier, J.F., and Furie, B.C. 1982. Computer-generated models of blood coagulation factor Xa, factor IXa and thrombin based upon structural homology with other serine proteases. J. Biol. Chem. 257 3875–3882. [PubMed] [Google Scholar]
- Gale, A.J., Tsavaler, A., and Griffin, J.H. 2002. Molecular characterization of an extended binding site for coagulation factor Va in the positive exosite of activated protein C. J. Biol. Chem. 277 28836–28840. [DOI] [PubMed] [Google Scholar]
- Inoue, K. and Morita, T. 1993. Identification of O-linked oligosaccharide chains in the activation peptides of blood coagulation factor X: The role of carbohydrate moieties in the activation of factor X. Eur. J. Biochem. 218 153–163. [DOI] [PubMed] [Google Scholar]
- Jackson, C.M. and Nemerson, Y. 1980. Blood coagulation. Annu. Rev. Biochem. 49 765–811. [DOI] [PubMed] [Google Scholar]
- Kolkman, J.A. and Mertens, K. 2000. Insertion loop 256–268 in coagulation factor IX restricts enzymatic activity in the absence but not in the presence of factor VIII. Biochemistry 39 7398–7405. [DOI] [PubMed] [Google Scholar]
- Le Bonniec, B.F., Guinto, E.R., and Esmon, C.T. 1992. The role of calcium ions in factor X activation by thrombin E192Q. J. Biol. Chem. 267 6970–6976. [PubMed] [Google Scholar]
- Manithody, C., Yang, L., and Rezaie, A.R. 2002. Role of basic residues of the autolysis loop in the catalytic function of factor Xa. Biochemistry 41 6780–6788. [DOI] [PubMed] [Google Scholar]
- Manithody, C., Fay, P.J., and Rezaie, A.R. 2003. Exosite-dependent regulation of factor VIIIa by activated protein C. Blood 101 4802–4807. [DOI] [PubMed] [Google Scholar]
- Mann, K.G., Nesheim, M.E., Church, W.R., Haley, P., and Krishnaswamy, S. 1990. Surface-dependent reactions of the vitamin K-dependent enzyme complexes. Blood 76 1–16. [PubMed] [Google Scholar]
- Mather, T., Oganessyan, V., Hof, P., Huber, R., Foundling, S., Esmon, C., and Bode, W. 1996. The 2.8 Å crystal structure of Gla-domainless activated protein C. EMBO J. 15 6822–6831. [PMC free article] [PubMed] [Google Scholar]
- Neuenschwander, P.F. and Morrissey, J.H. 1994. Roles of the membrane-interactive regions of factor VIIa and tissue factor. J. Biol. Chem. 269 8007–8013. [PubMed] [Google Scholar]
- Neuenschwander, P.F., Bianco-Fisher, E., Rezaie, A.R., and Morrissey, J.H. 1995. Phosphatidylethanolamine augments factor VIIa-tissue factor activity: Enhancement of sensitivity to phosphatidylserine. Biochemistry 34 13988–13993. [DOI] [PubMed] [Google Scholar]
- Padmanabhan, K., Padmanabhan, K.P., Tulinsky, A., Park, C.H., Bode, W., Huber, R., Blankenship, D.T., Cardin, A.D., and Kisiel, W. 1993. Structure of human des (1–45) factor Xa at 2•2 Å resolution. J. Mol. Biol. 232 947–966. [DOI] [PubMed] [Google Scholar]
- Persson, E., Hogg, P.J., and Stenflo, J. 1993. Effects of Ca2+ binding on the protease module of factor Xa and its interaction with factor Va. Evidence for two Gla- independent Ca2+-binding sites in factor Xa. J. Biol. Chem. 268 22531–22539. [PubMed] [Google Scholar]
- Pineda, A.O., Cantwell, A.M., Bush, L.A., Rose, T., and Di Cera, E. 2002. The thrombin epitope recognizing thrombomodulin is highly cooperative hot spot in exosite I. J. Biol. Chem. 277 32015–32019. [DOI] [PubMed] [Google Scholar]
- Rezaie, A.R. 1998. Reactivities of the S2 and S3 subsite residues of thrombin with the native and heparin-induced conformers of antithrombin. Protein Sci. 7 349–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ———. 2000. Identification of basic residues in the heparin-binding exosite of factor Xa critical for heparin and factor Va binding. J. Biol. Chem. 275 3320–3327. [DOI] [PubMed] [Google Scholar]
- ———. 2003. DX-9065a inhibition of factor Xa and the prothrombinase complex: Mechanism of inhibition and comparison with therapeutic heparins. Thromb. Haemost. 89 112–121. [PubMed] [Google Scholar]
- Rezaie, A.R. and Esmon, C.T. 1994. Asp-70 to Lys mutant of factor X lacks the high affinity Ca2+ binding site yet retains function. J. Biol. Chem. 269 21495–21499. [PubMed] [Google Scholar]
- Rezaie, A.R. and He, X. 2000. Sodium binding site of factor Xa: Role of sodium in the prothrombinase complex. Biochemistry 39 1817–1825. [DOI] [PubMed] [Google Scholar]
- Rezaie, A.R. and Yang, L. 2001. Probing the molecular basis of factor Xa specificity by mutagenesis of the serpin, antithrombin. Biochem. Biophys. Acta 1528 167–176. [DOI] [PubMed] [Google Scholar]
- Sabharwal, A.K., Padmanabhan, K., Tulinsky, A., Mathur, A., Gorka, J., and Bajaj, S.P. 1997. Interaction of calcium with native and decarboxylated human factor X. J. Biol. Chem. 272 22037–22045. [DOI] [PubMed] [Google Scholar]
- Sinha, U. and Wolf, D.L. 1993. Carbohydrate residues modulate the activation of coagulation factor X. J. Biol. Chem. 268 3048–3051. [PubMed] [Google Scholar]
- Smalley, D.M. and Preusch, P.C. 1988. Analysis of γ-carboxyglutamic acid by reverse phase HPLC of its phenylthiocarbamyl derivative. Anal. Biochem. 172 241–247. [DOI] [PubMed] [Google Scholar]
- Smirnov, M.D. and Esmon, C.T. 1994. Phosphatidylethanolamine incorporation into vesicles selectively enhances factor Va inactivation by activated protein C. J. Biol. Chem. 269 816–819. [PubMed] [Google Scholar]
- Underwood, M.C., Zhong, D., Mathur, A., Heyduk, T., and Bajaj, S.P. 2000, Thermodynamic linkage between the S1 site, the Na+ site, and the Ca2+ site in the protease domain of human coagulation factor Xa. J. Biol. Chem. 275 36876–36884. [DOI] [PubMed] [Google Scholar]
- Yang, L. and Rezaie, A.R. 2003. The fourth epidermal growth factor-like domain of thrombomodulin interacts with the basic exosite of protein C. J. Biol. Chem. 278 10484–10490. [DOI] [PubMed] [Google Scholar]