

Abstract
α1-Antitrypsin is the most abundant protease inhibitor in plasma and is the archetype of the serine protease inhibitor superfamily. Genetic variants of human α1-antitrypsin are associated with early-onset emphysema and liver cirrhosis. However, the detailed molecular mechanism for the pathogenicity of most variant α1-antitrypsin molecules is not known. Here we examined the structural basis of a dozen deficient α1-antitrypsin variants. Unlike most α1-antitrypsin variants, which were unstable, D256V and L41P variants exhibited extremely retarded protein folding as compared with the wild-type molecule. Once folded, however, the stability and inhibitory activity of these variant proteins were comparable to those of the wild-type molecule. Retarded protein folding may promote protein aggregation by allowing the accumulation of aggregation-prone folding intermediates. Repeated observations of retarded protein folding indicate that it is an important mechanism causing α1-antitrypsin deficiency by variant molecules, which have to fold into the metastable native form to be functional.
Keywords: α1-antitrypsin, conformational disease, folding, metastability, serpin
α1-Antitrypsin (α1AT) is the archetype of the serine protease inhibitor (serpin) superfamily (Huber and Carrell 1989). Members of this family include protease inhibitors found in blood plasma, such as α1-antichymotrypsin, antithrombin-III, C1 inhibitor, and plasminogen activator inhibitor-1, as well as noninhibitory proteins, such as ovalbumin and angiotensinogen. Serpins share a common tertiary structure based on a mobile reactive site loop (RSL), three β-sheets, and several α-helices (Elliott et al. 1996). Inhibitory serpins are unique in that the native form is metastable, and the RSL is mobile (Stratikos and Gettins 1997; Huntington et al. 2000). These features likely facilitate a conformational conversion during functional execution (Wiley and Skehel 1987; Huber and Carrell 1989; Stein and Carrell 1995; Carr et al. 1997; Im et al. 1999; Lee et al. 2000). In the active, native form of inhibitory serpins (Elliott et al. 1998), the RSL is exposed at one end of the molecule for protease binding (Fig. 1 ▶). Upon binding and cleavage of the RSL by the target protease, the acyl-enzyme intermediate is inserted into β-sheet A (Johnson and Travis 1978; Loebermann et al. 1984), the protease is translocated to the end of the serpin distal to the initial docking site (Huntington et al. 2000), and the catalytic triad of the target protease is distorted (Plotnick et al. 1996). These events markedly increase the stability of the serpin molecule (Bruch et al. 1988). In an alternate, “latent” serpin form, the RSL is inserted into β-sheet A without being cleaved (Mottonen et al. 1992). This latent form is more stable than the native form (Hekman and Loskutoff 1985; Wang et al. 1996), but is inactive. Another serpin conformation involves insertion of the RSL of one serpin molecule into a β-sheet of a second serpin molecule, resulting in the formation of loop–sheet serpin polymers (Lomas et al. 1992). Although the conformational versatility of serpins may be important for regulating their biological functions, it carries inherent disadvantages, such as vulnerability to protein misfolding and facile conformational conversion of the metastable native form into stable, inactive forms.
Figure 1.
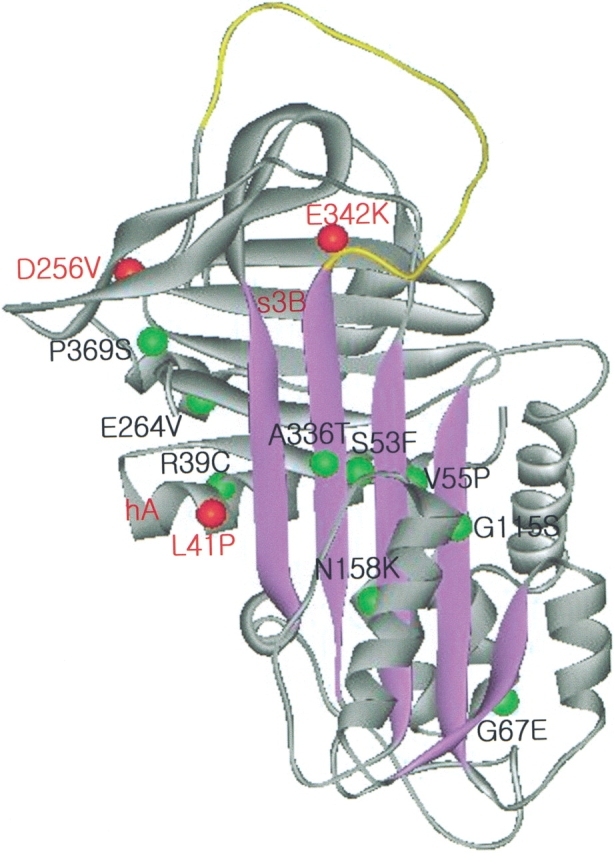
A schematic diagram of the native structure of α1AT (2PSI.PDB; Elliott et al. 1998) showing mutation sites carried by deficient α1AT variants. The β-sheet A is shown as purple strands, and the RSL is colored in yellow. Mutation sites that exhibited retarded protein folding are indicated with red beads, and other tested mutations that did not show retarded folding are indicated with green beads.
α1AT is synthesized in the liver and secreted into plasma to protect tissues against indiscriminate proteolytic attack by neutrophil elastase (Carrell et al. 1982). An imbalance between serum proteases and their inhibitors due to dysfunctional variant serpins or environmental factors causes serious clinical problems, such as tissue damage and bleeding disorders. Dozens of dysfunctional α1AT genetic variants have been reported, but, for most pathogenic α1AT molecules, the detailed structural basis for serpin deficiency has not been elucidated. They appear to form protein aggregates in the endoplasmic reticulum of hepatocytes, the site of α1AT biosynthesis, which lead to liver cirrhosis, and subsequent α1AT deficiency in plasma, causing pulmonary emphysema (Eriksson et al. 1986; Stein and Carrell 1995). The majority of dysfunctional α1AT proteins are probably conformationally labile, and are prone to subsequent intermolecular loop–sheet polymerization, leading to hepatic inclusions and plasma deficiency of α1AT (Lomas et al. 1993a, 1995). Indeed, loop–sheet polymers of α1AT have been found in vivo in patients carrying defective α1AT alleles, such as the I-type (Arg 39 → Cys) and Siiyama (Ser 53 → Phe) variants (Mahadeva et al. 1999). However, the mechanisms by which these variants form aggregates remain mostly unknown.
In the case of the best-studied Z-type (Glu 342 → Lys) α1AT variant, which is found at an allele frequency of 0.04 in Northern European populations, extremely retarded protein folding leads to the accumulation of an intermediate with a high tendency to aggregate. Once folded, the native Z-type protein is quite stable and shows inhibitory activity toward target proteases comparable to that of the wild-type molecule (Yu et al. 1995). Some variants show impaired protease inhibitory activity. Various biochemical (Wright and Scarsdale 1995; Gils et al. 1996; Huntington et al. 1997) and structural (Aertgeerts et al. 1995; Lukacs et al. 1996) studies have suggested that the rate of RSL insertion into β-sheet A is critical for inhibitory function. Hence, bulky substitutions on the inserted RSL would interfere with the kinetics of the inhibitory function of α1AT, converting the serpins into substrates rather than inhibitors of target proteases. Because the exposed RSL of serpins fits into a catalytic cleft on the protease, the serpin residues at sites P1 and P1′ (the residues before and after the scissile peptide bond, respectively) determine target protease specificity. α1ATPittsburgh has an amino acid substitution at site P1 on the RSL (Met 358 → Arg), and its target specificity is shifted from neutrophil elastase to thrombin, causing a bleeding disorder (Owen et al. 1983). Some variants may adopt conformations different from the native form. A variant α1-antichymotrypsin (Leu 55 → Pro) has a mutation in the so-called shutter domain underlying the opening of β-sheet A, which has to be mobilized on complex formation with the target protease. This variant is found not only in the native conformation, but also in the inactive latent conformation and in an inactive stable conformation that is an intermediate in the polymerization pathway (Gooptu et al. 2000). Several naturally occurring dysfunctional serpin mutations, such as Siiyama (53 Ser → Phe) and Mmalton (Phe 52-deleted) α1AT variants, are also clustered in the shutter domain. Perturbation of this region probably increases the flexibility of β-sheet A and favors the formation of different conformers. Indeed, the Mmalton α1AT variant spontaneously transforms into a latent-like conformation (Jung and Im 2003).
In this study, we examined the structural basis for the deficiency of a dozen α1AT variants with substitutions outside the RSL. Most variant α1AT proteins studied exhibited decreased stability as compared with the wild-type molecule. Conspicuously, protein folding of the D256V and L41P variants was markedly retarded, but, once folded, their stabilities and inhibitory activities were comparable to those of the wild-type molecule.
Results
Folding of some dysfunctional α1AT variants is markedly retarded
To study the structural bases for α1AT deficiency of genetic variants, a dozen mutant alleles carried by these variants were introduced separately into recombinant α1AT using oligonucleotide-directed mutagenesis. After expression and purification of the variant α1AT proteins, the conformation of the mutant serpins was analyzed by transverse urea gradient gel electrophoresis (Fig. 2 ▶). In this gel system, electrophoretic mobility of proteins depends on the hydrodynamic volumes of different conformational states, induced by urea-dependent denaturation. As urea concentration gradually increased, wild-type α1AT exhibited two cooperative unfolding transitions (Fig. 2 ▶). Most of the α1AT variants tested (R39C, S53F, V55P, G67E, G115S, N158K, E264V, A336T, and P369S) exhibited decreased stability, as shown by unfolding of their native forms at lower urea concentrations than that required to unfold wild-type α1AT (Fig. 2 ▶ for V55P; data not shown). In contrast, the D256V and L41P α1AT polypeptides folded into a molten globule-like intermediate that had a lower mobility than the native form. However, prolonged incubation of these species at 30°C induced a conformational change to a tightly folded form, which migrated to the same position as the native form on electrophoresis (Fig. 2 ▶). These results indicate that these α1AT variants can be folded into a native-like conformation, although the folding rate is very slow. A previously reported Z-type α1AT variant (Glu 342 → Lys) also exhibited retarded folding (Yu et al. 1995; Fig. 2 ▶). Although certain α1AT variants tested accumulate as low-mobility species, they do not convert to the tightly folded form on prolonged incubation; instead, they tend to aggregate (Fig. 2 ▶, V55P), possibly by facile loop insertion into a β-sheet of a neighboring molecule owing to destabilization of the native form.
Figure 2.

Transverse urea gradient gel electrophoresis showing retarded protein folding of α1AT variants. The wild-type (wt), D256V, L41P, Z-type (Z), and V55P α1AT were incubated in a buffer (10 mM phosphate at pH 6.5, 50 mM NaCl, 1 mM EDTA, and 1 mM β-mercaptoethanol) at 30°C for 24 h, and conformational changes were followed by transverse urea gradient gel electrophoresis (Goldenberg 1989). The transverse urea gradient gels contained a gradient of 0~8 M urea perpendicular to the direction of electrophoresis. Proteins were visualized by staining with Coomassie brilliant blue. Positions of the native and intermediate conformers are indicated.
Slow folding to the native form with inhibitory activity
The results of transverse urea gradient gel electrophoresis showed that, for some α1AT variants, a tightly folded conformation with electrophoretic mobility similar to that of the native form is generated upon prolonged incubation. However, it is not known whether this species is indeed the native form, with native biological functions, or is another misfolded conformation. To address this question, the ability of refolded α1AT samples to form an SDS-stable protease/inhibitor complex was monitored over time. Immediately after folding (0 h), very little of the D256V or L41P α1AT polypeptides was tightly folded (Fig. 3A ▶), and they were unable to form inhibitory complexes with the target protease, human neutrophil elastase (Fig. 3B ▶). Instead, these protein samples were fragmented into smaller pieces. This result is consistent with a loosely packed, molten globule-like conformation for the folding intermediate, which is vulnerable to protease attacks. On incubation at 30°C, however, the abilities of these variant proteins to form inhibitory complexes increased (Fig. 3B ▶) coincidently with the formation of the tightly folded form (Fig. 3A ▶). The Z-type variant behaved similarly, whereas wild-type α1AT folded immediately on the initiation of protein folding (Fig. 3 ▶). When the folding rates were quantitatively measured by acquisition of inhibitory activity (Fig. 3C ▶), the folding rates for the D256V, L41P, and Z-type variants were 0.48, 0.03, and 0.36 h−1, respectively. On the other hand, the wild-type molecules immediately obtained full inhibitory activity upon initiation of folding. When the folding rate of the wild-type α1AT molecules was measured by stopped-flow CD and fluorescence spectroscopy, most of the secondary structures were formed within a dead time of 2 msec (data not shown), and inhibitory activity was obtained within 210 sec (Kim and Yu 1996). The kinetics of inhibitory activity gain (Fig. 3C ▶) was compatible with the conversion rates from the folding intermediate into the tightly folded form shown in Figure 3A ▶. The results show that protein folding of these α1AT variants was, indeed, extremely retarded, but they could ultimately fold into their native, functional conformations.
Figure 3.


Protein folding time course of the α1AT variants. (A) The nondenaturing polyacrylamide gel electrophoresis showing retarded folding of α1AT variant proteins. Unfolded α1AT polypeptides were refolded at 30°C for the indicated times as described in Materials and Methods, and analyzed by nondenaturing gel electrophoresis. Proteins were visualized by staining with Coomassie brilliant blue. Migration positions of the native form, folding intermediate, and dimer are indicated. (B) Inhibitory complex formation during incubation of the refolded α1AT variants. Refolded α1AT variant proteins were sampled during the folding time course and incubated with human leukocyte elastase at a molar ratio of 1 : 0.2 (α1AT proteins to protease) for 10 min at 37°C. The reaction products were analyzed by 10% SDS-polyacrylamide gel electrophoresis, and proteins were visualized by staining with Coomassie brilliant blue. Migration positions of the inhibitory complex, intact α1AT, and the RSL-cleaved α1AT are indicated. MW, Molecular weight makers (Bio-Rad Co.; low range; 97.4, 66.2, 45, and 31 kD from the top); AT, the purified wild-type α1AT protein; wt, the refolded wild-type α1AT incubated for various times. (C) Measurement of refolding rates. The refolding rates to the native form during incubation of α1AT variants at 30°C were followed by the gain of inhibitory activity. Refolded α1AT variant proteins were sampled during the folding time course and incubated with 1 pmole of porcine pancreatic elastase for 10 min at 37°C. The residual enzyme activity was determined using 1 mM N-succinyl-Ala-Ala-Ala-p-nitroanilide as a substrate. The experimental data were fitted to a single exponential rise. (•) The wild-type α1AT, (○) D256V α1AT, (□) Z-type α1AT, (▵) L41P α1AT.
Stability of folded α1AT variants is comparable to the wild type
Because the inhibitory activity of the above α1AT variants increased gradually with time, a slow folding rate was implicated as the primary defect of these dysfunctional variants, and the stability and function of the folded native α1AT proteins might not be seriously impaired. To enrich the native form of α1AT variants sufficiently for detailed characterization, refolded α1AT proteins were incubated at 30°C for 1 d (for D256V and Z-type variants) or 2 d (for L41P). The native forms of the variant α1ATs were purified by FPLC and compared with that of the wild-type molecule. The thermostability of each variant during heat treatment was followed by nondenaturing gel electrophoresis. Surprisingly, the thermostability of these α1AT variants was comparable to that of the wild-type α1AT (Fig. 4 ▶). Neither the variant α1ATs nor the wild-type protein were observed to polymerize upon heat treatment to 40°C. Noticeable amounts of polymers were first detected upon heat treatment at 45°C for variant α1ATs and at 50°C for the wild-type protein.
Figure 4.

Thermostability of the native α1AT variants. The folded α1AT variant proteins were purified and heat-treated for 30 min at various temperatures ranging from 30°C to 50°C. The protein in each reaction was 4 μg in 30 μL of buffer (10 mM phosphate, 50 mM NaCl at pH 6.5). The degree of polymerization was analyzed on nondenaturing gels containing 10% polyacrylamide. Lane C contains nonheated α1AT proteins. Migration positions of the monomer and dimer of α1AT molecules are indicated.
The conformational stability of the folded α1AT variants was quantified by equilibrium unfolding in the presence of urea. When monitored at 360 nm by fluorescence spectroscopy, emission fluorescence intensity drastically increases upon transition from the native form to an unfolding intermediate (corresponding to the first unfolding transition on transverse urea gradient gels). The unfolding transition midpoint was 1.8 M urea for the wild-type protein. The native D256V and L41P α1AT proteins showed unfolding transition midpoints at 1.2 M and 1.3 M urea, corresponding to a free energy change (ΔΔG) of 1.0 and 0.9 kcal/mole, respectively. The Z-type variant has a mutation (Glu 342 → Lys) located at the head of β-sheet A, very close to Trp 194, which is responsible for the intrinsic fluorescence of native α1AT (Tew and Bottomley 2001). Because of the effect of the E342K mutation on the microenvironment near Trp 194, the fluorescence intensity of the native Z-type α1AT was almost as high as that of the unfolding intermediate, and unfolding transition of the native Z-type protein could not be monitored by intrinsic fluorescence changes. Therefore, the conformational stability of this variant was measured by equilibrium unfolding using circular dichroism spectroscopy. The native Z-type protein unfolded with a transition midpoint of 1.2 M urea, compared with 1.8 M urea for the wild-type α1AT. The results of the thermostability and the conformational stability examinations were consistent with each other and indicate that the stability of the folded variant proteins were only marginally decreased as compared with that of the wild-type α1AT.
Inhibitory activity of folded α1AT variants is comparable to the wild type
The inhibitory activities of the native forms of the α1AT variants were examined by monitoring formation of a covalently bound, inhibitory complex with a target protease, porcine pancreatic elastase. The folded α1AT variants formed amounts of SDS-resistant inhibitory complexes comparable to that of wild-type α1AT (Fig. 5 ▶). Consistent with previous studies (Lee et al. 1998; Im and Yu 2000), slightly more portions of α1AT molecules partitioned into the cleaved form when incubated with porcine pancreatic elastase, compared with those incubated with human neutrophil elastase. When the ratios of α1AT present in inhibitor–protease complexes to α1AT cleaved by the protease were compared, the variant α1AT molecules showed a slight increase in cleavage as compared with the wild-type protein, especially for Z-type variant proteins. These results may have been caused by the slightly diminished inhibitory activities of the variant proteins or by small amounts of residual folding intermediates remaining in the preparation. However, these results confirm that the inhibitory activity of the native conformation, once formed, is not significantly affected by these mutations.
Figure 5.

Inhibitory complex formation by the folded α1AT variant proteins with elastase. The native wild-type (wt), D256V, L41P, and Z-type (Z) α1AT proteins were incubated with porcine pancreatic elastase at the indicated molar ratios of the protease to α1AT (E/I ratios). After incubation for 10 min at 37°C in the assay buffer (30 mM phosphate, 160 mM NaCl, 0.1% PEG 8000, 0.1% Triton X-100 at pH 7.4), formation of the SDS-resistant α1AT–elastase complex was analyzed by 10% SDS-polyacrylamide gel electrophoresis. The protein bands were visualized by staining with Coomassie brilliant blue. Migration positions of the inhibitory complex, intact α1AT, the RSL-cleaved α1AT, and elastase are indicated. MW, Molecular weight makers (Bio-Rad Co.; low range; 97.4, 66.2, 45, and 31 kD from the top); PPE, porcine pancreatic elastase.
Discussion
Retarded folding of D256V and L41P variants may cause α1AT deficiency
We have examined the detailed structural basis for the deficiency of a dozen α1AT variants. Unlike most α1AT variants, the D256V and L41P α1AT polypeptides initially fold into a loosely packed intermediate, and then convert spontaneously into the native conformation under physiological conditions (Figs. 2 ▶, 3 ▶). Because the native form of these variants, once folded, has comparable stability and inhibitory activity (Figs. 4 ▶, 5 ▶), extremely slow folding is indicated as a cause for α1AT deficiency by D256V and L41P variants.
Although the L41P α1AT protein level in plasma is not sufficient to protect the lower respiratory tract, the variant protein in plasma exhibits inhibitory activity comparable to that of wild-type α1AT (association rate constants of 7.0 × 106 and 9.3 × 106 M−1 sec−1, respectively, for human neutrophil elastase), and the half-life of this variant in plasma is similar to that of the wild-type α1AT (Takahashi et al. 1988). The low plasma level of this protein was puzzling, given the normal behavior of plasma L41P α1AT protein and its normal promoter region regulating α1AT gene expression (Takahashi et al. 1988). There is a possibility that the extremely retarded folding of L41P polypeptide causes accumulation of folding intermediates, which are prone to aggregation and clearance at the site of biosynthesis, rather than being secreted into plasma as the functional, native form. The comparable stability and activity of the folded D256V protein indicate that slow folding may cause the deficiency of this variant as well. Retarded protein folding has also been suggested as the predominant cause of aggregation of Z-type α1AT in liver cells (Yu et al. 1995). However, the plasma form of Z-type α1AT protein exhibited a reduced association rate constant with neutrophil elastase (1.2 × 107 M−1 sec−1 for Z-type versus 5.3 × 107 M−1 sec−1 for wild-type α1AT; Lomas et al. 1993b). The increased cleavage of Z-type variant molecules as compared with wild-type α1AT molecules (Fig. 5 ▶) is consistent with this observation. Many dysfunctional α1AT proteins easily adopt a loop–sheet polymeric conformation, and especially a high body temperature during inflammation is likely to exacerbate α1AT polymerization and develop the clinical symptoms of α1AT deficiency (Lomas et al. 1992). However, the native form of slow-folding α1AT variants has comparable thermostability to the wild-type molecule, and remains intact for 30 min even at 40°C (Fig. 4 ▶). Therefore, these variants are likely to have a different mechanism for α1AT deficiency; that is, retarded folding accumulates folding intermediates prone to polymerization prior to being secreted into plasma.
Structural basis for retarded folding
In the native structure of wild-type α1AT (2psi.pdb; Elliott et al. 1998), Leu 41 is located on helix A (Fig. 1 ▶). Distortion of helix A by the L41P substitution may delay compact packing of secondary structures into a tightly folded tertiary structure, the last step in α1AT folding. Interestingly, the Cδ1 atom of Leu 41 interacts with the Cδ2 atom of Phe 52 at a distance of 3.5 Å, and deletion of Phe 52 also causes α1AT deficiency (Lomas et al. 1995). Asp 256 is located in the turn between s3B (the third strand in β-sheet B) and helix G, and forms a salt bridge to His 231 in s1B. Loss of the salt bridge by the D256V substitution might affect the folding rate. Likewise, in the Z-type variant (Glu 342 → Lys), a salt bridge between Glu 342 and Lys 290 is broken, and the charge repulsion between the newly introduced Lys 342 and Lys 290 is likely to interfere with normal packing.
Conformational versatility and disease
This study has provided significant insight into the structure–function relationships of serpins. The native state of serpins is not their most thermodynamically stable state, but is a strained metastable state. The native metastability of these proteins is important for regulating their biological functions (Wiley and Skehel 1987; Huber and Carrell 1989; Stein and Carrell 1995; Wright and Scarsdale 1995; Carr et al. 1997), and the final, stable state of these proteins is normally reached only when the function is executed. Therefore, the metastability is probably needed for facile conformational conversion during formation of the inhibitory complex with a protease (Lee et al. 2000). However, the structural aspects that cause the native metastability of inhibitory serpins also expose serpin proteins to easily adopt several abnormal conformations, such as the RSL-inserted latent form and loop–sheet polymers. Structural conversion to these inactive, stable conformations seems to occur at physiologically significant rates in many deficient serpin variants.
The precise site and nature of the mutations determine etiology of the serpin variants. Destabilizing α1AT substitutions in the “shutter” domain, such as V55P and S53F, also accumulated as intermediate-like species with low mobility, as determined by transverse urea-gradient gel electrophoresis. However, they did not convert into the native state upon prolonged incubation; rather, they formed protein aggregates (Fig. 2 ▶). Therefore, instability of the native proteins appears to be the main cause of inhibitory deficiency for these α1AT variants. A previous study suggested that the hydrophobic region underneath β-sheet A is overpacked, because mutations that reduce the size of the side chain in this region, for example, F51L, stabilize the α1AT molecule (Kwon et al. 1994). Indeed, Siiyama (Ser 53 → Phe), which increases the volume of the side chain in this region, induces conformational instability and subsequent polymerization. A genetic variant of α1-antichymotrypsin (L55P) accumulated as a species that may be an intermediate in the formation of the latent state (Gooptu et al. 2000), as well as in the native and latent forms. However, V55P variation of α1AT, which is equivalent to L55P substitution of α1-antichymotrypsin, did not accumulate as a similar intermediate or as the latent form, indicating that subtle structural differences among inhibitory serpins affect the etiology of serpin deficiency. Unlike most α1AT variants, whose major defect may be conformational instability of the native form, leading to exacerbated protein polymerization, the D256V, L41P, and Z-type variants suffer from retarded folding, although once folded, their native form behaves quite normally. Retarded folding arrests the polypeptide at the stage of an incompletely folded intermediate that cannot be properly channeled through the secretory pathways. This phenomenon may lead to accumulation of the intermediate, which has a high tendency to polymerize, leading to hepatic inclusions. Retarded protein folding seems to be a recurrent theme associated with dysfunctional α1AT variants, because three of 12 α1AT variants tested displayed retarded folding (Fig. 1 ▶). It will be of interest in future studies to identify other types of variant proteins that adopt versatile conformations.
Materials and methods
Chemicals
Ultrapure urea was purchased from ICN Biochemicals. Porcine pancreatic elastase, human leukocyte elastase, and N-succinyl-(Ala)3-p-nitroanilide were purchased from Sigma. All other chemicals were reagent grade.
Mutagenesis, expression, and purification of recombinant α1AT
Substitution mutations were introduced on pFEAT30, the plasmid for α1AT expression in Escherichia coli (Kwon et al. 1994), by oligonucleotide-directed mutagenesis (Kunkel et al. 1987). Recombinant α1AT was expressed as inclusion bodies in E. coli and refolded as described previously (Kwon et al. 1994). Either immediately or after incubation at 30°C for the indicated time, α1AT proteins were quickly purified on a Q Sepharose Fast Flow column (2.5 × 0.7 cm; Amersham Bioscience Ltd.) in 10 mM phosphate, 1 mM β-mercaptoethanol, and 1 mM EDTA (pH 6.5). This purification process takes less than 1 h at 4°C. To characterize the native conformation of α1AT variants, it was enriched by incubation at 30°C, and the folded conformation was purified by ion exchange chromatography by FPLC on a ResourceQ column (Amersham Bioscience Ltd.) in 10 mM phosphate, 1 mM β-mercaptoethanol, and 1 mM EDTA (pH 6.5). Concentrations of α1AT were determined in 6 M guanidine hydrochloride using a value of A1 cm1% = 4.3 at 280 nm, calculated from the tyrosine and tryptophan content of the α1AT protein (Edelhoch 1967) and based upon Mr = 44,250 (Kwon et al. 1994).
Conformational analysis by gel electrophoresis
Conformation of α1AT proteins was analyzed by transverse urea gradient gel electrophoresis (Goldenberg 1989). Transverse urea gradient gels were prepared with a gradient of 0~8 M urea perpendicular to the direction of electrophoresis with an opposing gradient of acrylamide from 15% to 11%. Four slab gels (100 × 80 mm) were prepared simultaneously in a multigel caster (Hoefer Scientific Instruments) by using a gradient maker and a single-channel peristaltic pump. The α1AT protein (20 μg in 100 μL) was applied across the top of the gel. The electrode buffer was 50 mM Tris-acetate, and 1 mM EDTA (pH 7.5). The gels were run at a constant current of 6 mA for 3 h at a controlled temperature of 20°C. The protein bands were visualized by staining with Coomassie brilliant blue.
Folding of α1AT proteins was followed using 10% nondenaturing polyacrylamide gel with Tris-glycine buffer system. Inclusion bodies from E. coli cells overexpressing α1AT proteins were dissolved in 8 M urea in a buffer (50 mM Tris-Cl at pH 8, 50 mM NaCl, 1 mM EDTA, and 1 mM β-mercaptoethanol), and refolded by dilution 10-fold in 10 mM phosphate (pH 6.5), 1 mM EDTA, and 1 mM β-mercaptoethanol. Refolded α1AT protein was incubated at 30°C for various times, and their conformation was analyzed by nondenaturing gel electrophoresis.
Thermostability
To measure thermostability of α1AT variants, purified α1AT protein was incubated for 30 min at various temperatures ranging from 30°C to 50°C. The protein concentration was 0.15 mg/mL in a buffer (10 mM phosphate, 50 mM NaCl, 1 mM EDTA, and 1 mM β-mercaptoethanol at pH 6.5). Disappearance of monomers and formation of oligomers of α1AT molecules were monitored on 10% nondenaturing gels containing Tris-glycine buffer system.
Denaturant-induced unfolding transition
To measure the stability of α1AT variants, equilibrium unfolding of the native α1AT as a function of urea (ICN Biomedicals, Inc.) was monitored by changes in intrinsic fluorescence of α1AT. The protein concentration was 10 μg/mL in a buffer (10 mM phosphate, 50 mM NaCl, 1 mM EDTA, and 1 mM β-mercaptoethanol at pH 6.5). The native protein was incubated in the buffer containing various concentrations of urea for 2 h at 25°C. Equilibrium unfolding was monitored by fluorescence spectroscopy (λex = 280 nm and λem = 360 nm, excitation and emission slit widths = 5 nm for both), using a Shimadzu RF-5301PC spectrophotometer as described previously (Kwon et al. 1994; Lee et al. 1996). Intrinsic fluorescence emission at 360 nm markedly increases upon transition from the native form to an unfolding intermediate (the first transition), but remains almost constant during transition from the unfolding intermediate to the unfolded state (the second transition). Therefore, experimental data of the fluorescence measurement at 360 nm were fitted to a two-state unfolding model to measure stability of the native form. For the Z-type variant, unfolding of the native form was monitored at 25°C by circular dichroism spectroscopy at 220 nm using a Jasco J715 spectropolarimeter. The protein concentration was 10 μg/mL in a buffer (10 mM phosphate, 50 mM NaCl at pH 6.5) containing various concentrations of urea.
Determination of the inhibitory activity
During the refolding time course, inhibitory activity of α1AT variants was followed by monitoring the ability to form SDS-stable complexes with human leukocyte elastase. Active concentration of human leukocyte elastase was determined by trypsin-titration reactions as described (Hopkins et al. 1993). For this, 4 μg of α1AT samples from refolding time courses was incubated with human leukocyte elastase at a molar ratio of 1 : 0.2 (α1AT : protease) for 10 min at 37°C. The buffer was 30 mM phosphate (pH 7.4), 160 mM NaCl, 0.1% PEG 6000, and 0.1% Triton X-100. The inhibitory complex formation of α1AT proteins with target proteases was examined by 10% SDS-polyacrylamide gel electrophoresis.
The refolding rates to the native form during incubation of α1AT variants at 30°C were followed by the gain of inhibitory activity. Active concentration of porcine pancreatic elastase, a target protease, was determined by measuring the initial rates of hydrolysis of 1 mM N-succinyl-Ala-Ala-Ala-p-nitroanilide (Bieth et al. 1974). α1AT samples were taken at various time points and incubated with 1 pmole of the protease in 50 μL of elastase assay buffer (30 mM phosphate, 160 mM NaCl, 0.1% PEG 6000, and 0.1% Triton X-100 at pH 7.4). After incubation with the protease for 10 min at 37°C, the reaction mixture was diluted 20-fold with the assay buffer, and the residual enzyme activity was determined. The experimental data were fitted to a single exponential rise.
To measure inhibitory activity of the native form, 4 μg of purified folded α1AT protein was incubated with porcine pancreatic elastase at various molar ratios for 10 min at 37°C. Appearance of the SDS-resistant α1AT–proteinase complex was monitored by 10% SDS-polyacrylamide gel electrophoresis. The protein bands were visualized by staining with Coomassie brilliant blue.
Acknowledgments
This work was supported by Korea Research Foundation Grant (KRF-2002-015-CP0343).
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Article published online ahead of print. Article and publication date are at http://www.proteinscience.org/cgi/doi/10.1110/ps.03356604.
References
- Aertgeerts, K., De Bondt, H.L., De Ranter, C.J., and Declerck, P.J. 1995. Mechanism contributing to the conformational and functional flexibility of plasminogen activator inhibitor-1. Nat. Struct. Biol. 2 891–897. [DOI] [PubMed] [Google Scholar]
- Bieth, J., Spiess, B., and Wermuth, C.G. 1974. The synthesis and analytical use of a highly sensitive and convenient substrate of elastase. Biochem. Med. 11 350–357. [DOI] [PubMed] [Google Scholar]
- Bruch, M., Weiss, V., and Engel, J. 1988. Plasma serine proteinase inhibitors (serpins) exhibit major conformational changes and a large increase in conformational stability upon cleavage at their reactive sites. J. Biol. Chem. 263 16626–16630. [PubMed] [Google Scholar]
- Carr, C.M., Chaudhry, C., and Kim, P.S. 1997. Influenza hemagglutinin is spring-loaded by a metastable native conformation. Proc. Natl. Acad. Sci. 94 14306–14313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrell, R.W., Jeppsson, J.-O., Laurell, C.-B., Brennan, S.O., Owen, M.C., Vaughan, L., and Boswell, D.R. 1982. Structure and variation of human α1-antitrypsin. Nature 298 329–334. [DOI] [PubMed] [Google Scholar]
- Edelhoch, H. 1967. Spectroscopic determination of tryptophan and tyrosine in proteins. Biochemistry 6 1948–1954. [DOI] [PubMed] [Google Scholar]
- Elliott, P.R., Lomas, D.A., Carrell, R.V., and Abrahams, J.P. 1996. Inhibitory conformation of the reactive loop of α1-antitrypsin. Nat. Struct. Biol. 3 676–681. [DOI] [PubMed] [Google Scholar]
- Elliott, P.R., Abrahams, J.P., and Lomas, D.A. 1998. Wild-type α1-antitrypsin is in the canonical inhibitory conformation. J. Mol. Biol. 275 419–425. [DOI] [PubMed] [Google Scholar]
- Eriksson, S., Carlson, J., and Velez, R. 1986. Risk of cirrhosis and primary liver cancer in α1-antitrypsin deficiency. New Engl. J. Med. 314 736–739. [DOI] [PubMed] [Google Scholar]
- Gils, A., Knockaert, I., and Declerck, P.J. 1996. Substrate behavior of plasminogen activator inhibitor-1 is not associated with a lack of insertion of the reactive site loop. Biochemistry 35 7474–7481. [DOI] [PubMed] [Google Scholar]
- Goldenberg, D.P. 1989. Analysis of protein conformation by gel electrophoresis. In Protein structure: A practical approach (ed. T.E. Creighton), pp. 225–250. IRL Press at Oxford University Press, Oxford, UK.
- Gooptu, B., Hazes, B., Chang, W.S., Dafform, T.R., Carrell, R.W., Read, R.J., and Lomas, D.A. 2000. Inactive conformation of the serpin α1-antichymotrypsin indicates two-stage insertion of the reactive loop: Implications for inhibitory function and conformational disease. Proc. Natl. Acad. Sci. 97 67–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hekman, C.M. and Loskutoff, D.J. 1985. Endothelial cells produce a latent inhibitor of plasminogen activators that can be activated by denaturants. J. Biol. Chem. 260 11581–11587. [PubMed] [Google Scholar]
- Hopkins, P.C.R., Carrell, R.W., and Stone, S.R. 1993. Effects of mutations in the hinge region of serpins. Biochemistry 32 7650–7657. [DOI] [PubMed] [Google Scholar]
- Huber, R. and Carrell, R.W. 1989. Implications of the three-dimensional structure of α1-antitrypsin for structure and function of serpins. Biochemistry 28 8951–8966. [DOI] [PubMed] [Google Scholar]
- Huntington, J.A., Fan, B., Karlsson, K.E., Deinum, J., Lawrence, D.A., and Gettins, P.G.W. 1997. Serpin conformational change in ovalbumin: Enhanced reactive center loop insertion through hinge region mutations. Biochemistry 36 5432–5440. [DOI] [PubMed] [Google Scholar]
- Huntington, J.A., Read, R.J., and Carrell, R.W. 2000. Structure of a serpin–protease complex shows inhibition by deformation. Nature 407 923–926. [DOI] [PubMed] [Google Scholar]
- Im, H. and Yu, M.-H. 2000. Role of Lys335 in the metastability and function of inhibitory serpins. Protein Sci. 9 934–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Im, H., Seo, E.J., and Yu, M.-H. 1999. Metastability in the inhibitory mechanism of human α1-antitrypsin. J. Biol. Chem. 274 11072–11077. [DOI] [PubMed] [Google Scholar]
- Johnson, D. and Travis, J. 1978. Structural evidence for methionine at the reactive site of human α1-proteinase inhibitor. J. Biol. Chem. 253 7142–7144. [PubMed] [Google Scholar]
- Jung, C.-H. and Im, H. 2003. A recombinant human α1-antitrypsin variant, Mmalton, undergoes a spontaneous conformational conversion into the latent form. J. Microbiol. 41 335–339. [Google Scholar]
- Kim, D.Y. and Yu, M.-H. 1996. Folding pathway of human α1-antitrypsin: Characterization of an intermediate that is active but prone to aggregation. Biochem. Biophy. Res. Comm. 226 378–384. [DOI] [PubMed] [Google Scholar]
- Kunkel, T.A., Roberts, J.D., and Zakour, R.A. 1987. Rapid and efficient site-specific mutagenesis without phenotypic selection. Methods Enzymol. 154 367–382. [DOI] [PubMed] [Google Scholar]
- Kwon, K.-S., Kim, J., Shin, H.S., and Yu, M.-H. 1994. Single amino acid substitutions of α1-antitrypsin that confer enhancement in thermal stability. J. Biol. Chem. 269 9627–9631. [PubMed] [Google Scholar]
- Lee, K.N., Park, S.D., and Yu, M.-H. 1996. Probing the native strain in α1-antitrypsin. Nat. Struct. Biol. 3 497–500. [DOI] [PubMed] [Google Scholar]
- Lee, K.N., Im, H., Kang, S.W., and Yu, M.-H. 1998. Characterization of a human α1-antitrypsin variant that is as stable as ovalbumin. J. Biol. Chem. 273 2509–2516. [DOI] [PubMed] [Google Scholar]
- Lee, C., Park, S.-H., Lee, M.-Y., and Yu, M.-H. 2000. Regulation of protein function by native metastability. Proc. Natl. Acad. Sci. 97 7727–7731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loebermann, H., Tokuoka, R., Deisenhofer, J., and Huber, R. 1984. Human α1-antitrypsin: Crystal structure analysis of two crystal modifications, molecular model and preliminary analysis of the implications for function. J. Mol. Biol. 177 531–556. [PubMed] [Google Scholar]
- Lomas, D.A., Evans, D.L., Finch, J.T., Seyama, K., and Carrell, R.W. 1992. The mechanism of Z α1-antitrypsin accumulation in the liver. Nature 357 605–607. [DOI] [PubMed] [Google Scholar]
- Lomas, D.A., Finch, J.T., Seyama, K., Nukiwa, T., and Carrell, R.W. 1993a. α1-Antitrypsin Siiyama (Ser53 → Phe): Further evidence for intracellular loop–sheet polymerization. J. Biol. Chem. 268 15333–15335. [PubMed] [Google Scholar]
- Lomas, D.A., Evans, D.L., Stone, S.R., Chang, W.S., and Carrell, R.W. 1993b. Effect of the Z mutation on the physical and inhibitory properties of α1-antitrypsin. Biochemistry 32 500–508. [DOI] [PubMed] [Google Scholar]
- Lomas, D.A., Elliott, P.R., Sidhar, S.K., Foreman, R.C., Finch, J.T., Cox, D.W., Whisstock, J.C., and Carrell, R.W. 1995. α1-Antitrypsin Mmalton (Phe52-deleted) forms loop–sheet polymers in vivo: Evidence for the C sheet mechanism of polymerization. J. Biol. Chem. 270 16864–16870. [DOI] [PubMed] [Google Scholar]
- Lukacs, C.M., Zhong, J.Q., Plotnick, M.I., Rubin, H., Cooperman, B.S., and Christianson, D.W. 1996. Arginine substitutions in the hinge region of antichymotrypsin affect serpin β-sheet rearrangement. Nat. Struct. Biol. 3 888–893. [DOI] [PubMed] [Google Scholar]
- Mahadeva, R., Chang, W.S., Dafforn, T.R., Oakley, D.J., Foreman, R.C., Calvin, J., Wight, D.G., and Lomas, D.A. 1999. Heteropolymerization of S, I, and Z α1-antitrypsin and liver cirrhosis. J. Clin. Invest. 103 999–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mottonen, J., Strand, A., Symersky, J., Sweet, R.M., Danley, D.E., Geoghegan, K.F., Gerard, R.D., and Goldsmith, E.J. 1992. Structural basis of latency in plasminogen activator inhibitor-1. Nature 355 270–273. [DOI] [PubMed] [Google Scholar]
- Owen, M.C., Brennan, S.O., Lewis, J.H., and Carrell, R.W. 1983. Mutation of antitrypsin to antithrombin: α1-Antitrypsin Pittsburgh (358 Met leads to Arg), a fatal bleeding disorder. New Engl. J. Med. 309 694–698. [DOI] [PubMed] [Google Scholar]
- Plotnick, M.I., Mayne, L., Schecter, N.M., and Rubin, H. 1996. Distortion of the active site of chymotrypsin complexed with a serpin. Biochemistry 35 7586–7590. [DOI] [PubMed] [Google Scholar]
- Stein, P.E. and Carrell, R.W. 1995. What do dysfunctional serpins tell us about molecular mobility and disease? Nat. Struct. Biol. 2 96–113. [DOI] [PubMed] [Google Scholar]
- Stratikos, E. and Gettins, P.G.W. 1997. Major proteinase movement upon stable serpin–proteinase complex formation. Proc. Natl. Acad. Sci. 94 453–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi, H., Nukiwa, T., Satoh, K., Ogushi, F., Brantly, M., Fells, G., Stier, L., Courtney, M., and Crystal, R.G. 1988. Characterization of the gene and protein of the α1-antitrypsin “deficiency” allele Mprocida. J. Biol. Chem. 263 15528–15534. [PubMed] [Google Scholar]
- Tew, D.J. and Bottomley, S.P. 2001. Probing the equilibrium denaturation of the serpin α1-antitrypsin with single tryptophan mutants: Evidence for structure in the urea unfolded state. J. Mol. Biol. 313 1161–1169. [DOI] [PubMed] [Google Scholar]
- Wang, Z., Mottonen, J., and Goldsmith, E.J. 1996. Kinetically controlled folding of the serpin plasminogen activator inhibitor 1. Biochemistry 35 16443–16448. [DOI] [PubMed] [Google Scholar]
- Wiley, D.C. and Skehel, J.J. 1987. The structure and function of the hemagglutinin membrane glycoprotein of influenza virus. Ann. Rev. Biochem. 56 365–394. [DOI] [PubMed] [Google Scholar]
- Wright, H.T. and Scarsdale, J.N. 1995. Structural basis for serpin inhibitor activity. Proteins 22 210–225. [DOI] [PubMed] [Google Scholar]
- Yu, M.-H., Lee, K.N., and Kim, J. 1995. The Z type variation of human α1-antitrypsin causes a protein folding defect. Nat. Struct. Biol. 2 363–367. [DOI] [PubMed] [Google Scholar]