

Abstract
Antimicrobial peptides are key components of the innate immune response in most multicellular organisms. These molecules are considered as one of the most innovative class of anti-infective agents that have been discovered over the last two decades, and therefore, as a source of inspiration for novel drug design. Insect cystein-rich antimicrobial peptides with the CSαβ scaffold (an α-helix linked to a β-sheet by two disulfide bridges) represent particularly attractive templates for the development of systemic agents owing to their remarkable resistance to protease degradation. We have selected heliomicin, a broad spectrum antifungal CSαβ peptide from Lepidoptera as the starting point of a lead optimization program based on phylogenic exploration and fine tuned mutagenesis. We report here the characterization, biological activity, and 3D structure of heliomicin improved analogs, namely the peptides ARD1, ETD-135, and ETD-151. The ARD1 peptide was initially purified from the immune hemolymph of the caterpillars of Archeoprepona demophoon. Although it differs from heliomicin by only two residues, it was found to be more active against the human pathogens Aspergillus fumigatus and Candida albicans. The peptides ETD-135 and ETD-151 were engineered by site-directed mutagenesis of ARD1 in either cationic or hydrophobic regions. ETD-135 and ETD-151 demonstrated an improved antifungal activity over the native peptides, heliomicin and ARD1. A comparative analysis of the 3D structure of the four molecules highlighted the direct impact of the modification of the amphipathic properties on the molecule potency. In addition, it allowed to characterize an optimal organization of cationic and hydrophobic regions to achieve best antifungal activity.
Keywords: insect immunity, antifungal, antimicrobial, heliomicin, NMR structure
The growing problem of resistance of microorganisms to current antibiotics has fostered the development of innovative approaches to antimicrobial therapy (Breithaupt 1999). In the antifungal field, the need for novel drugs has also been driven by the expanding population of immunocompromised patients at risk of invasive fungal infections (De Lucca and Walsh 1999; Dixon and Edwards 2000). The quest for new antifungals is even more critical, given the dearth of existing safe and effective compounds. In that regard, natural antimicrobial peptides offer entirely new opportunities for the development of therapeutic agents (Hoffmann et al. 1999; Zasloff 2002).
Antimicrobial peptides are key components of the innate immune system, and are widely distributed among multicellular organisms (Bulet et al. 1999). The last two decades have witnessed the discovery of an impressive number of antimicrobial structures reported from the animal and plant kingdoms. They are small cationic amphipathic proteins comprising 20 to 50 amino acids, with a broad spectrum of activity against bacterial and fungal cells (Hoffmann et al. 1999). Undeniably, insects offer the largest and most diversified source of cationic antimicrobial peptides (Dimarcq et al. 1998). In most insects the hallmark of the humoral response to a septic challenge is the rapid synthesis of antimicrobial peptides by the fat body and systemic release of these factors into the hemolymph. Paramount among these peptides are a family of compact cysteine-rich molecules that harbor the CSαβ fold (Hoffmann 1995). This motif, consisting of a single α-helix connected to a twisted antiparallel β-sheet by two disulfide bridges, confers to the peptide a highly compact structure and a remarkable resistance to protease degradation. The CSαβ motif is also found in peptides with other functions including toxins (Bontems et al. 1991), protease inhibitors (Zhao et al. 2002), and elicitor of the sweet-taste response (Caldwell et al. 1998), and served as a template for protein engineering (Vita et al. 1998).
Insect defensins are CSαβ antibacterial peptides with activity against Gram-positive bacteria (Matsuyama and Natori 1988; Lambert et al. 1989). They are widely distributed among phylogenetically diverse insect orders, and have also been found in scorpions (Cociancich et al. 1993) and in molluscs (Yang et al. 2000). By contrast to the wealth of antibacterial insect defensins, only three antifungal CSαβ peptides have been characterized in insects: drosomycin, termicin, and heliomicin. Drosomycin was the first inducible antifungal peptide to be isolated from an insect species (Fehlbaum et al. 1994). To date, it has only been reported in Drosophila, where it represents one of the major peptides produced during the immune response. Several gene sequences coding for drosomycin analogues have now been identified in the Drosophila genome, but the exact function of the gene products has not been yet elucidated (Khush and Lemaitre 2000). Interestingly, drosomycin exhibits striking similarities with cysteine rich antifungal peptides found in plants (Landon et al. 1997). Termicin was characterized from the termite Pseudacanthotermes spiniger (Lamberty et al. 2001b). It is constitutively produced and stored in hemocyte granules and only released into the hemolymph after immune challenge. Heliomicin was isolated from the immune hemolymph of the lepidopteran Heliothis virescens (Lamberty et al. 1999). The structure of this 44-residue peptide displays a marked dichotomy in the positioning of hydrophobic and cationic clusters at the surface of the molecule (Lamberty et al. 2001a).
Heliomicin exhibits a broad spectrum of antifungal activity against fungal strains responsible for severe nosocomial diseases (Lamberty et al. 2001a) and is not toxic to human cells. In addition, its activity is retained at physiological ionic strength that makes it a good candidate for further lead optimization. This report describes the characterization of new heliomicin analogs, both natural and laboratory-designed, with significantly improved biological activity over the parent molecule. One of these peptides, called ARD1, was isolated from the larvae of the Lepidoptera Archeoprepona demophoon. Although ARD1 differs from heliomicin by only two residues, it displays enhanced activity against the two human pathogens A. fumigatus and Candida albicans, including C. albicans strains resistant to fluconazole. We have determined the three-dimensional structure of ARD1 and compared the 3D structures and the surface properties of heliomicin and ARD1. Based on these comparisons, we designed new analogs, in which the hydrophobic and/or the cationic regions of ARD1 were modified (Fig. 1 ▶). Among these analogs, we selected ETD135 and ETD151 for their particularly interesting spectrum of activity, and therefore, their potential as new agents for therapeutic applications. We have determined the three-dimensional structure of ETD135 and ETD151, and compared their molecular properties to those deduced from the previously determined solution structure of heliomicin and ARD1, in an attempt to explain the differences in the antifungal activities.
Figure 1.
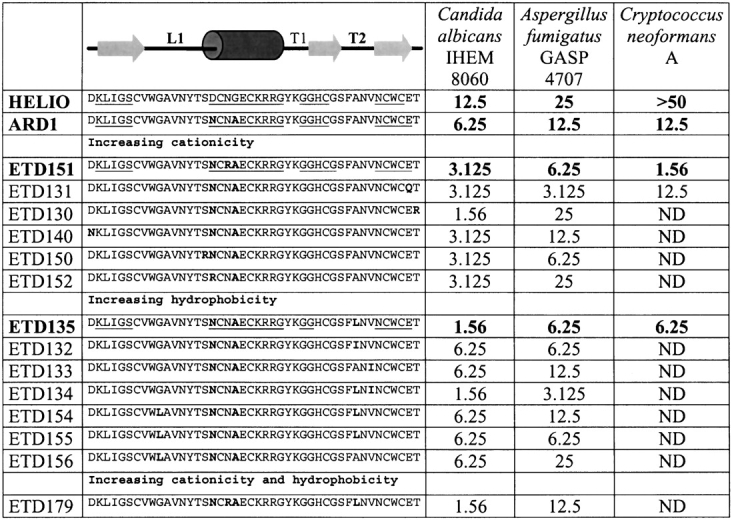
Sequence alignment and antifungal activity: Heliomicin, ARD1, and the 14 analogs designed to increase the hydrophobicity and/or the cationicity of the peptides. Sequence differences are in bold, and the secondary structure elements of heliomicin, ARD1, ETD135, and ETD151 are underlined. Activities against C. albicans, A. fumigatus, and C. neoformans (MIC50, see text) are expressed in μg/mL (ND: not determined).
Results
Isolation of the ARD1 peptide
Eighty fourth instar larvae of A. demophoon provided 13 mL of hemolymph. After reverse-phase HPLC purification, the fraction eluting at 27% acetonitrile displayed the strongest antifungal activity. Purification to homogeneity in two extra HPLC steps leads to an isolated compound with a molecular mass of 4803.4 found by MALDI-TOF-MS. Reduction and S-Pyridylethylation of the antifungal molecule enabled the identification of 6 cysteine residues. Edman degradation of the S-pyridylethylated peptide provided the sequence displayed on Figure 1 ▶. ARD1 sequence differs from that of heliomicin by two residues (D17N and G20A), and can thus be classified in the same group of antifungal peptides.
Expression and purification of recombinant ARD1 and its analogs
Recombinant peptides were expressed in the yeast Saccharomyces cerevisiae. Constructs obtained after modification of the pSEA2 plasmid to include the different mutations (in particular D17N and G20A for ARD1; D17N, G20A, and A36L for ETD135; D17N, N19R, and G20A for ETD151) were controlled by sequencing. The usage of these constructs, coding for fusion proteins carrying at its N termini the pre-BGL2 and the pro sequence derived from the precursor of the yeast pheromone mating factor α1, allowed the secretion of the biologically active peptides directly in the broth medium. Production and purification of the peptides from broth supernatants were conducted as described in Materials and Methods. The integrity and purity of the compounds were assessed by MALDI-TOF, and the results were in perfect agreement with the expected theoretical masses. Fermentation in shake flasks provided enough material for activity evaluation of all analogs. Peptides chosen to undergo the NMR structural study (Heliomicin, ARD1, ETD135, and ETD151) were produced in fermentor. Final yields after purification were very satisfactory, ranging from 3 to 8 mg/L of culture broth depending on the peptide.
In vitro antifungal activities
ARD1 proves to be twice as potent as heliomicin against both C. albicans and A. fumigatus (Table 1). This enhanced activity arises from two mutations (D17N and G20A), both located in the helical part of heliomicin. Fourteen mutants of ARD1 were designed to increase the positive charge of the cationic pole (helix and C terminus) and/or the hydrophobicity of the hydrophobic one (essentially T2 turn; Fig. 1 ▶). Most variants display an improved activity against at least one pathogen. Increasing the charge, in the helix or in the C-terminal part, allowed to improve the activity against C. albicans. ETD151, ETD131, and ETD150 are also more active against A. fumigatus. Within this series, ETD151, showing efficacy against Cryptococcus neoformans and a high level of expression, was chosen for structural analysis. Changing the hydrophobicity, especially in the T2 turn affects both the activity against C. albicans and A. fumigatus. Within this series, ETD134 is the most potent peptide against C. albicans and A. fumigatus, but was very poorly expressed, so we chose ETD135 for the structural study. ETD135 and ETD151 also present the advantage of a high potency against some rare filamentous fungi of Fusarium and Scedosporium species, for which conventional treatments, with azolated derivatives or amphtericin B, are rather ineffective (Table 1).
Table 1.
Antifungal activities of Heliomicin analogs
| MIC50 (μg/ml) | ||||||
| Compound | C. albicans IHEM 8060 | C. albicans No. 245962 | C. neoformans A | A. fumigatus GASP4707 | F. solani FFUS 8591 | S. prolificans FSSP 8591 |
| Heliomicin | 12.5 | 12.5 | >50 | 25 | ND | ND |
| ARD1 | 6.25 | 1.56 | 12.5 | 12.5 | ND | ND |
| ETD135 | 1.56 | 1.56 | 6.25 | 6.25 | 1.56 | 0.2 |
| ETD151 | 3.125 | 0.4 | 1.56 | 6.25 | 0.4 | 0.1 |
| ETD179 | 1.56 | ND | ND | 12.5 | ND | ND |
| Fluconazole | 0.25 | >64 | 4 to 8 | ND | >64 | ND |
| Itraconazole | ND | 0.25 | ND | 0.125 | >16 | ND |
| Amphotericin B | 0.03 | 0.125 | 0.25 | 0.5 | >16 | >16 |
MIC50 are defined as the concentration that inhibits 50% of fungal growth.
NOE and molecular modeling
The proton resonances of ARD1 and its analogs were assigned using sequential assignment methods developed by Wüthrich (1986). The proton resonances were quasi-totally assigned except for some η(NH2) of arginine residues, γ(OH) of serines or threonines, ζ(CH) of phenylalanines and ζ(OH) of tyrosines. Tables containing the proton chemical shifts of ARD1, ETD151, and ETD135 are available as supplementary material.
NOE peaks were picked and integrated in NMRView. A first set of about 200–300 intraresidue, sequential and easily determined longer range peaks were assigned. Additional assignments were progressively proposed during the following ARIA runs, and manually validated. The final number of NOEs (more than 15 restraints per residue) allowed to determine the 3D structure with a very good precision. The distance restraints used in the last run of ARIA calculations are detailed in Table 2. The number of ambiguous NOEs is low: below 5% for ARD1 and ETD151, and 7% for ETD135. This percentage was about 27% at the first ARIA run, indicating that nonresolved peaks were correctly assigned.
Table 2.
Summary of the data on NOE restraints and calculated structures
| NOE restraints | ARD1 | ETD151 | ETD135 |
| Intraresidue |j − i| = 0 | 371.3 | 364.0 | 325.2 |
| Sequential |j − i| = 1 | 182.7 | 213.3 | 169.6 |
| Medium range 2 ≤ |j − i| ≤ 4 | 97.7 | 121.0 | 89.7 |
| Long-range |j − i| > 5 | 195.3 | 230.7 | 139.3 |
| Total | 847 | 929 | 724 |
| Unambiguous | 806 | 888 | 694 |
| Ambiguous | 41 | 41 | 30 |
| Quality of the calculated structures | |||
| Distance violation >0.3 Å | 0 | 0 | 0 |
| Pairwise RMS differences (Å) | |||
| all-atoms | 1.90 ± 0.22 | 1.79 ± 0.14 | 2.54 ± 0.29 |
| backbone Cα atoms | 0.89 ± 0.26 | 0.78 ± 0.11 | 1.43 ± 0.25 |
| secondary structures (Cα atoms) | 0.64 ± 0.15 | 0.48 ± 0.08 | 0.79 ± 0.12 |
| Helix (Cα atoms) | 0.36 ± 0.08 | 0.24 ± 0.09 | 0.36 ± 0.10 |
| β-sheet (Cα atoms) | 0.46 ± 0.11 | 0.44 ± 0.09 | 0.69 ± 0.11 |
| L1 loop (Cα atoms) | 0.45 ± 0.14 | 0.64 ± 0.18 | 0.42 ± 0.19 |
| T2 turn (Cα atoms) | 0.42 ± 0.19 | 0.42 ± 0.19 | 1.83 ± 0.53 |
The solution structures of ARD1, ETD151, and ETD135 are represented by the 10 lowest energy conformers of each peptide refined in a shell of water. All these structures are in agreement with the experimental data, there is no violation of distance restraints larger than 0.3 Å. Most residues fall in the most favorable, or additionally allowed regions, of the Ramachandran map. The few residues in the less favorable regions belong to the T2 turn. The structures are well defined by the NMR restraints, as evidenced by the low values of the pairwise RMS deviation calculated for different structure selections (Table 2). Larger RMSD values found for ETD135 T2 turn are discussed below (Fig. 2 ▶).
Figure 2.
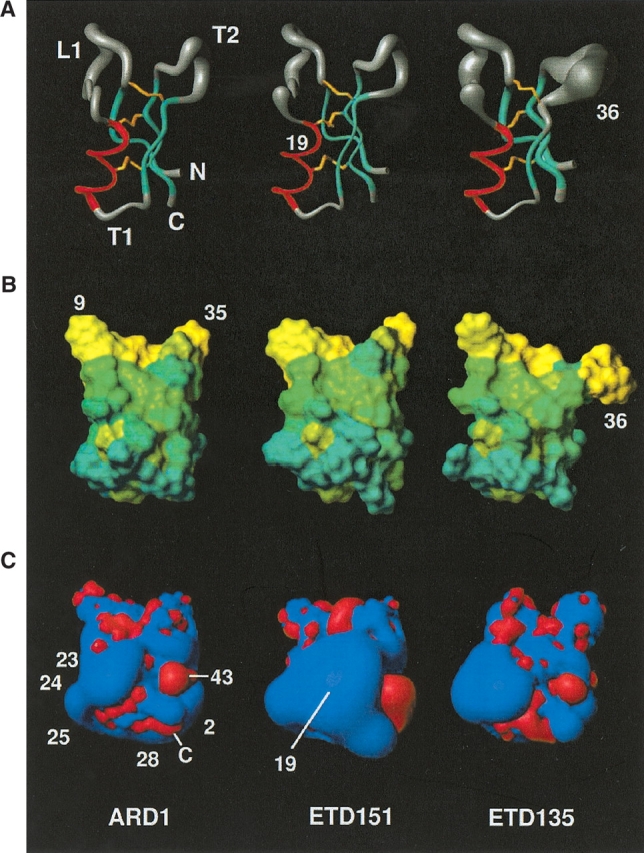
Schematic representation of the structure of ARD1 (left) and its two mutants ETD151 (middle) and ETD135 (right) in the same orientation. (A) Sausage representation of the backbones drawn using MOLMOL (Koradi et al. 1996). The radius of the cylinder is proportional to the RMSD on the backbone atoms calculated on the 10 best structures. (B) Representation of the molecular surface with the residues color-coded according to the Eisenberg hydrophobicity scale. (C) Electrostatic isopotential surfaces drawn using MOLMOL. In red, negative contours at −1 eV, in blue, positive contours at +1 eV. Residues cited in the text are labeled.
Structure descriptions
The global fold of ARD1, ETD135, and ETD151 displays the same general features: a helix and a three-stranded antiparallel β-sheet forming the CSαβ motif (Cysteine stabilized α-helix β-sheet motif; Cornet et al. 1995). The secondary structure elements determined using backbone φ, ψ angles and characteristic hydrogen bond network are reported in Figure 1 ▶ for each molecule. The first strand of the sheet and the helix are separated by a long loop (L1), the helix and the second strand by a turn (T1), and the second and third strands of the β-sheet are linked by a further turn (T2; Fig. 2 ▶).
The RMSD differences between the lowest energy structure of ARD1 and its two mutants ETD151 and ETD135 are grouped in Table 3. The two closest molecules (0.77 Å for Cα atoms) are clearly ARD1 and ETD151, which differ by the N19R mutation only. This modification occurring in the helix does not induce structural changes; the RMSD between the Cα atoms of the helices is only 0.34 Å (in comparison, this RMSD is 0.51 Å between ARD1 and heliomicin, which differ by residues 17 and 20 in the helix). In contrast, the single substitution of a small residue alanine by a γ-branched cumbersome leucine at position 36 in ETD135 considerably affects the T2 turn, and to a smaller extent the β-sheet (1.61 Å between all Cα atoms, 0.96 in the T2 turn, 0.76 in the β-sheet). Most hydrophobic and aromatic side chains show well-defined positions, characterized by χ1 and χ2 circular variances smaller than 0.2. The only exceptions are W9, showing a large variability in all the peptides but ETD151, and F35 in ETD135 structure. Concerning the hydrophilic side chain, only H31 shows a unique position in the ensemble of structures for all molecules. Remarkably, the ring of H31 and of W41 display a similar orientation with respect to the backbone in the four peptide structures. The cysteine residues and the three disulphide bridges show diverse behaviors. The C7–C32 bridge, which is the most accessible to the solvent, is the least well defined, showing left- or right-handed conformations, whatever the molecule; the C18–C40 bridge, with intermediate accessibility, adopts a single conformation in the ETD151 structure but is more variable on the others. The last bridge (C22–C44), embedded in the structure, is always found in a left-handed conformation in ARD1 and ETD151 structures, while it generally adopts a right-handed hook conformation in ETD135.
Table 3.
RMSD differences (Å) between the two best structures of ARD1 and ETD151 (middle) or ETD135 (right)
| RMSD (Å) | ARD1/ETD151 | ARD1/ETD135 |
| All Cα | 0.77 | 1.61 |
| Secondary structure Cα | 0.54 | 0.79 |
| α-Helix Cα | 0.34 | 0.35 |
| β-sheet Cα | 0.51 | 0.76 |
| L1 loop Cα | 1.02 | 0.75 |
| T2 turn Cα | 0.71 | 0.96 |
Structural comparison
A superimposition of the structure of heliomicin, ARD1, ETD151, and ETD135 (Fig. 3 ▶) highlights the very good fit between the secondary structure elements. The differences principally concern the orientation of turn T2 and loop L1. On the four structures, the helix starts at residue 17 (D/N), and is interrupted at G26 (Fig. 1 ▶) that adopts a left-handed conformation. The canonical hydrogen bond network is formed up to that residue. An additional hydrogen bond between the C = O of C22 and the NH of Y27 is present in all structures. Although most amino acid substitutions are located in the helix, no structural modifications are observed in this zone. In fact, these substitutions concern solvent-exposed residues and do not introduce residues unfavorable to a helix formation, such as β-branched amino acids. In heliomicin, the D17 side chain position is very well defined and a hydrogen bond is formed in a majority of structures between one of its Oδ atoms and the NH amide proton of G20 (a very slowly exchangeable proton in amide exchange experiment). These two residues are precisely those differing between heliomicin and ARD1 (D17N and G20A). A hydrogen bond between the side chain carbonyl group of N17 and the NH proton of A20 cannot be formed due to steric hindrance caused by the alanine methyl group. As a result, the side chain position of N17 is rather ill-defined.
Figure 3.

Superimposition of structures: ARD1 (green), ETD151 (gray), ETD135 (blue), and heliomicin (red).
The first strand (β1) of the β-sheet is identical in the four molecules. It is characterized by the presence of a bulge at positions 4 and 5, leading to an interruption in the standard hydrogen bond network with the third strand. The second (β2) and third (β3) strands of ARD1 and ETD151 are very similar and a little longer than what is observed in heliomicin. In contrast, the second strand in ETD135 is shorter following the A36L mutation occurring downstream in the T2 turn, while the third strand remains identical.
Residues F35, A36, and N37 in the T2 turn of ARD1 and ETD151 adopt a 310 helix conformation in a majority of structures that is not found for ETD135 and heliomicin. The T2 turn for ETD135 is, in fact, better described as an 8-residue ill-defined loop.
The L1 loop in the four molecules displays a similar overall shape (Fig. 3 ▶). Its fold is stabilized by a hydrogen bond, found in all four structures, between the NH of S6 and the C=O of N13. Another hydrogen bond appears between the C=O of S6 and the NH of T15 in ARD1, ETD151, and heliomicin structures but is absent in ETD135. A hydrogen bond is also formed between the NH group of C7 and the carbonyl of V38 in all four structures and contributes to the stabilization of the β-sheet. All NH protons involved in the hydrogen bond show intermediate or very slow exchange rates.
Comparison of hydrophobic and electrostatic properties
ARD1 displays an amphipatic structure. All the mutants, and specially ETD151 and ETD135, were engineered to enhance this amphiphily either by increasing the hydrophoby of the hydrophobic region (ETD135), or by enhancing the cationicity of the opposite side (ETD151). These amphipathic properties are illustrated in Figure 2B ▶, where residues are colored according to a hydrophobicity scale (Eisenberg et al. 1984), and in Figure 2C ▶, showing electrostatic isopotential contours at the surface of the molecules. One pole of the molecules, comprising L1 loop and T2 turn, is hydrophobic with the presence of numerous hydrophobic and aromatic residues. This character is reinforced in ETD135 where A36 is mutated into a more hydrophobic amino acid (L36). The other pole (comprising the N terminus, T1 turn, and the C terminus) is clearly more hydrophilic, while the rest of the structure presents an intermediate hydrophobicity. The hydrophilic pole includes positively charged residues: K2, K23, R24, R25, and K28 leading to a highly cationic surface, except for negative patches around E43 and at the carboxy C terminus. This pole is enlarged towards the top of the helix in ETD151 following the N19R substitution, and this could be correlated with its improved antifungal activity.
Discussion
The CSαβ motif is a well-defined scaffold adopted by various peptides that fulfill a large variety of biological function in plants, insects, and scorpions. Because the array of disulphide bridges provides most of the stabilization energy, these natural peptides constitute interesting candidates for protein engineering. This has been successfully applied in the case of the scorpion toxin charybdotoxin by the design of a chimera with a new bioactivity (Drakopoulou et al. 1996), and the grafting of new functional sites onto the CSαβ motif (Martin and Vita 2000). Similarly, heliomicin scaffold constitutes a particularly well-suited platform to design peptides with enhanced antifungal activity for industrial development as far as substitutions comply with the folding of the peptide. In this respect, we have determined the three-dimensional structure of ARD1, an antifungal peptide from A. demophoon differing from H. virescens heliomicin by only two residues located in the helix, but which is twice as potent against C. albicans and A. fumigatus. Based on comparative analysis of the 3D structures and the surface properties of the two natural peptides, we designed several mutants of ARD1 in which the hydrophobic and/or the cationic character was enhanced. Two of these mutants, ETD151 (N19R) and ETD135 (A36L), were selected according to their activities, for structural analysis. Compared to heliomicin, the activities of these two mutants are enhanced by a factor of 4–8 against C. albicans and by a factor of 4 against A. fumigatus. In addition, they are both against rare filamentous fungi such as Scedosporium and Fusarium for which no effective treatment exists at the moment.
As previously shown for a mutant of heliomicin, in which two residues of the helix (K23 and L24) were mutated into leucines (Lamberty et al. 2001a), the helix geometry is not affected by the mutation N19R in ETD151. These mutations are restricted to solvent exposed residues and avoid the introduction of β-branched residues into the helix. In contrast, the substitution in ETD135 of a small residue (A36) by a cumbersome one (L) in the T2 turn significantly affects not only the T2 turn but also the second strand of the β sheet. This single conservative mutation noticeably alters the structure, showing that mutations in some part of the molecule can affect the whole structure. Mutation A36L is also responsible for an increased flexibility in that part of the molecule, indicated by the variability observed in the final set of NMR structures. Obviously, that flexibility does not adversely affect the activity, ETD135 being as potent as ETD151.
Our study reveals a strong correlation between the distribution of hydrophobic and charged residues at the surface of the molecule and the antifungal activity. We have shown previously that antifungal defensins from plant and insect display a common amphiphilic character (Landon et al. 1997, 2000; Lamberty et al. 2001a; Da Silva et al. 2003). This is also the case for the heliomicin family of analogs (ARD1, ETD151, and ETD135) in which the L1 loop and the T2 turn make a hydrophobic pole, while the other side of the molecule is largely hydrophilic (T1, N-, and C-term). Enhancing this bipolar character by making T2 turn more hydrophobic, as in ETD135 (A36L), increased the antifungal activity. Although the hydrophobic zone is rather neutral, the hydrophilic one is highly positively charged. When increasing the cationicity of the electropositive pole as in ETD151 (N19R), the activity against A. fumigatus and C. albicans increases. In addition, the molecule becomes very potent against Scedosporium and Fusarium. The double mutant ETD179 (A36L, N19R) affecting both the hydrophobic and cationic poles does not exhibit superiority to the single mutants (Table 1). As a result, further improvement of ETD135 or ETD151 by single-point mutations is not straightforward.
Although more potent against sensitive species, the presently reported analogs have a spectrum of activity similar to that of heliomicin. In particular, they are devoid of antibacterial properties and they do not affect yeasts from S. cerevisiae (thus enabling their production in this host) or Candida glabrata species. The degree of specificity observed in the spectrum of activity suggests the peptides interact specifically with a targeted component of the fungus, most likely the membrane surface as a whole or a specific membraneous constituent. This is in marked contrast with what is found for small antimicrobial peptides such as thanatin (Mandard et al. 1998), tachyplesin (Tamamura et al. 1993), protegrin (Aumelas et al. 1996), gomesin (Mandard et al. 2002a), etc. The latter molecules, adopting an amphiphilic β-hairpin like structure, are active at low concentrations against a large variety of bacterial and fungal strains due to tensioactive properties on biological membranes (Mandard et al. 2002b). The mode of action of the peptides from the heliomicin family is not known yet, but owing to sequence and structural similarities with plant defensins, and more specifically with morphogenic plant defensin such as Rs-AFP1 and Rs-AFP2 (Fant et al. 1998), it seems likely that they exhibit an analogous mode of action (Thevissen et al. 1996, 1997, 1999, 2000a,b).
A number of studies have attempted to elucidate the mode of action of plant defensin (Thevissen et al. 1996, 1997, 1999, 2000a,b). It has been shown that they induce membrane permeabilization without forming voltage dependant ion-permeable pores in artificial membranes, nor changing the electrical properties of artificial membranes. In addition, for several plant defensins, high-affinity binding sites were identified on fungal cells or microsomal membranes, suggesting a two step interaction mode in which the binding of the peptide to a specific membraneous component facilitates the insertion of the peptide in the membrane and the formation of pores.
Assuming that H33 is positively charged, the global charge of ARD1 and ETD135 is +3 and it is +4 for ETD151. These molecules are not very cationic, relative to their size, so that a direct interaction with the membrane is unlikely. A mechanism where insertion in the membrane is mediated through a specific binding site could explain the specificity of heliomicins against various fungal strains. Different classes of peptides would target different specific components and thus display different activity spectrum, the strain sensitivity being driven by the presence of the targeted site acting as an “entrance key”. Under this assumption, the activity can be modulated by affecting one or the other step: improve the binding to the “primary” target, or facilitate the subsequent insertion in the membrane. In the present work, where we essentially modified the global properties of the molecule, we probably modulated the second step, the final insertion into the membrane. Such assumption could explain the nonimproved activity of ETD179 ARD1 analog (which combines the two mutations N19R and A36L) over ETD151 (single mutation N19R) and ETD135 (single mutation A36L) if binding to the specific membraneous component became the limiting factor.
Concerning the interaction site, mutational analysis of the plant defensin Rs-AFP2 from radish Raphanus sativus L. revealed two adjacent sites important for antifungal activity (De Samblanx et al. 1997). In particular, a positive charge (K44) within a predominantly hydrophobic cluster is important for antifungal activity, showing that the interaction of the protein and its putative receptor on fungal hyphae is based both on ionic and nonspecific interactions. The antifungal defensin from the insect Drosophila melanogaster, which presents the same morphogenic characteristic than Rs-AFP2 (Bulet et al. 1999), also possesses a positive residue (K38) embedded within a hydrophobic cluster, certainly constituting its active site (Landon et al. 2000). This site was also characterized recently in NaD1, the first floral antifungal defensin from Nicotiana alata (Lay et al. 2003), but is not conserved in nonmorphogenic antifungal plant defensins (Landon et al. 2000). Heliomicin from H. virescens, although morphogenic as well, is devoid of positive charges in the area of the molecule. However. heliomicin and drosomycin have different modes of action towards the membrane of pathogens (Banzet et al. 2002); they thus have presumably different interaction sites. Characterization of the interaction site in heliomicin will require specific studies.
In conclusion, the knowledge of the global features of the 3D structure of antifungal peptides found in insect allowed the design of new analogs preserving the architecture of the parent molecules but with improved activity against human pathogens, which are one of the leading causes of nosocomial infections. Current work focuses on the in vivo evaluation of the best analogs in an aim to deliver candidate compounds for preclinical and clinical development.
The coordinates of ARD1, ETD151, and ETD135 have been deposited in the Protein Data Bank (accession numbers 1OZZ, 1P00, 1P0A, respectively).
Materials and methods
ARD1 peptide isolation and identification
Fourth instar larvae of the lepidoptera A. demophoon (Nymphalidae family) were individually immunized by two injections of 20 μL of a phosphate-buffered saline (PBS) solution containing the Gram-positive bacteria Micrococcus luteus and Staliphococcus aureus, the Gram-negative bacteria Pseudomonas aeruginosa, the yeast C. albicans and the filamentous fungus A. fumigatus. Bacteria were prepared from overnight cultures grown at 37°C in Luria-Bretani medium. C. albicans was prepared from overnight cultures incubated at 30°C in Sabouraud medium, whereas for A. fumigatus, fungal spores were taken from a stock frozen at −80°C. After 24 h, the insects were chilled on a bed of ice and hemolymph was collected (about 160 μL/larva) by sectioning an abdominal appendix and gently squeezing the abdomen. The total hemolymph (13 mL) was pooled in ice-cold polypropylene tubes containing aprotinine as a protease inhibitor (20 μg/mL, final concentration) and phenylthiourea as a melanization inhibitor (40 μM, final concentration). It was centrifuged (4°C) for 1 min at 8000 rpm, and the resulting supernatant was then centrifuged (4°C) for 20 min at 12000 rpm. Cell-free hemolymph was frozen at 80°C until use.
Five milliliters of the hemolymph were acidified to pH 3 with water containing 0.05% TFA (trifluoroacetic acid). Extraction was performed by gentle shaking for 30 min on an ice-bath. After centrifugation (4°C) at 10000 × g for 30 min, the supernatant was loaded onto a 5-g C18 cartridge (Sep-Pak from Waters) equilibrated with water containing 0.05% TFA. A washing step with water/TFA 0.05% was used to eliminate salts and other hydrophilic material. Peptides of interest were then eluted with 60% acetonitrile in water/TFA 0.05%. This fraction was dried in a vacuum centrifuge (Speed-Vac, Savant) and reconstituted with water/TFA 0.05%.
Subsequent HPLC purification was performed on a preparative C8 column (Aquapore RP-300 from Brownlee, 220 × 10 mm, 300 Å), applying a linear gradient of acetonitrile/0.05% TFA against water, from 2% to 10% in 5 min, from 10% to 25% in 30 min, from 25% to 35% in 40 min, and from 35% to 60% in 50 min, for a total run time of 125 min, at a constant flow of 2.5 mL/min. Fractions were manually collected according to UV absorbance at 225 nm, dried, reconstituted in MilliQ water, and tested for antifungal activity as described in ref (Lamberty et al. 1999).
The fraction displaying antifungal activity was eluted with 27% of acetonitrile and was further purified on an analytical C8 column (Aquapore RP-300 from Brownlee, 220 × 4.6 mm, 300 Å), via the application of a biphasic linear gradient of acetonitrile/0.05% TFA against water, from 2% to 23% in 5 min, then from 23% to 31% in 50 min at a constant flow of 0.8 mL/min. The resulting fractions were collected manually again and treated as above, before testing.
Finally, the fraction with antifungal activity was purified to homogeneity on a narrowbore C18 column (Delta Pak HPI from Waters, 150 × 2 mm, 300 Å). Elution was performed with a biphasic linear gradient of acetonitrile/0.05% TFA against water, from 2% to 24% in 10 min, then from 24% to 44% in 100 min at a constant flow of 0.2 mL/min. Peptide purity was assessed by Maldi-TOF mass spectral analysis, as described in ref (Uttenweiler-Joseph et al. 1997), on a Bruker Biflex instrument. Reduction of disulfide bridges followed by S-Pyridylethylation was performed as described in ref (Bulet et al. 1992). Sequencing of the reduced S-Pyridylethylated peptide was performed by N-terminal amino acid Edman degradation on a pulse liquid automatic sequencer ABI 473A from Applied Biosystem. Confirmation of the C-terminal sequence was obtained by sequencing after enzymatic cleavage, as described in ref (Lamberty et al. 1999).
Production of heliomicin analogues in the yeast S. cerevisiae
The heliomicin expression vector pSEA2 (Lamberty et al., 1999) was used as a template for PCR amplification of a DNA fragment with the mutations A17N, and G20A encoding for ARD1 sequence: the two wished mutations (in bold) and a SacII restriction site (underlined) were introduced in the EM89 primer (5′-TTTTTTCCGCGGCGCTTGCACTCGGCGTTGCAGTTACTA GTGTAGTTGACGGCGC-3′). EM89 was used in conjuction with EM72 (5′-GTAAATGCATGTATACTAAACTCACA-3′) to amplify the MF-α1, the pre-BGL2 and pro-MF-α1 sequences as well as the coding sequence up to the heliomicin SacII site. The PCR amplified fragment was then digested with Sph1 and SacII restriction enzymes and cloned in frame into the pSEA2 vector that had been cleaved by the same enzymes and dephosphorylated with alkaline phosphatase. The resulting pEM2 plasmid was checked by restriction analysis and DNA sequencing. A similar strategy was followed to generate expression vectors for the other variants, in particular, ETD135 and ETD151 carrying the additional A36L and N19R substitutions.
The yeast strain TGY48.1 (Reichhart et al. 1992) was transformed by the expression plasmids using the lithium acetate method (Ito et al. 1983), and transformants were selected on YNBG medium supplemented with 0.5% casamino acids.
Production of the peptides was carried out in shake flasks under selective conditions (50-mL YNBG medium supplemented with 0.5% casamino acids) for 72 h at 29°C. Peptides chosen for the NMR structural study were subsequently produced in a 5-L working volume bench-top fermentor (New Brunswick Scientific, bioflo 3000). Seed culture was started from a fresh culture on solid medium and inoculated directly into a 500-mL shake flask containing 50 mL of Kappeli medium, and incubated for 24 h at 30°C. The entire volume of inoculum was transfered into the fermentation vessel containing 3.7 L of basal medium (14.25 g KH2PO4; 4 g MgSO4; 1.65 mL H3PO4) supplemented with trace elements (80 mg MnSO4, H2O; 2 mg CuSO4, 5 H2O; 75 mg ZnSO4, 7 H2O; 14 mg CoCl2, 6H2O; 12.4 mg Na2MoO4, 2 H2O; 37.5 mg H3BO3; 1 mg citric acid; 5 mg KCl; 12.5 mg NiSO4, 6 H2O; 125 mg trisodium citrate 2 H2O). Fermentation was initiated by adding 10% of fed-batch medium (200 g glucose; 100 g casamino acids; 0.25 g FeCl3; 0.25 g CaCl2; 5 mg thiamine HCl; 12.5 mg pyridoxine HCl; 20 mg nicotinic acid; 0.125 mg D-biotine; 25 mg Ca-D-pantothenate; 200 mg meso inositol).
Fermentation was then conducted at 30°C with a constant dissolved oxygen rate maintained above 80% saturation. The impeller speed varied between 200 and 600 rpm. The pH of the culture was not regulated and, due to spontaneous acidification, dropped to pH 3 at the end of the fermentation.
Purification of recombinant peptides
After centrifugation at 4000g for 30 min at 4°C, the supernatant was filtered on a 0.2-μm membrane. The pH was adjusted to 3 before the first purification step, consisting in an ion exchange. Capture on a strong cation exchanger membrane (Sartobind-S100, Sartorius) allows rapid concentration of cationic material within the peptide of interest, and was applied to the broth of peptides produced in fermentor; this step was skipped for production carried out in shake flasks. The membrane was equilibrated with 25 mM citrate buffer at pH 3, prior to the loading of 4 to 5 liters of supernatant at a flow rate of 30 mL/min. Elution was performed with 1 M NaCl in water. Removal of salt was achieved using Sep-Pak C18 cartridges (Waters) as reverse-phase media. The material was acidified at pH 3 with acetic acid, then loaded on cartridges equilibrated with water/TFA 0.05%. The elution achieved with 60% acetonitrile in acidified water was submitted to the final step of purification consisting in reverse phase HPLC on a preparative C8 column (Dynamax-300 Å, 21.4 × 250 mm), equilibrated with 10% acetonitrile in acidified water. The peptide was eluted with a linear gradient of acetonitrile in acidified water from 10% to 60% over 50 min at a flow rate of 10 mL/min. The purified peptide was recovered by lyophilization.
Antimicrobial assays
The antimicrobial properties of the peptides against the yeasts C. albicans and C. neoformans (C. albicans IHEM 8060, C. albicans No. 245962 and C. neoformans A; gift from H. Koenig, Hôpital Civil de Strasbourg, France) and the filamentous fungus A. fumigatus, Fusarium solani and Scedosporium prolificans (A. fumigatus GASP 4707, gift from H. Koenig, Hôpital Civil de Strasbourg, France; F. solani FFUS 8591 and S. prolificans FSSP 7902, gift from Dr. J. FGM Meis, Canisius-Wilhelmina Hospital, Nijmegen, The Netherlands) were performed following a growth inhibition assay in liquid medium in 96-well microplates.
The MIC determination against yeasts and fungi was performed respectively in Sabouraud-Chloramphenicol and YPG (1 g peptone, 1 g yeast extract, and 3 g glucose per liter). The final inoculum was of 5 103/mL for filamentous fungi and 2.5 103/mL for yeasts. The microdilution trays were incubated for 24 to 48 h at 30°C for yeasts and 48 to 72 h at 37°C for filamentous fungi. A visual check of the plates gives us the MIC, which corresponds to the concentration that inhibits 50% of the fungal growth, compared to the control without drug.
Nuclear magnetic resonance experiments
A standard set of 2D 1H-NMR experiments (DQF-COSY, 80 msec TOCSY, and 120 msec NOESY) of the three peptides—ARD1, ETD135, and ETD151—were performed on 2-mM solutions prepared in sodium acetate buffer at pH 4.3, at 293 K (as for heliomicin from H. virescens; Lamberty et al. 2001a). An additional set of data, recorded at 303 K, was used to resolve assignment ambiguities due to spin system overlaps. Exchange kinetics of amide protons were estimated from 1D and short 2D-TOCSY spectra recorded at 293 K, after dissolution in D2O. All spectra were recorded on a VARIAN INOVA NMR spectrometer equipped with a z-axis field-gradient unit and operating at a proton frequency of 600 MHz. The NMR data sets were processed using the NMRPipe/NMRDraw software package (Delaglio et al. 1995).
Structure calculations
1H chemical shifts were assigned according to classical procedures (Wüthrich 1986). NOE cross-peaks were integrated and assigned within the NMRView software (Johnson and Blevins 1994). Because the disulfide pairings are already known, covalent bonds were built between the sulfur atoms of the paired cysteine, Cys7–Cys32, Cys18–Cys40, and Cys22–Cys42. Structure calculations were performed with the ARIA 1.1 software (Linge et al. 2001). ARIA implements a semiautomated method in the software CNX 2000.1 (MSI; Brünger et al. 1998) combining structure calculations and NOE assignments in an iterative process. This is completed by treating ambiguous NOEs as the sum of contributions from all possible assignments and using floating assignments for prochiral groups. ARIA calculations were initiated using default parameters and a first set of easily assigned NOEs. Additional assignments were proposed in the following runs of ARIA. Each ARIA run included nine iterations in which the nonredundant set of NOE-based distance restraints obtained at the preceding iteration were used in a simulated annealing step to generate 50 structures except for iteration 0, used for the purpose of calibrating the NOEs. The 20 structures with the lowest energy were used for NOE assignments and conserved for the following iteration. Finally, the 20 best structures were further refined by molecular dynamics steps in explicit solvent to remove artefacts due to the simplifications of the force field used in the previous steps. At the end of each run, the new assignments proposed by ARIA were checked manually and introduced (or not) in the following run. Rejected constraints and residual violations were also analyzed, and assignments were corrected if needed. This iterative process was repeated until complete assignment of the NOESY map. A last run of 100 structures was then performed with the final list of NOE-derived distance restraints in which no restraint can be rejected. The 10 structures with the lowest energy were considered as characteristic of the peptide structures. Representation and quantitative analysis of the calculated structures were performed using MOLMOL (Koradi et al. 1996) and in-house programs.
Electronic supplemental material
1H chemical shifts (ppm) tables of ARD1, ETD151, and ETD135.
Acknowledgments
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Supplemental material: see www.proteinscience.org
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.03404404.
References
- Aumelas, A., Mangoni, M., Roumestand, C., Chiche, L., Despaux, E., Grassy, G., Calas, B., and Chavanieu, A. 1996. Synthesis and solution structure of the antimicrobial peptide protegrin-1. Eur. J. Biochem. 237 575–583. [DOI] [PubMed] [Google Scholar]
- Banzet, N., Latorse, M.-P., Bulet, P., Francois, E., Derpierre, C., and Dubald, M. 2002. Expression of insect cystein-rich antifungal peptides in transgenic tobacco enhances resistance to fungal disease. Plant Sci. 162 995–1006. [Google Scholar]
- Bontems, F., Roumestand, C., Gilquin, B., Menez, A., and Toma, F. 1991. Refined structure of charybdotoxin: Common motifs in scorpion toxins and insect defensins. Science 254 1521–1523. [DOI] [PubMed] [Google Scholar]
- Breithaupt, H. 1999. The new antibiotics. Nat. Biotechnol. 17 1165–1169. [DOI] [PubMed] [Google Scholar]
- Brünger, A.T., Adams, P.D., Clore, G.M., DeLano, W.L., Gros, P., Grosse-Kunstleve, R.W., Jiang, J.-S., Kuszewski, J., Nilges, M., Pannu, N.S., et al. 1998. Crystallography and NMR systems: A new software for macromolecular structure determination. Acta Crystallogr. D 54 905–921. [DOI] [PubMed] [Google Scholar]
- Bulet, P., Cociancich, S., Reuland, M., Sauber, F., Bischoff, R., Hegy, G., Van Dorsselaer, A., Hetru, C., and Hoffmann, J. 1992. A novel insect defensin mediates the inducible antibacterial activity in larvae of the dragonfly Aeschna cyanea (Paleoptera, Odonata). Eur. J. Biochem, 209 977–984. [DOI] [PubMed] [Google Scholar]
- Bulet, P., Hetru, C., Dimarcq, J.-L., and Hoffman, D. 1999. Antimicrobial peptides in insects; Structure and function. Dev. Comp. Immunol. 23 329–344. [DOI] [PubMed] [Google Scholar]
- Caldwell, J.E., Abildgaard, F., Dzakula, Z., Ming, D., Hellekant, G., and Markley, J.L. 1998. Solution structure of the thermostable sweet-tasting protein brazzein. Nat. Struct. Biol. 5 427–431. [DOI] [PubMed] [Google Scholar]
- Cociancich, S., Goyffon, M., Bontems, F., Bulet, P., Bouet, F., Menez, A., and Hoffmann, J. 1993. Purification and characterization of a scorpion defensin, a 4kDa antibacterial peptide presenting structural similarities with insect defensins and scorpion toxins. Biochem. Biophys. Res. Commun. 194 17–22. [DOI] [PubMed] [Google Scholar]
- Cornet, B., Bonmatin, J.M., Hetru, C., Hoffmann, J.A., Ptak, M., and Vovelle, F. 1995. Refined three-dimensional solution structure of insect defensin A. Structure 3 435–448. [DOI] [PubMed] [Google Scholar]
- Da Silva, P., Jouvensal, L., Lamberty, M., Bulet, P., Caille, A., and Vovelle, F. 2003. Solution structure of termicin, an antimicrobial peptide from the termite Pseudacanthotermes spiniger. Protein Sci. 12 438–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delaglio, F., Grzesiek, S., Vuister, G.W., Zhu, G., Pfeifer, J., and Bax, A. 1995. NMRPipe: A multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6 277–293. [DOI] [PubMed] [Google Scholar]
- De Lucca, A.J. and Walsh, T.J. 1999. Antifungal peptides: Novel therapeutic compounds against emerging pathogens. Antimicrob. Agents Chemother. 43 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Samblanx, G.W., Goderis, I.J., Thevissen, K., Raemaekers, R., Fant, F., Borremans, F., Acland, D.P., Osborn, R.W., Patel, S., and Broekaert, W.F. 1997. Mutational analysis of a plant defensin from radish (Raphanus sativus L.) reveals two adjacent sites important for antifungal activity. J. Biol. Chem. 272 1171–1179. [DOI] [PubMed] [Google Scholar]
- Dimarcq, J.L., Bulet, P., Hetru, C., and Hoffmann, J. 1998. Cysteine-rich antimicrobial peptides in invertebrates. Biopolymers 47 465–477. [DOI] [PubMed] [Google Scholar]
- Dixon, D.M. and Edwards, J.E. 2000. Summary draft report of the antifungal working group. Summit on Development of Infectious Disease Therapeutics, September 26–27, 2000, Bethesda, MD. http://www.niaid.nih.gov/dmid/drug/antifungalreport.htm.
- Drakopoulou, E., Zinn-Justin, S., Guenneugues, M., Gilqin, B., Menez, A., and Vita, C. 1996. Changing the structural context of a functional β-hairpin. Synthesis and characterization of a chimera containing the curaremimetic loop of a snake toxin in the scorpion α/β scaffold. J. Biol. Chem. 271 11979–11987. [DOI] [PubMed] [Google Scholar]
- Eisenberg, D., Schwarz, E., Komaromy, M., and Wall, R. 1984. Analysis of membrane and surface protein sequences with the hydrophobic moment plot. J. Mol. Biol. 179 125–142. [DOI] [PubMed] [Google Scholar]
- Fant, F., Vranken, W., Broekaert, W., and Borremans, F. 1998. Determination of the three-dimensional solution structure of Raphanus sativus antifungal protein 1 by 1H NMR. J. Mol. Biol. 279 257–270. [DOI] [PubMed] [Google Scholar]
- Fehlbaum, P., Bulet, P., Michaut, L., Lagueux, M., Broekaert, W.F., Hetru, C., and Hoffmann, J.A. 1994. Insect immunity. Septic injury of Drosophila induces the synthesis of a potent antifungal peptide with sequence homology to plant antifungal peptides. J. Biol. Chem. 269 33159–33163. [PubMed] [Google Scholar]
- Hoffmann, J.A. 1995. Innate immunity of insects. Curr. Opin. Immunol. 7 4–10. [DOI] [PubMed] [Google Scholar]
- Hoffmann, J.A., Kafatos, F.C., Janeway, C.A., and Ezekowitz, R.A. 1999. Phylogenetic perspectives in innate immunity. Science 284 1313–1318. [DOI] [PubMed] [Google Scholar]
- Ito, H., Fukuda, Y., Murata, K., and Kimura, A. 1983. Transformation of intact yeast cells treated with alkali cations. J. Bacteriol. 153 163–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson, B.A. and Blevins, R.A. 1994. NMRView: A computer program for the visualisation and analysis of NMR data. J. Biomol. NMR 4 603–614. [DOI] [PubMed] [Google Scholar]
- Khush, R.S. and Lemaitre, B. 2000. Genes that fight infection: What the Drosophila genome says about animal immunity. Trends Genet. 16 442–449. [DOI] [PubMed] [Google Scholar]
- Koradi, R., Billeter, M., and Wuthrich, K. 1996. MOLMOL: A program for display and analysis of macromolecular structures. J. Mol. Graph. 14 51–55. [DOI] [PubMed] [Google Scholar]
- Lambert, J., Keppi, E., Dimarcq, J.L., Wicker, C., Reichhart, J.M., Dunbar, B., Lepage, P., Van Dorsselaer, A., Hoffmann, J., Fothergill, J., et al. 1989. Insect immunity: Isolation from immune blood of the dipteran Phormia terranovae of two insect antibacterial peptides with sequence homology to rabbit lung macrophage bactericidal peptides. Proc. Natl. Acad. Sci. 86 262–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamberty, M., Ades, S., Uttenweiler-Joseph, S., Brookhart, G., Bushey, D., Hoffmann, J.A., and Bulet, P. 1999. Insect immunity. Isolation from the lepidopteran Heliothis virescens of a novel insect defensin with potent antifungal activity. J. Biol. Chem. 274 9320–9326. [DOI] [PubMed] [Google Scholar]
- Lamberty, M., Caille, A., Landon, C., Tassin-Moindrot, S., Hetru, C., Bulet, P., and Vovelle, F. 2001a. Solution structures of the antifungal heliomicin and a selected variant with both antibacterial and antifungal activities. Biochemistry 40 11995–12003. [DOI] [PubMed] [Google Scholar]
- Lamberty, M., Zachary, D., Lanot, R., Bordereau, C., Robert, A., Hoffmann, J.A., and Bulet, P. 2001b. Insect immunity. Constitutive expression of a cysteine-rich antifungal and a linear antibacterial peptide in a termite insect. J. Biol. Chem. 276 4085–4092. [DOI] [PubMed] [Google Scholar]
- Landon, C., Sodano, P., Hetru, C., Hoffmann, J., and Ptak, M. 1997. Solution structure of drosomycin, the first inducible antifungal protein from insects. Protein Sci. 6 1878–1884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landon, C., Pajon, A., Vovelle, F., and Sodano, P. 2000. The active site of drosomycin, a small insect antifungal protein, delineated by comparison with the modeled structure of Rs-AFP2, a plant antifungal protein. J. Pept. Res. 56 231–238. [DOI] [PubMed] [Google Scholar]
- Lay, F.T., Schirra, H.J., Scanlon, M.J., Anderson, M.A., and Craik, D.J. 2003. The Three-dimensional solution structure of NaD1, a new floral defensin from Nicotiana alata and its application to a homology model of the crop defense protein alfAFP. J. Mol. Biol. 325 175–188. [DOI] [PubMed] [Google Scholar]
- Linge, J.P., O’Donoghue, S.I., and Nilges, M. 2001. Automated assignment of ambiguous nuclear overhauser effects with ARIA. Methods Enzymol. 339 71–90. [DOI] [PubMed] [Google Scholar]
- Mandard, N., Sodano, P., Labbe, H., Bonmatin, J.M., Bulet, P., Hetru, C., Ptak, M., and Vovelle, F. 1998. Solution structure of thanatin, a potent bactericidal and fungicidal insect peptide, determined from proton two-dimensional nuclear magnetic resonance data. Eur. J. Biochem. 256 404–410. [DOI] [PubMed] [Google Scholar]
- Mandard, N., Bulet, P., Caille, A., Daffre, S., and Vovelle, F. 2002a. The solution structure of gomesin, an antimicrobial cysteine-rich peptide from the spider. Eur. J. Biochem. 269 1190–1198. [DOI] [PubMed] [Google Scholar]
- Mandard, N., Bulet, P., Hetru, C., Landon, C., and Vovelle, F. 2002b. Solution structure of antimicrobial peptides with a β-hairpin fold. structure–activity relationships. In Membrane interacting peptides and proteins (ed. F. Heitz), pp. 155–171. Research signpost.
- Martin, L. and Vita, C. 2000. Engineering novel bioactive mini-proteins from small size natural and de novo designed scaffolds. Curr. Protein Pept. Sci. 1 403–430. [DOI] [PubMed] [Google Scholar]
- Matsuyama, K. and Natori, S. 1988. Molecular cloning of cDNA for sapecin and unique expression of the sapecin gene during the development of Sarcophaga peregrina. J. Biol. Chem. 263 17117–17121. [PubMed] [Google Scholar]
- Reichhart, J.-M., Petit, I., Legrain, M., Dimarcq, J.-L., Keppi, E., Lecocq, J.-P., Hoffmann, J.A., and Achstetter, T. 1992. Expression and secretion in yeast of active insect defensin, an inducible antibacterial peptide from the flesh fly Phormia terranovae. Invert. Reprod. Dev. 21 15–24. [Google Scholar]
- Tamamura, H., Kuroda, M., Masuda, M., Otaka, A., Funakoshi, S., Nakashima, H., Yamamoto, N., Waki, M., Matsumoto, A., Lancelin, J.M., et al. 1993. A comparative study of the solution structures of tachyplesin I and a novel anti-HIV synthetic peptide, T22 ([Tyr5,12, Lys7]-polyphemusin II), determined by nuclear magnetic resonance. Biochim. Biophys. Acta 1163 209–216. [DOI] [PubMed] [Google Scholar]
- Thevissen, K., Ghazi, A., De Samblanx, G.W., Brownlee, C., Osborn, R.W., and Broekaert, W.F. 1996. Fungal membrane responses induced by plant defensins and thionins. J. Biol. Chem. 271 15018–15025. [DOI] [PubMed] [Google Scholar]
- Thevissen, K., Osborn, R.W., Acland, D.P., and Broekaert, W.F. 1997. Specific, high affinity binding sites for an antifungal plant defensin on Neurospora crassa hyphae and microsomal membranes. J. Biol. Chem. 272 32176–32181. [DOI] [PubMed] [Google Scholar]
- Thevissen, K., Terras, F.R., and Broekaert, W.F. 1999. Permeabilization of fungal membranes by plant defensins inhibits fungal growth. Appl. Environ. Microbiol. 65 5451–5458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thevissen, K., Osborn, R.W., Acland, D.P., and Broekaert, W.F. 2000a. Specific binding sites for an antifungal plant defensin from Dahlia (Dahlia merckii) on fungal cells are required for antifungal activity. Mol. Plant Microbe Interact. 13 54–61. [DOI] [PubMed] [Google Scholar]
- Thevissen, K., Cammue, B.P., Lemaire, K., Winderickx, J., Dickson, R.C., Lester, R.L., Ferket, K.K., Van Even, F., Parret, A.H., and Broekaert, W.F. 2000b. A gene encoding a sphingolipid biosynthesis enzyme determines the sensitivity of Saccharomyces cerevisiae to an antifungal plant defensin from dahlia (Dahlia merckii). Proc. Natl. Acad. Sci. 97 9531–9536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uttenweiler-Joseph, S., Moniatte, M., Lambert, J., Van Dorsselaer, A., and Bulet, P. 1997. A matrix-assisted laser desorption ionization time-of-flight mass spectrometry approach to identify the origin of the glycan heterogeneity of diptericin, an O-glycosylated antibacterial peptide from insects. Anal. Biochem 247 366–375. [DOI] [PubMed] [Google Scholar]
- Vita, C., Vizzavona, J., Drakopoulou, E., Zinn-Justin, S., Gilquin, B., and Menez, A. 1998. Novel miniproteins engineered by the transfer of active sites to small natural scaffolds. Biopolymers 47 93–100. [DOI] [PubMed] [Google Scholar]
- Wüthrich, K. 1986. NMR of proteins and nucleic acids. John Wiley & Sons, New York.
- Yang, Y.S., Mitta, G., Chavanieu, A., Calas, B., Sanchez, J.F., Roch, P., and Aumelas, A. 2000. Solution structure and activity of the synthetic four-disulfide bond Mediterranean mussel defensin (MGD-1). Biochemistry 39 14436–14447. [DOI] [PubMed] [Google Scholar]
- Zasloff, M. 2002. Antimicrobial peptides of multicellular organisms. Nature 415 389–395. [DOI] [PubMed] [Google Scholar]
- Zhao, Q., Chae, Y.K., and and Markley, J.L. 2002. NMR solution structure of ATTp, an Arabidopsis thaliana trypsin inhibitor. Biochemistry 41 12284–12296. [DOI] [PubMed] [Google Scholar]