

Abstract
Using only hard-sphere repulsion, we investigated short polyalanyl chains for the presence of sterically imposed conformational constraints beyond the dipeptide level. We found that a central residue in a helical peptide cannot adopt dihedral angles from strand regions without encountering a steric collision. Consequently, an α-helical segment followed by a β-strand segment must be connected by an intervening linker. This restriction was validated both by simulations and by seeking violations within proteins of known structure. In fact, no violations were found within an extensive database of high-resolution X-ray structures. Nature’s exclusion of α-β hybrid segments, fashioned from an α-helix adjoined to a β-strand, is built into proteins at the covalent level. This straightforward conformational constraint has far-reaching consequences in organizing unfolded proteins and limiting the number of possible protein domains.
Keywords: protein folding, unfolded protein, protein secondary structure, Ramachandran plot
The hard sphere model (Richards 1977) has been an invaluable tool in characterizing fundamental aspects of protein molecules, including their accessible surface area (Lee and Richards 1971; Eriksson et al. 1992), packing (Richards 1977, 1979), and fitting errors (Word et al. 1999). Clearly, atoms are not simply hard spheres, but, quoting Richards,
“For chemically bonded atoms the distribution is not spherically symmetric nor are the properties of such at oms isotropic. In spite of all this, the use of the hard sphere model has a venerable history and an enviable record in explaining a variety of different observable properties (Richards 1977).”
Arguably, the most important application of the hard sphere model in biochemistry is the now famous φ,ψ-plot for a dipeptide, developed by Sasisekharan, Ramakrishnan, and Ramachandran (Ramachandran et al. 1963; Ramachandran and Sasisekharan 1968). This simple idea has been applied to nucleic acids as well (Murthy et al. 1999). In proteins, the hard sphere model identifies two major populated regions for an alanine dipeptide; backbone dihedral angles in these regions resemble those of an α-helix or a β-strand. Despite their remarkable structural diversity, protein molecules have mainchain conformations that lie almost entirely within these two regions.
In this paper, we explore additional steric constraints on polypeptide chains beyond the dipeptide level. We find that an α-helix cannot be followed by a β-strand without an intervening linker. This restriction is a consequence of unavoidable collisions between backbone atoms, and it derives experimental support from the paucity of exceptions among high-resolution protein structures (Berman et al. 2000).
We use several conventions for describing regions of the φ,ψ-map. Figure 1 ▶ shows a φ,ψ-distribution of α-helix (green), β-strand (red), and polyproline II helix (blue), determined from structures in the Protein Data Bank (PDB; Berman et al. 2000); the more populated the region, the darker the color. Throughout this paper, β refers to the region given by −135.0° ≤ φ ≤ −105.0° and 120.0° ≤ ψ ≤ 150.0°, and PII refers to −80.0° ≤ φ ≤ −55.0° and 130.0° ≤ ψ ≤ 155.0°. We define both a relaxed helical region α′, where −75.0° ≤ φ ≤ −45.0° and −60.0° ≤ ψ ≤ −30.0°, and a strict helical region α, as a circle of radius 7.0° centered about φ = −63.0° and ψ = −45.0°. Finally, κ represents the entirety of sterically accessible φ,ψ-space for the alanine dipeptide.
Figure 1.

(A) The distribution of secondary structure from the PDB using 5° by 5° bins (see Materials and Methods). Colors: β-strand, red; polyproline II, blue; α-helix, green. The regions β, PII, and α′, defined in the text, are shown as black boxes embedded in the colored regions. (B) Smaller 1° by 1° bins were used to determine the size and location of the α-region, shown as a black circle overlaid on the PDB distribution, in green.
An experimental observation raises a question
Hybrid segments consisting of an α-helix followed by a β-strand are rarely observed in the PDB. Instead, helices and strands are interconnected by a transition region—a turn, a loop, or some other linker. Only seven occurrences of three or more α′ residues followed by a single β residue were found in a representative set of PDB structures (Hobohm and Sander 1994), but a pattern consisting of three or more α′ residues followed by a non-α′ residue was detected 37,563 times in this data set. Why is the direct transition from α to β so rare?
Materials and methods
Ramachandran plots
Ramachandran plots were generated from φ,ψ-distributions by subdividing φ,ψ-space into a 5° by 5° grid; each of the 72 by 72 grid squares corresponds to a bin. These bins were ranked according to the number of φ,ψ-pairs they contain and then grouped into three categories representing the top 33%, 66%, and 90% of the data. The three groups are plotted in Figure 1 ▶.
Mining the PDB
In all cases, the chains selected from the PDB were that subset of PDBSelect (Hobohm and Sander 1994) structures determined by X-ray diffraction. In all, 1455 chains at the 25% aligned sequence identity level and 5378 chains at the 90% level were included.
Idealized secondary structures
Secondary structure was assigned using PROSS (Srinivasan and Rose 1999), a method based solely on φ,ψ-angles. Unlike the more familiar DSSP (Kabsch and Sander 1983), PROSS does not include hydrogen bonding in its assignment criteria. Distributions of φ,ψ-values were grouped into three secondary structure categories: α-helix, β-strand, and polyproline II (PII; Fig. 1A ▶). The helical region was further subdivided into 1° grid squares (Fig. 1B ▶). Idealized ranges for α, β, and PII were then defined, guided by those bins that represent the top 33% of the data. The α′ region is a relaxed definition of α, similar in size to β and PII. These definitions agree well with textbook classifications of secondary structure (Creighton 1984).
Simulations
Monte Carlo simulations of polyalanyl peptides were performed to determine how steric factors influence chain conformation. Polyalanine was chosen as a model for the peptide backbone. Simulated peptides had lengths ranging from 9 to 12 residues. Hydrogen atoms were not included. For each simulation, the φ,ψ-distributions of all sterically allowed conformers were collected so as to accumulate up to 5000 clash-free structures from a maximum of five million attempts.
Generation of sterically allowed structures was accomplished by sampling backbone torsion angles at random from α, β, PII, or κ, as appropriate. ω-Torsions were varied at random in the range [−175.0°, −185.0°] and assigned in conjunction with backbone torsions. Each attempt was checked for collisions; if none were found, the conformer was accepted and its torsion angles were retained. Otherwise, the conformer was rejected. Rejected structures with a single atomic collision occur at the boundaries between regions; these were cataloged by φ,ψ-angle and collision type for use in assembling a collision map. Structures with multiple atomic collisions are not localized at boundary regions and were ignored.
Hard sphere atomic radii from Word et al. (1999) are among the most conservative in the literature and were adopted for this study (Table 1). These radii were further scaled by a factor of 0.95, ensuring that the observed collisions are not methodological artifacts. The overall robustness of our results was tested extensively by determining the degree to which steric restrictions persist as radii diminish (described below). The collisions identified in this study do not include those with hydrogen atoms. Inclusion of hydrogens would have enlarged the effective radii and, consequently, imposed further restrictions on available conformational space.
Table 1.
Hard sphere radii
| Atom type | Radius (Å)a | Scaled radius (Å)b |
| Carbon | 1.75 | 1.66 |
| Carbonyl Carbon | 1.65 | 1.57 |
| Nitrogen | 1.55 | 1.47 |
| Oxygen | 1.40 | 1.33 |
a Atomic radii taken from Word et al. (1999).
b Radii shown use the scaling factor of 0.95.
Results
Flexibility of a central wild-card residue in an α-helical peptide: α4-κ-α4
A series of host–guest simulations was performed; each consisted of a wild-card κ-residue (the guest) in the middle of an eight-residue polyalanyl peptide that was constrained to be helical (the host). In every case, sterically disallowed patterns identified in simulations were validated against X-ray-elucidated structures by searching for exceptions.
The resulting distribution of the guest residue (Fig. 2 ▶) plots 5000 allowed structures (from 418,213 attempts). Both raw (Fig. 2A ▶) and binned (Fig. 2B ▶) data exhibit a Y-shaped plot for the guest residue. A comparison of the two figures (Fig. 2A,B ▶) shows that the binning method captures the distribution successfully. Notably, the Y-shape encompasses α-helix, but both β-strand and PII are excluded. This conclusion is highlighted in Figure 2B ▶ by superimposing the 66% contour for β-strand from Figure 1A ▶ on the binned simulation data. In short, a single β-guest residue cannot avoid a steric collision in an α-helical host.
Figure 2.

The φ,ψ-distribution in polyalanine for a central κ-residue flanked on either side by four consecutive α-residues. (A) Raw φ,ψ-values. Each point represents a sterically allowed structure when all residues were assigned random values of φ and ψ. (B) Same data as A but grouped into 5° × 5° bins. Sterically disfavored regions fall outside the 90% boundaries; the most favorable regions are the most intensely colored. The 66% contour line of the observed β-strand distribution (from Fig. 1A ▶) is shown in black outline.
The collision maps in Figure 3 ▶ rationalize this restriction, which is a consequence of a steric clash between the carbonyl oxygens of the guest i-residue (Oi) and the i−3 α-residue (Oi−3). The distribution of points for this collision (Fig. 3 ▶, green) fits precisely into the void region of Figure 2 ▶.
Figure 3.

Collision map for a single κ-residue in a sequence of α-residues. Atom collisions responsible for the Y-shape in Fig. 2 ▶ are color coded: Oi−3 − Oi, green; Oi−3 − CBi+3, red; Oi−1 − Ci, blue; Oi−3 − Oi+1, brown; Ci−1 − Ni+1, purple; Ci−1 − Ni+1, cyan; Oi−1 − CBi, yellow. The most conspicuous collision, between Oi and Oi−3, is responsible for the void in the strand region, in Fig. 2 ▶.
Additional collisions from this simulation are also shown in Figure 3 ▶. Of particular note is the collision between Oi−3 and Cβi+3 (in red) which is responsible for exclusion of the PII region. These two atoms are brought into juxtaposition when the guest κ-residue, at i, samples the relevant region in φ,ψ-space.
Two different methods were used to test the robustness of these results. First, the atomic radii were reduced well beyond any plausible van der Waals limit by successively decrementing the scaling factor from 0.95 to 0.92, 0.90, 0.88, and 0.85 (Fig. 4A–D ▶). Reduction of the atomic radii results in expansion of the Y-shaped boundaries of the guest residue. However, substantial strand exclusion survives even the most extreme reduction. Similar behavior was also observed when the α-region was expanded to a radius of 14° or 21° (Fig. 4E,F ▶). Again, the Y-shaped boundary expands, yet persists. Thus, steric exclusion of a β-residue in a sequence of α-residues is a robust finding, not an artifact of our helix definition or hard sphere radii.
Figure 4.

(A–D) Reducing the hard sphere scaling factor. Same experiment as Fig. 2B ▶, but with scaling factors of 0.92, 0.90, 0.88, and 0.85, respectively. Even at the extreme of 0.85, a remnant of the original Y-shape survives. (E,F) Relaxing the definition of α. A radius of 14° around φ = −63.0° and ψ = −45.0° (E) and 21° (F). On all plots, the 66% contour line from the observed β-strand distribution (in Fig. 1A ▶) is overlaid in black.
As further validation, helices were excised from proteins of known structure and used as starting structures in simulations. Specifically, forty 12-residue helices were selected at random from X-ray-elucidated structures in the PDB (Table 2), and all side chain atoms beyond Cβ were eliminated. Simulations were then performed as before, except that the definition of α was varied for each helical residue, using a radius of 7.0° centered about its experimentally determined φ,ψ-value: α6-κ-α5. With a radial scaling factor of 0.95, all but three helices were found to be sterically incompatible with β-values for the central residue. The β-region was largely, but not entirely, excluded in these three exceptions as well (Fig. 5 ▶); in each case, the φ,ψ-values of flanking residues were well outside the high-confidence α-region (Fig. 1B ▶), sometimes extending into 310 helix.
Table 2.
Twelve-residue helices used in testing robustness
| PDB IDa | Res (Å) | R factor | Residue start | Residue end | PDB ID | Res (Å) | R factor | Residue start | Residue end |
| 1AIHA | 2.5 | 0.21 | 176 | 187 | 1HBKA | 2.0 | 0.20 | 51 | 62 |
| 1ALN | 2.3 | 0.19 | 14 | 25 | 1HNNB | 2.4 | 0.23 | 604 | 615 |
| 1B16A | 1.4 | 0.18 | 109 | 120 | 1HQ6B | 2.7 | 0.25 | 123 | 134 |
| 1CXQA | 1.0 | 0.13 | 185 | 196 | 1IXH | 1.0 | 0.12 | 298 | 309 |
| 1D2HA | 3.0 | 0.20 | 247 | 258 | 1JSYF | 2.0 | 0.23 | 4 | 15 |
| 1D6JA | 2.0 | 0.21 | 110 | 121 | 1J9LAa | 1.9 | 0.20 | 14 | 25 |
| 1DI2B | 1.9 | 0.23 | 113 | 124 | 1JD22 | 3.0 | 0.25 | 167 | 178 |
| 1DSZAa | 1.7 | 0.20 | 1158 | 1164 | 1JK7A | 1.9 | 0.20 | 146 | 157 |
| 1EG9E | 1.6 | 0.19 | 592 | 603 | 1JMVA | 1.9 | 0.22 | 64 | 75 |
| 1EJ0A | 1.5 | 0.20 | 34 | 45 | 1JN0A | 3.0 | 0.21 | 252 | 263 |
| 1EJ3A | 2.3 | 0.22 | 162 | 173 | 1KPGA | 2.0 | 0.19 | 185 | 196 |
| 1EXJB | 3.0 | 0.24 | 51 | 62 | 1MUN | 1.2 | 0.12 | 30 | 41 |
| 1F0JBa | 1.8 | 0.20 | 191 | 202 | 1POC | 2.0 | 0.19 | 61 | 72 |
| 1F0JB | 1.8 | 0.20 | 261 | 272 | 1POC | 2.0 | 0.19 | 77 | 88 |
| 1F4LA | 1.9 | 0.18 | 536 | 547 | 1QTWA | 1.0 | 0.12 | 268 | 279 |
| 1FSGA | 1.1 | 0.00 | 153 | 164 | 1XROa | 3.0 | 0.20 | 64 | 75 |
| 1FUIA | 2.5 | 0.16 | 65 | 76 | 1YGE | 1.4 | 0.20 | 159 | 170 |
| 1G9ZA | 1.8 | 0.20 | 99 | 110 | 2ACY | 1.8 | 0.17 | 22 | 33 |
| 1GAL | 2.3 | 0.18 | 29 | 40 | 4HB1 | 2.9 | 0.23 | 11 | 22 |
| 1GRCB | 3.0 | 0.19 | 12 | 23 | 8OHM | 2.3 | 0.23 | 506 | 517 |
a The set of 40 12-residue helical segments simulated using experimentally determined φ, ψ-values (see text). Table entries marked with superscript represent those in which the β-region was not excluded completely; even in these four cases, the distribution maintains a distinct Y-shape (see Fig. 5 ▶). The chain identifier is listed as the fifth character of the PDB ID.
Figure 5.
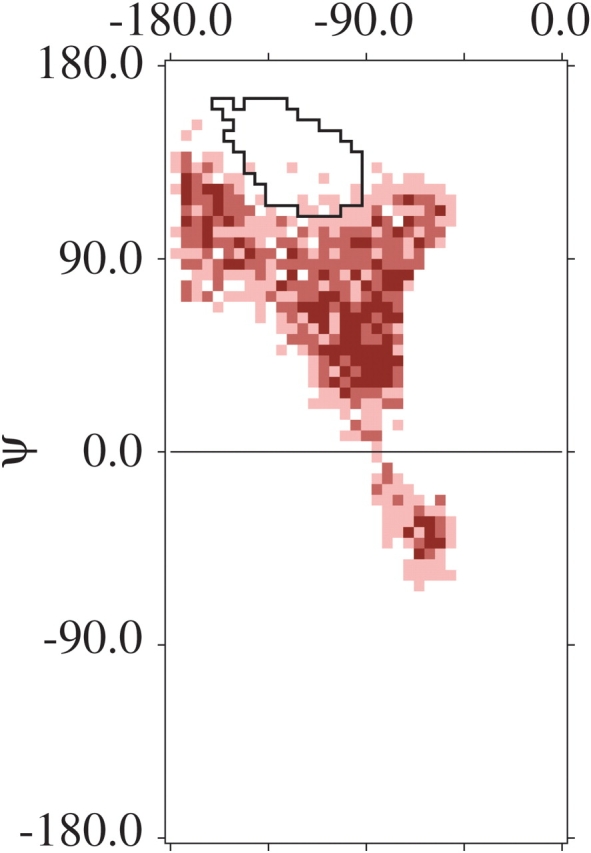
One of three extreme examples from the set of 40 12-residue helical segments (Table 2, PDB entry 1J9L, chain A, residues 14–25) in which simulations used experimentally determined φ’s and ψ’s to define α (see text). The resulting distribution still maintains a Y-shape, but a slight overlap with the 66% β-strand contour is evident.
In all, these results culminate in a prediction that a β-residue cannot follow three or more consecutive α-residues, a testable hypothesis using the PDB. In the list of 5378 chains with sequence identity of 90% or less, a series of three or more α-residues was found 19,062 times; none was followed by a β-residue. However, as mentioned earlier, seven exceptions were found when α′ is used instead of α (Table 3). For two of these structures, a small but real overlap is indicated between carbonyl oxygens as assessed by either our unscaled radii or contact dots (Word et al. 1999). In a third case, backbone clash is avoided by an unusual progression of ω-torsions. The final four cases may be legitimate, albeit marginal, exceptions. One of the four cases involves a single β-residue that follows the α′-residues; the other three cases involve type III turns and do not represent an intermixing of helix and strand.
Table 3.
Violations of relaxed helical complementarity in the PDB
| PDB IDa | Res (Å) | R factor | Residue start | Residue end | O-O Distd (Å)b | Explanation |
| 1CERO | 2.5 | 0.20 | 43 | 46 | 2.8 | Collisionc |
| 1PHK | 2.2 | 0.21 | 197 | 200 | 2.9 | Tight packingd |
| 1QCIA | 2.0 | 0.23 | 177 | 180 | 2.8 | Collisionc |
| 1RCD | 2.0 | 0.19 | 128 | 131 | 3.1 | Tight packingd |
| 1DSSG | 1.9 | 0.17 | 43 | 46 | 3.1 | Omega anglese |
| 1IFT | 1.8 | 0.22 | 178 | 181 | 3.1 | Tight packingd |
| 1HFUA | 1.7 | 0.18 | 474 | 477 | 3.1 | Tight packingd |
a The chain identifier is listed as the fifth character of the PDB ID.
b Distance between the carbonyl oxygens of the first and last residues.
c A collision between carbonyl oxygens is observed using unscaled radii (Table 1).
d No collision observed, but packing is tight and perturbation of any torsion angle would lead to a collision.
e Violation occurs because of devations from planarity in ω-torsions; deviations were greater than 5.0° for all four residues.
In sum, a direct transition from canonical α-helix to β-strand is disallowed: A single β-residue adjoined to a helical peptide results in a steric collision (Fig. 6 ▶). Further, this collision only affects residues N-terminal to the β-residue. Therefore, an N-terminal to C-terminal transition from helix to strand must pass through at least one “buffer” residue from the turn region. This finding rationalizes the familiar observation that many α-helices terminate in a 310 helix (Richardson 1981), a progression that both satisfies helix capping requirements (Aurora and Rose 1998) and facilitates the transition from helix to strand, turn, or loop.
Figure 6.

A β-residue cannot be added to three or more residues of α-helix without encountering a steric clash. Ball-and-stick backbone atoms for three residues of an α-helix (αi−3 − αi−1) are shown superimposed on a longer helical ribbon, followed by a single β-residue (βi). This conformation forces a substantial overlap between Oi and Oi−3, shown here as transparent van der Waals spheres. Atoms are rendered using conventional CPK colors: carbon, black; nitrogen, blue; oxygen, red.
Discussion
More than four decades ago, Sasisekharan, Ramakrishnan and Ramachandran (Ramachandran et al. 1963; Ramachandran and Sasisekharan 1968) elucidated the steric map for an alanyl dipeptide (more precisely, the compound Cα-CO-NH-CαHR-CO-NH-Cα, which, has two degrees of backbone freedom, like a dipeptide). Similar ideas about the importance of sterics as an organizing force in proteins were also implicit in space-filling models (Koltun 1965), developed during this same era. Such ideas have been validated repeatedly in proteins (Berman et al. 2000) and are now invoked routinely when assessing the quality of experimentally determined structures (Laskowski et al. 1993).
Today, the restrictions that sterics impose on the conformation of a dipeptide are widely accepted. Yet, hard sphere models have played a comparatively small role in both protein structure prediction and analysis of the unfolded state. Why?
The perceived problem is one of scale. If each φ,ψ-pair is independent of its neighbors (Flory 1969), then conformational space grows exponentially, despite dipeptide restrictions. Accordingly, the conformations accessible to a peptide backbone—even a short one—can quickly overwhelm constraints imposed by dipeptide sterics. This view is often invoked by alluding to the “Levinthal paradox” (Levinthal 1969): How does a protein find its unique native conformation among the more-than-astronomical number of conformational possibilities? For Levinthal, this conundrum was a demonstration, not a paradox, indicating that additional conformational constraints must exist. But what additional constraints might have been overlooked in this well cultivated field?
Earlier work using explicit counting showed that the size of conformational space is smaller than previously believed (Pappu et al. 2000) because local steric interactions exert influence beyond the dipeptide, winnowing the number of accessible conformations. Here, we focused specifically on steric restrictions in the α-helix.
Unfolded proteins
It has been proposed that the coil library—defined as the set of all nonhelical, nonstrand structures in the PDB—can be used to model the unfolded state of proteins (Swindells et al. 1995; Avbelj and Baldwin 2003). Therefore, our steric rules, which were validated against the PDB, including the coil library, would also hold for this model of the unfolded state. Plausibly so, because van der Waals repulsive forces will be unaffected by whether or not the protein is folded or unfolded.
Repulsive forces can have an organizing influence on the folding reaction, N(ative) ⇄ U(nfolded), and related order-disorder transitions, such as helix-coil theory (Zimm and Bragg 1959; Van Holde et al. 1998), where each residue is characterized by initiation and propagation constants. For example, any “coil” conformation with φ,ψ-angles in the β-region would exert a cooperative influence on the helix-coil equilibrium, making it harder to initiate a helix from the coil state by constricting the size of conformational space accessible to a residue that follows a helix nucleation site. Conversely, once nucleated it would also be harder to melt a helix, because the helical conformer would inhibit introduction of a central coil residue with φ,ψ-angles in either the β- or PII-regions.
The number of protein domains
Our analysis of short polyalanyl chains demonstrates that a β-conformer cannot be introduced into an α-helix without an accompanying steric clash. This restriction maintains the structural homogeneity of α-helices by excluding heterogeneous conformers consisting of a turn of α-helix followed by one or more β-residues. Exclusion of folds in which there is an immediate transition from helix to strand eliminates many conceivable protein domains.
In particular, when a protein folds, backbone polar groups removed from solvent will participate in compensatory intramolecular hydrogen bonds. To do so, they form segments of α-helix or strands of β-sheet, the only regular, repeating hydrogen-bonded protein structures that are sterically available (Aurora et al. 1997). Proteins are largely supramolecular complexes of helices and strands (Levitt and Chothia 1976), and their intramolecular recognition and self-assembly are facilitated by the sterically imposed elimination of α-β hybrids.
Acknowledgments
We thank Rajgopal Srinivasan, Rohit Pappu, Teresa Przytycka, Patrick Fleming, Nicholas Panasik, Timothy Street, Haipeng Gong, and Robert L. Baldwin for formative discussion, and Ross Shiman for his critical reading of this manuscript. Support from the NIH and the Mathers Foundation is gratefully acknowledged.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Article published online ahead of print. Article and publication date are at http://www.proteinscience.org/cgi/doi/10.1110/ps.03503304.
References
- Aurora, R. and Rose, G.D. 1998. Helix capping. Protein Sci. 7 21–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aurora, R., Creamer, T.P., Srinivasan, R., and Rose, G.D. 1997. Local interactions in protein folding: Lessons from the α-helix. J. Biol. Chem. 272 1413–1416. [DOI] [PubMed] [Google Scholar]
- Avbelj, F. and Baldwin, R.L. 2003. Role of backbone solvation and electrostatics in generating preferred peptide backbone conformations: Distributions of φ. Proc. Natl. Acad. Sci. 100 5742–5747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman, H.M., Westbrook, J., Feng, Z., Gilliland, G., Bhat, T.N., Weissig, H., Shindyalov, I.N., and Bourne, P.E. 2000. The Protein Data Bank. Nucleic Acids Res. 28 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creighton, T.E. 1984. Proteins: Structures and molecular principles, p. 515. H. Freeman, New York.
- Eriksson, A.E., Baase, W.A., Zhang, X.J., Heinz, D.W., Blaber, M., Baldwin, E.P., and Matthews, B.W. 1992. Response of a protein structure to cavity-creating mutations and its relation to the hydrophobic effect. Science 255 178–183. [DOI] [PubMed] [Google Scholar]
- Flory, P.J. 1969. Statistical mechanics of chain molecules, pp. 250–255. Wiley, New York.
- Hobohm, U. and Sander, C. 1994. Enlarged representative set of protein structures. Protein Sci. 3 522–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch, W. and Sander, C. 1983. Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 22 2577–2637. [DOI] [PubMed] [Google Scholar]
- Koltun, W.L. 1965. Precision space-filling atomic models. Biopolymers 3 665–679. [DOI] [PubMed] [Google Scholar]
- Laskowski, R.A., MacArthur, M.W., Moss, D.S., and Thornton, J.M. 1993. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Cryst. 26 283–291. [Google Scholar]
- Lee, B. and Richards, F.M. 1971. The interpretation of protein structures: Estimation of static accessibility. J. Mol. Biol. 55 379–400. [DOI] [PubMed] [Google Scholar]
- Levinthal, C. 1969. In Mössbauer spectroscopy in biological systems (eds. P. Debrunner et al.), pp. 22–24. University of Illinois Press, Urbana, IL.
- Levitt, M. and Chothia, C. 1976. Structural patterns in globular proteins. Nature 261 552–558. [DOI] [PubMed] [Google Scholar]
- Murthy, V.L., Srinivasan, R., Draper, D.E., and Rose, G.D. 1999. A complete conformational map for RNA. J. Mol. Biol. 291 313–327. [DOI] [PubMed] [Google Scholar]
- Pappu, R.V., Srinivasan, R., and Rose, G.D. 2000. The flory isolated-pair hypothesis is not valid for polypeptide chains: Implications for protein folding. Proc. Natl. Acad. Sci. 97 12565–12570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramachandran, G.N. and Sasisekharan, V. 1968. Conformation of polypeptides and proteins. Adv. Prot. Chem. 23 283–438. [DOI] [PubMed] [Google Scholar]
- Ramachandran, G.N., Ramakrishnan, C., and Sasisekharan, V. 1963. Stereochemistry of polypeptide chain configurations. J. Mol. Biol. 7 95–99. [DOI] [PubMed] [Google Scholar]
- Richards, F.M. 1977. Areas, volumes, packing, and protein structure. Ann. Rev. Biophys. Bioeng. 6 151–176. [DOI] [PubMed] [Google Scholar]
- ———. 1979. Packing defects, cavities, volume fluctuations, and access to the interior of proteins. Including some general comments on surface area and protein structure. Carlsberg Res. Commun. 44 47–63. [Google Scholar]
- Richardson, J.S. 1981. The anatomy and taxonomy of protein structure. Adv. Prot. Chem. 34 168–340. [DOI] [PubMed] [Google Scholar]
- Srinivasan, R. and Rose, G.D. 1999. A physical basis for protein secondary structure. Proc. Natl. Acad. Sci. 96 14258–14263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swindells, M.B., MacArthur, M.W., and Thornton, J.M. 1995. Intrinsic φ, ψ propensities of amino acids, derived from the coil regions of known structures. Nat. Struct. Biol. 2 596–603. [DOI] [PubMed] [Google Scholar]
- Van Holde, K.E., Johnson, W.C., and Ho, P.S. 1998. Principles of physical biochemistry, pp. 156–162. Prentice Hall, Upper Saddle River, NJ.
- Word, J.M., Lovell, S.C., LaBean, T.H., Taylor, H.C., Zalis, M.E., Presley, B.K., Richardson, J.S., and Richardson, D.C. 1999. Visualizing and quantifying molecular goodness-of-fit: Small-probe contact dots with explicit hydrogen atoms. J. Mol. Biol. 285 1711–1733. [DOI] [PubMed] [Google Scholar]
- Zimm, B.H. and Bragg, J.K. 1959. Theory of the phase transition between helix and random coil in polypeptide chains. J. Chem. Phys. 31 526–535. [Google Scholar]