

Abstract
RNase PH is a member of the family of phosphorolytic 3′ → 5′ exoribonucleases that also includes polynucleotide phosphorylase (PNPase). RNase PH is involved in the maturation of tRNA precursors and especially important for removal of nucleotide residues near the CCA acceptor end of the mature tRNAs. Wild-type and triple mutant R68Q-R73Q-R76Q RNase PH from Bacillus subtilis have been crystallized and the structures determined by X-ray diffraction to medium resolution. Wild-type and triple mutant RNase PH crystallize as a hexamer and dimer, respectively. The structures contain a rare left-handed βαβ-motif in the N-terminal portion of the protein. This motif has also been identified in other enzymes involved in RNA metabolism. The RNase PH structure and active site can, despite low sequence similarity, be overlayed with the N-terminal core of the structure and active site of Streptomyces antibioticus PNPase. The surface of the RNase PH dimer fit the shape of a tRNA molecule.
Keywords: crystal structure, maturation of tRNA, ribonuclease, RNase PH, tRNA precursor
Six exoribonuclease families have been identified in Escherichia coli based on their sequence and catalytic properties (Zuo and Deutscher 2001). RNase PH, EC 2.7.7.56, is a member of the PDX family of phosphate-dependent 3′ → 5′ exoribonucleases that also includes PNPase, EC 2.7.7.8 (Mian 1997; Zuo and Deutscher 2001). These two enzymes are the only known examples of phosphorolytic exoribonucleases, which catalyze the cleavage of RNA substrates using inorganic orthophosphate. The reaction releases nucleoside diphosphates rather than the nucleoside monophosphates produced by hydrolytic RNases. The best characterized biological function of RNase PH is the trimming of tRNA precursors at their 3′ ends. This process involves the action of several exo- and endonucleases (Deutscher 1995). The exonucleases have overlapping specificities. Deleting a single gene encoding one of these enzymes in bacteria does not affect growth (Kelly and Deutscher 1992a). Despite the overlapping specificities of these enzymes, Li and Deutscher (1996) have shown that RNase PH together with RNase T are the prime enzymes responsible for removing the 2 nt closest to the CCA acceptor end of the tRNAs. They can substitute each other’s function, but the lack of both enzymes in E. coli leads to accumulation of 3′-extended tRNAs in the cells and severe retardation of cell growth (Kelly et al. 1992).
Other, but less understood functions of RNase PH are indicated by the observation that the lack of both RNase PH and PNPase in E. coli leads to very slow growth of the cells and a defect in ribosome assembly (Zhou and Deutscher 1997) and by the observation that high expression of Bacillus subtilis RNase PH in E. coli suppresses several cold-sensitive phenotypes (Craven et al. 1992) by a mechanism that is not at all understood.
Most of the biochemical characterization of RNase PH has been done with the enzyme isolated from E. coli (Poulsen et al. 1984; Jensen et al. 1992). In our hands, this enzyme did only generate poorly diffracting crystals. Instead we produced and characterized the enzyme from B. subtilis (Craven et al. 1992). The sequence of this enzyme is 50% identical to the sequence of the E. coli RNase PH. Recently, the structure of RNase PH from Aquifex aeolicus has been published (Ishii et al. 2003). In the present work, we report the crystal structures of wild-type and triple mutant R68Q-R73Q-R76Q RNase PH from B. subtilis.
Results
Description of the structure
The structure of B. subtilis RNase PH has been obtained from three different crystal forms, numbered I, II, and III as obtained in chronological order. Due to disorder in the crystals, none of the crystal forms has led to a complete model of the 245 residues of B. subtilis RNase PH. Crystal form II, containing a hexamer of residues Met 1-Pro 242 and two SO42− ions per monomer, yielded the most complete model and is used for the illustrations and comparisons. The more incomplete crystal forms I and III contain a hexamer of Met 1-Leu 66/Ser 84-Glu 243 and two SO42− ions per monomer and a dimer of the segments Met 1-Asp 19/Glu 25-Met 65/Gly 85-Glu 243, respectively (see also Table 1). Within experimental uncertainty, the monomeric structures of crystal forms I, II, and III are identical where they overlap. The 245-residue RNase PH monomer is a single domain subunit with a mixture of α-helix and β-strand secondary structure elements ordered in layers as a βαβα-sandwich. We have assigned nine β-strands and four α-helices numbered in sequential order (Figs. 1 ▶, 4 ▶). Strands β1 to β5 and β6 to β9 form a mixed and an antiparallel sheet, respectively. The two β-sheets are structurally separated by helices α1, α2, and α3 and helix α4 packs across the other face of sheet β6–β9. The hexameric form displays P32 symmetry; that is, it has three twofold axes perpendicular to a threefold axis. This symmetry creates two types of interfaces between the monomers. For one of the interfaces, the interactions between subunits A and B are clearly closer than for the other between subunits A and F. It is therefore natural to consider the hexamer as a trimer of dimers. The dimerization interface (AB) contains as a key feature an edge-to-edge antiparallel hydrogen bond interaction between the two β9 strands from both subunits A and B, resulting in the formation of a β6–β9:β9–β6 eight-stranded antiparallel β-sheet across the dimer (Fig. 2A ▶). In addition, four salt bridges contribute to the dimerization: Glu 89A/B-Arg 92B/A and Glu 89A/B-Arg 96B/A. The trimerization of dimers involves face-to-face interactions between sheets β1–β5 from subunits A and F (Fig. 2B ▶). In crystal form II, 10 salt bridges are observed (see after next section) and are partly responsible for this interaction. One of the two SO42− ions per monomer, in crystal forms I and II, is found at the N-terminal end of helix α2, where it binds to the main-chain atoms Thr 125-N and Arg 126-N and to the side-chain atoms T125-Oγ1 and Arg 126-Nη2. The second SO42− ion binds to side-chain atoms Trp 58-Nɛ1, Thr 60-Oγ1 and Arg 99-Nη1. The structures have been deposited with the Protein Data Bank with accession codes 1OYP, 1OYR, and 1OYS.
Table 1.
Data collection and refinement statistics
| Crystal form/PDB entry | |||
| I/1OYP | II/1OYR | III/1OYS | |
| Data collectiond | |||
| Space-group | P212121 | P212121 | P41212 |
| Cell dimensions (Å) | a = 51.3 | a = 99.7 | a = 63.2 |
| b = 163.6 | b = 146.5 | b = 63.2 | |
| c = 166.8 | c = 152.3 | c = 203.0 | |
| Temperature (K) | 120 | 263 | 120 |
| Wavelength (Å) | λ = 1.0924 | λ = 1.1024 | λ = 1.0689 |
| Resolution (Å)a | 30–2.75 (2.80–2.75) | 25–3.10 (3.15–3.10) | 20–2.40 (2.44–2.40) |
| # Unique reflections | 36,688 (1753) | 40,776 (2021) | 16,768 (817) |
| Completeness (%) | 99.2 (96.6) | 99.1 (99.8) | 98.8 (100.0) |
| Redundancy | 5.9 (4.9) | 5.2 (4.1) | 4.4 (5.5) |
| Rmerge (%)b | 9.7 (33.2) | 11.6 (37.2) | 4.7 (20.6) |
| I/σ(I) | 15.8 (3.9) | 10.1 (3.1) | 22.8 (7.0) |
| Refinement | |||
| # Copies/asym. unit | 6 | 6 | 1 |
| Vol./monomer (Å)c | 58,329 | 92,780 | 101,353 |
| Multimeric state | hexamer | hexamer | dimer |
| Model refined | 6 × (Met1–Leu 66, | 6 × (Met 1–Pro 242, | Met 1–Asp 19, |
| (full monomer comprises Met 1–Lys 245) | Ser 84–Glu 243, | 2 SO42− | Glu 25–Met 65, |
| 2 SO42− | + 3 Cd2+ | Gly 85–Leu 237 | |
| Rwork/Rfree (%)c | 28.5/30.6 | 27.8/28.9 | 25.8/28.4 |
a Outermost resolution shells and statistics for these shells are given in parentheses.
b Rmerge = |∑I − Iave|/∑ I, where I = observed intensity and Iave = average intensity obtained from multiple observations of symmetry related reflections.
c The R-factor is defined as R = ∑|Fobs − Fcalc|/∑Fobs; “work” refers to 95% of the data used in refinement and “free” is for 5% of the data excluded from refinement.
d Data set for crystal form I was collected at the MAX-lab synchrotron in Lund, Sweden, mounted with a MAR CCD detector. For crystal forms II and III, the data were collected at the DESY synchrotron in Hamburg, Germany, mounted with a MAR345 imaging plate and a MAR CCD detector, respectively.
Figure 1.
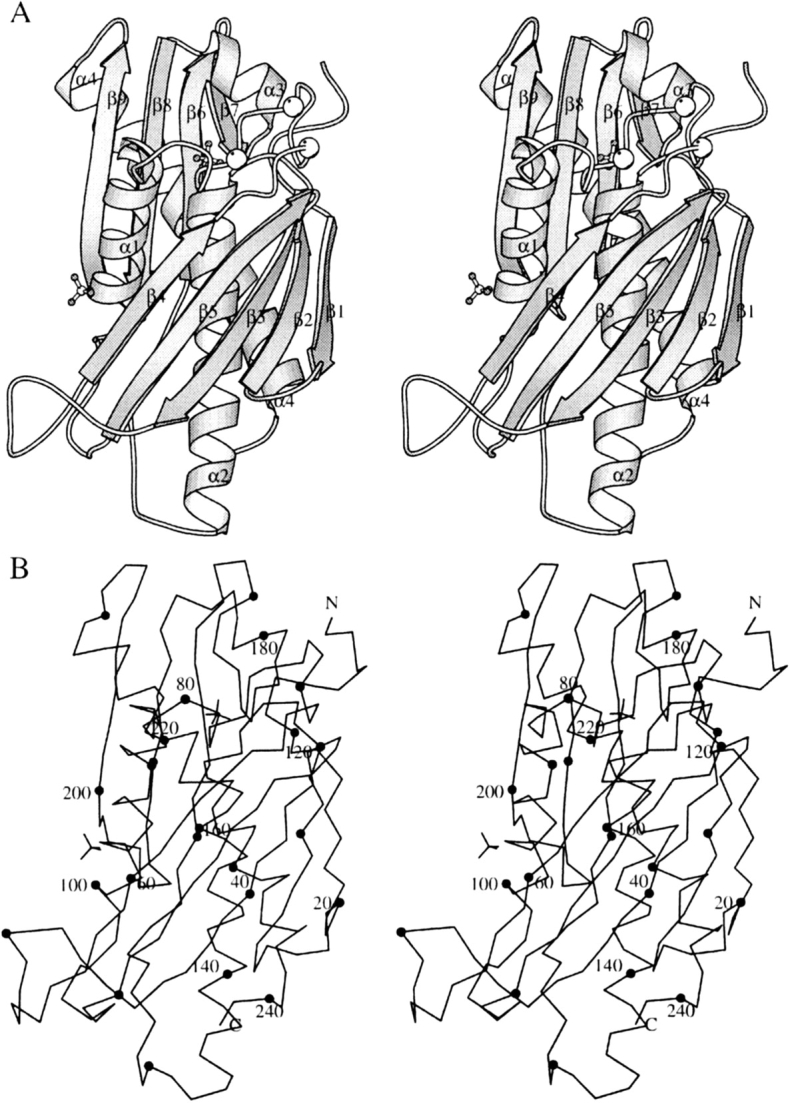
Stereo views of the B. subtilis RNase PH monomer. (A) Ribbon representation showing the secondary structure elements with labels and ball-and-stick representation of sulfate ions. Cα positions for mutated residues Arg68, Arg73, and Arg76 are marked with white balls. (B) Cα trace of the monomer with dots every 10 residues and labels every 20 residues. This figure and Figure 2 ▶ were made using the program MOLSCRIPT (Kraulis 1991).
Figure 4.

Alignments of B. subtilis RNase PH with A. aeolicus RNase PH and B. subtilis PNPase sequence. Secondary structure elements are shown in gray bars (β-strands) and streaked bars (α-helices). Conserved residues (in more than 90% of the sequences) are in dark gray (white letters) and semiconserved residues are in light gray (dark letters). (A) Amino acid sequence of B. subtilis RNase. Comparison with 33 other known bacterial RNase PH sequences. (B) Comparison between B. subtilis and A. aeolicus RNase PH amino acids sequences. (C) Sequence alignment of second core domain of PNPase from S. antibioticus (amino acids 338–571) and B. subtilis RNase PH (amino acids 1–245).
Figure 2.
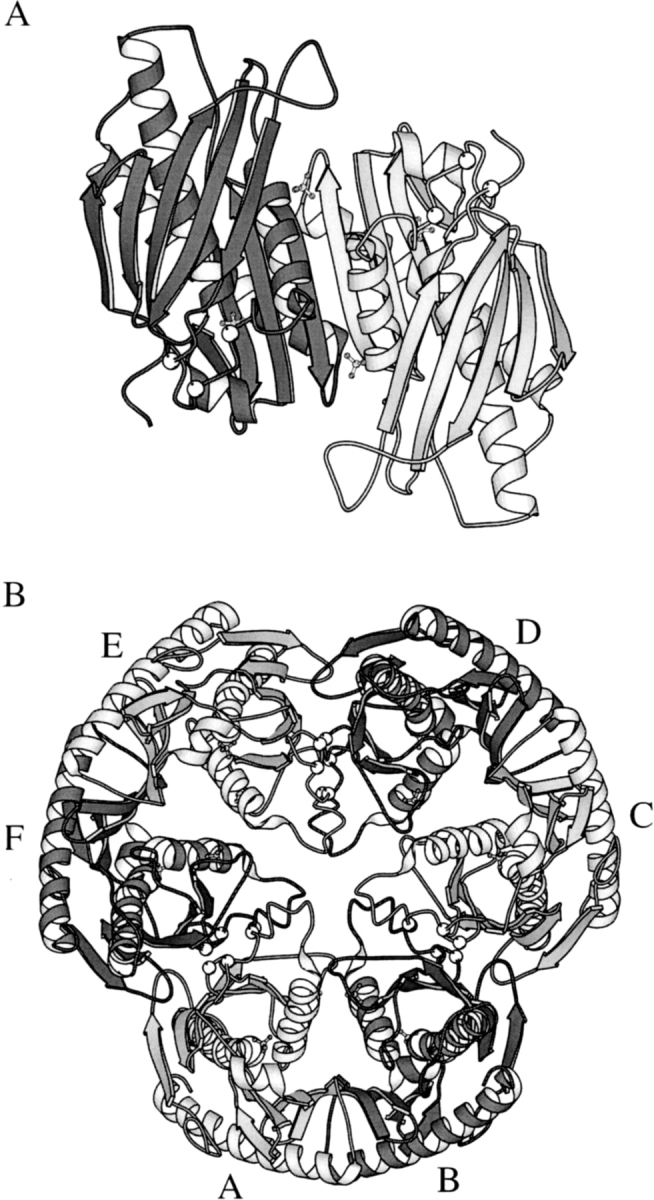
Ribbon representations of RNase PH multimers. Cα positions for mutated residues Arg68, Arg73, and Arg76 are marked with white balls. (A) View of the dimer along a twofold axis from the inside of the hexamer. The orientation is almost the same as for Figure 1 ▶. The most characteristic interactions between subunits A and B are β-backbone hydrogen bonds forming an antiparallel β6–β9:β9–β6 eight-stranded β-sheet across the dimer. Also four salt bridges (not shown) contribute to the dimer stabilization. (B) View along the threefold axis from the top of the hexamer. The hexamer interaction between subunits A and F is held together by 10 salt bridges (not shown).
Comparison with RNase PH from Aquifex aeolicus
The structure of A. aeolicus RNase PH has recently been published (Ishii et al. 2003; PDB entry: 1UDN). A sequence alignment of B. subtilis and A. aeolicus RNase PH showed 62% identity between the two enzymes (Fig. 4B ▶). The high identity between the two enzymes was also reflected in an excellent root-mean-square deviation of 0.77 Å using 228 pairs of Cα atoms with a maximum distance of 2.0 Å, when overlayed. Although the connectivity of the secondary structure elements is very similar, the A. aeolicus enzyme contains two additional short α-helices (α1, α2) and three extra β-strands (β10, β11, β12). The extra β-strands are located at the C terminus of the enzyme, which is 10 amino acid residues longer than the B. subtilis enzyme. The α-helix α2 is located in the central region of the hexameric structure, which, in crystal forms I and III of B. subtilis, are disordered. We show that this region contains conserved arginine residues that are important for maintaining the hexameric structure and propose that upon binding of the substrate RNA the hexamer is destablized.
Conserved arginine residues are important in assembly of the hexameric structure
The structure of crystal form II shows a long loop, Leu 66-Ser 84, which is unresolved in crystal forms I and III. During purification of the protein, this region was recognized to contain a cleavage site (probably a trypsinlike protease site) resulting in a heterogeneous sample (less than 5% were cleaved). The amino acid sequence in this region of the protein contains three arginyl residues, Arg 68, Arg 73, and Arg 76, of which the latter two are conserved among all RNase PH sequences. The three arginyl residues appear to be structurally important, stabilizing the hexamer by forming electrostatic interactions with acidic amino acid residues from the neighboring dimers. In crystal form II, the following 10 salt bridges between dimers are observed: Arg 68A/F-Glu 25F/A, Arg 73A/F-Glu 62F/A, Arg 73A/F-Asp 115F/A, Arg 76A/F-Glu 62F/A, and Arg 76A/F-Asp 117 F/A. We have substituted the arginyl residues in the loop region with glutaminyl residues by site-directed mutagenesis, resulting in the triple mutant enzyme R68Q-R73Q-R76Q. The spontaneous cleavage during purification of the wild-type enzyme is not observed in the R68Q-R73Q-R76Q enzyme. In contrast to wild-type RNase PH, the mutant enzyme crystallized as a dimer with nothing reminiscent of the hexamer form, yielding crystal form III.
The change in quaternary structure was consistent with results from size exclusion chromatography experiments, which also revealed a native molecular weight of the triple mutant enzyme corresponding to that of a dimer (data not shown). Thus, the substitution of the arginyl residues with glutaminyl residues in the triple mutant R68Q-R73Q-R76Q enzyme results in an enzyme with an apparently altered quaternary structure and clearly demonstrates that the three arginyl residues, Arg 68, Arg 73, and Arg 76, are important for maintaining the hexameric structure.
Comparison of RNase PH with the more distantly related polynucleotide phosphorylase
The crystal structure of another member of the RNase PH superfamily, Streptomyces antibioticus PNPase, has been solved by Symmons et al. (2000). In addition to the 3′ → 5′ phosphorylase activity shared with RNase PH, the S. antibioticus PNPase has a guanosine 3′-diphosphate-5′-triphosphate pppGpp synthetase activity. The monomer of S. antibiotocus PNPase consists of 757 amino acids and is thus considerably larger than the RNase PH monomer. The structural core of PNPase has two homologous domains interrupted by an all helical domain. Two RNA-binding domains are located at the C-terminal end of the protein, the KH (Burd and Dreyfuss 1994) and the S1 (Bycroft et al. 1997) domains. These domains have been shown to be dispensable for catalytic activity. Comparison of the two PNPase core domain sequences shows that they have diverged extensively since presumed gene duplication gave origin to the PNPase family (Symmons et al. 2002). The binding of the inhibitory orthophosphate analog tungstate to the second core domain suggests that the phosphorolytic activity resides in this domain. It had earlier been shown that the second core domain of PNPase aligns to RNase PH (Mian 1997). Despite relatively low sequence similarity of 17%, the connectivity of the secondary structure elements is similar in the two enzymes (Fig. 4C ▶) and both enzymes form an overall structure of a bulky disk with a central channel (Fig. 2B ▶). The quaternary structure is maintained by forming a trimer of monomers in PNPase and by forming a trimer of dimers in RNase PH. This places the three PNPase active sites on the same face of the disk, whereas in RNase PH the twofold symmetry of the dimer places three active sites on either side of the disk, as also has been noted by Symmons et al. (2002). We have superimposed the entire RNase PH structure with the second core domain of PNPase. The structures could be overlayed with a root-mean-square deviation of 1.1 Å using 184 pairs of Cα atoms with a maximum distance of 2.0 Å. It should be mentioned that as the two core domains of S. antibioticus PNPase have arisen from gene duplication, the first core domain of S. antibioticus PNPase also shows structural similarity to RNase PH.
The N-terminal part of RNase PH contains a fold common to other enzymes in RNA metabolism
The RNase PH structure was submitted to the Dali-server (Holm and Sander 1993) that searches for structural homology in the Protein Data Bank. This search revealed that the N-terminal portion: Gly 26 to Ala 147 of RNase PH has structural similarity to other enzymes involved in RNA metabolism. Two structures were found with very low Z scores: the protein part of the ribozyme ribonuclease P (Z = 3.2; Stams et al. 1998) and domain IV of elongation factor G Ef-G. (Z = 2.2; Al-Karadaghi et al. 1996). These structures are shown in Figure 3 ▶. The common fold consists of a four-stranded β-sheet covered on one side with two α-helices and displays a rare left-handed βαβ crossover motif. This topology has also been observed in the second domain of DNA gyrase B, a type II topoisomerase, and the C-terminal domain of ribosomal protein S5 (Murzin 1995), and argues for the evolution of a module adapted for polynucleotide binding. Photo-cross-linking studies of precursor tRNA molecules to RNase P holoenzyme have shown that the β-sheet forms a platform for binding of the tRNA molecules (Niranjanakumari et al. 1998). However, in RNase PH, the β-sheet is used for trimerization contacts between dimers to form the hexamer and can thus only be accessible to tRNA binding when the hexamer dissociates to dimers.
Figure 3.

Schematic representation of the structures of elongation factor G (PDB entry 1DAR), ribonuclease P protein (PDB entry 1A6F) and RNase PH. The structurally related parts indicating an RNA-binding module are highlighted in red, blue, and yellow. For RNase PH, the motif consists of the secondary structure elements β2, β3, β4, α1, β5, and α2. Also related structures are ribosomal protein S5 (PDB entry 1PKP) and DNA gyrase B (PDB entry 1EI1; Murzin 1995). This figure and Figure 6 ▶ were made using TURBO-FRODO (Roussel and Cambillau 1992).
The putative RNase PH active site and tRNA binding
Two sulfate ions per monomer are present in the RNase PH crystal forms I and II, and indicate possible sites for binding of Pi in the active site and binding of the phosphodiester bonds in the substrate tRNA. One of the sulfate ions binds to the conserved residues Thr 125 (Thr 462 of PNPase, which binds WO42−; see Fig. 4C ▶) and Arg 126 at the N terminus of helix α2. This corresponds to the binding of the phosphate ion in the A. aeolicus structure (binds to Thr 125 at the N terminus of α4) and thus strongly confirms the identification of the active site. This region also contains the highly conserved Gly 123 of RNase PH (Gly 460 of PNPase). Close by is helix α3 with the acidic motif DX4EDX5D (Zuo and Deutscher 2001), which is completely conserved in RNase PH (D175, E180, D181, D187) and PNPase (D508, E513, D514, D520) families. In the more distantly related members of the RNase PH family only the first aspartic acid is completely conserved (Mian 1997). In the RNase PH crystal form II, the carboxylate groups of Asp 181 and Asp 187 are pointing towards each other with a poorly resolved residual density in between (Fig. 5 ▶). This may represent a partially occupied Cd2+ ion site (not included in the model) as the crystals were grown in its presence. The role of these residues could therefore be binding of the divalent metal ion Mg2+, which is required for enzymatic activity (Kelly and Deutscher 1992b). However, Asp 181 and Asp 187 could also be the cleaving site of the phosphodiester bond by carrying one common catalytic proton. The active site residues, Thr 125, Arg 126, Asp 181, Asp 187, and the sulfate ion are shown in Figure 5 ▶. Connected to the DX4EDX5D motif is the RX3RX5R motif (Zuo and Deutscher 2001), which is also completely conserved in RNase PH (R2, R6, R12) and PNPase (R339, R343, R349). It has been proposed by Symmons et al. (2002) that this motif in PNPase could be involved in RNA binding. In RNase PH, however, this is only possible in an unfolded mode of the protein, as salt-bridge interactions are observed between Arg 2-Glu 180, Arg 6-Asp 4, and Arg 12-Asp 175, indicating a role as structural stabilization of helix α3.
Figure 5.

Close-up stereo view at the active site residues with sixfold averaged electron density. Nitrogen, oxygen, sulfur, and carbon atoms are dark gray, medium gray, light gray, and white, respectively. The 2mFobs − DFcalc total density is shown with weak line contours at a 1σ level and the mFobs − DFcalc difference density is shown with strong lines and contoured at 4σ. The carboxylate groups of Asp 181 and Asp 187 are unexpectedly close but residual density present in between may indicate the presence of a positive countercharge, for example, a partially occupied Cd2+ ion (not included in the model). This figure was made with BOBSCRIPT (Esnouf 1999).
The second sulfate ion binds to conserved residues Trp 58, Thr 60, and Arg 99 and could constitute the binding region for the phosphate backbone in the tRNA. In Figure 6 ▶ we have monitored conserved residues in a space-filling model of the RNase PH dimer. A groove extends from the active site in molecule A (marked by the active site sulfate ion), then passes by the second sulfate ion of molecule B and continues over the RNA binding βαβ-motif also located in molecule B. The groove conforms to the shape of a tRNA molecule with the 3′ end (CCA sequence) pointing down into the active site. Not only does this strongly argue that the dimer is the active form, but we also envisage that the hexamer is displaced upon substrate binding, due to interactions between the βαβ-motif and the substrate tRNA. This is consistent with the studies on the R68Q-R73Q-R76Q mutant. This model is different from the model of tRNA binding by Ishii et al. (2003), in which only the tRNA acceptor stem interacts with the dimer.
Figure 6.
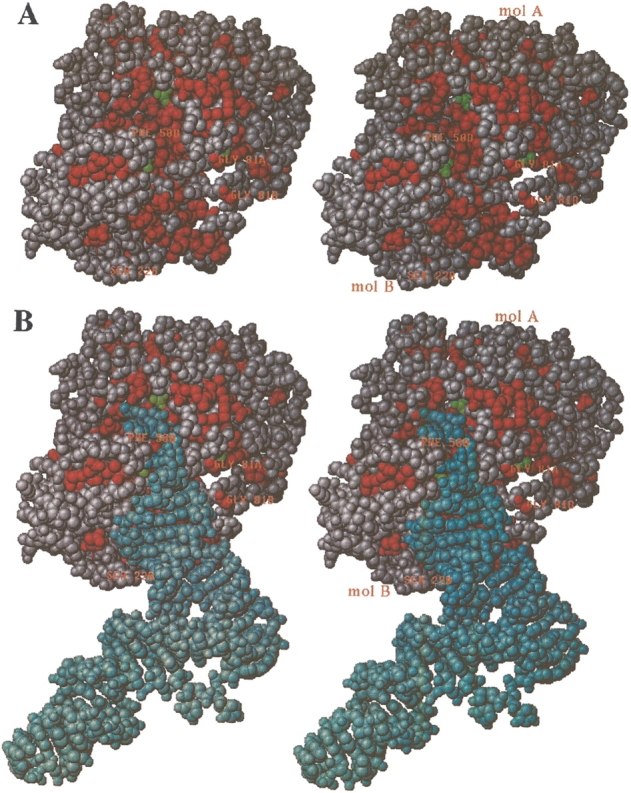
Stereo space-filling model of the dimer with conserved (among bacterial RNases PHs) residues colored in red. (A) View down the active site groove, which extends vertically from the active site (up top near Thr 125, containing a green colored sulfate ion) in molecule A, passes by the second sulfate south to molecule B, where the bottom of the groove is created by residues from secondary structure elements α1, β4, β5, and β3. The right side of the groove is flanked by the loops β4α1 containing the mutation sites Arg68, Arg73, Arg76 from both molecules A and B (marked with Gly 81A and Gly 81B, respectively). The left side is made of loop β3β4 (marked with Phe 50B) and loop β1β2 (marked with Ser 22B), both from the B molecule. (B) Same as in A, but now with a tRNA molecule docked into the active site groove. The docking was performed manually by restraining the 3′ end to be close to the active site sulfate (Thr 125) and then matching the complementary faces of tRNA and RNase PH. The tRNA coordinates used were taken from Nissen et al. (1999; PDB entry 1B23).
Discussion
The crystal structure of B. subtilis RNase PH, a member of the PDX superfamily of exoribonucleases, has been determined to medium resolution. Sequence similarity between the second core domain of S. antibioticus PNPase and RNase PH led Symmons et al. (2002) to suggest a structure of RNase PH similar to this PNPase domain. The structure of the second core domain of PNPase can be superimposed with the entire structure of RNase PH, resulting in a low root-mean-square deviation of 1.1 Å. The similar reactions catalyzed by these two enzymes also argue for a similar mechanism. However, the two enzymes have different substrate specificities in vivo. RNase PH is involved in the maturation process of precursor tRNA molecules, in particular removing the nucleotide residue at the +2 position following the CCA sequence. Because the +2 nucleotide is close to the stem structure formed by base pairing of the 3′ and 5′ends of the tRNA molecule, the enzyme likely recognizes short single-stranded RNA as well as the tertiary structure of the tRNA. PNPase involved in the degradation of mRNA is stalled by RNA tertiary structure (Littauer and Grunberg-Manago 1998) and recognizes longer stretches of single-stranded RNA compared to RNase PH. In vitro RNase PH and PNPase, however, both catalyze the degradation of structured and unstructured RNA although with different specificities, which is reflected in the ratio of poly(A) phosphorolysis over tRNA-CCA-Cn phosphorolysis, which is 11 and 1400 for RNase PH and PNPase, respectively (Ost and Deutscher 1991). The difference in substrate specificity may be correlated with the addition of the KH and S1 RNA-binding domains in course of evolution of PNPase. The location of the active site and the RNA-binding site on opposite sides of the disk in PNPase forms an arrangement where the RNA substrate can be retained on the enzyme for subsequent rounds of catalysis resulting in a processive mode of degradation. Symmons et al. (2000) have proposed two models for the binding of substrate RNA to PNPase. One model involves binding of RNA at the accessory KH and S1 domains at one side of the disk, down to the active site on the other side of the disk, by wrapping the RNA substrate around the trimeric molecule. According to the other model, the RNA is bound at a conserved loop region, the FFRR loop, of the first core domain of PNPase located near the KH and S1 domains, down through the central channel to the active site. These models are consistent with earlier observations that argue for two subsites on the enzyme (Godefroy 1970; Sulewski et al. 1989).
Substrate RNA binding
We have no biochemical data addressing the number of subsites present on RNase PH for the binding of tRNA. However, the density of a sulfate ion in proximity to the conserved residues Trp 58, Thr 60, and Arg 99 could be a potential binding site for phosphate groups in the RNA phosphodiester backbone. In addition, the highly conserved region of helix α1, RX5RX3RX2R (Arg 86, Arg 92, Arg 96, Arg 99), could also be thought as stabilizing the phosphodiester backbone of the tRNA substrate. Actually helix α1 contributes to the walls of the tRNA binding groove shown in Figure 6 ▶. The RX5RX3RX2R region is comparable to the RNR signature motif, RNX3RX2R, in ribozyme RNase P, which also has been argued to be implicated in RNA binding by recognizing the tertiary structure of the catalytic RNA (Spitzfaden et al. 2000). As mentioned above, it is likely that RNase PH recognizes the tertiary structure of the precursor tRNA molecule, and, thus, comparable to RNase P, the conserved arginines of helix α1 could be involved in recognizing the tertiary structure of the precursor tRNA molecules. Interestingly, the arginines of helix α1 in RNase PH and the RNR motif of RNase P are located in the “RNA-binding domain” found in both proteins, where they are located in the left-handed βαβ-crossover. The recognition of tRNA tertiary structure by RNase PH through the interactions of amino acids in the left-handed crossover could argue for the existence of this domain in the RNase PH superfamily.
The active site
The sulfate ion at positions Thr 125 and Arg 126 superimposes well with the partially occupied phosphate analog, tungstate in S. antibioticus PNPase and the phosphate ion in A. aeolicus RNase PH and thus establishes the location of the active site. In support, we have found a poorly resolved residual density between Asp 181 and Asp 187 in helix α3 close to the active site that may represent the binding of a divalent metal ion Mg2+ that is required for catalysis (Fig. 5 ▶). Arg 86, which Ishii et al. (2003) showed by structural and biochemical analysis played an important role in the phosphorolysis reaction, is placed in the same Cα position in the B. subtilis enzyme; the side-chain, however, does not bind in a well ordered way to the sulfate ion and thus did not provide us with any information.
Quaternary structure
Both B. subtilis and A. aeolicus RNase PH crystallize as a hexamer. Docking of a substrate tRNA molecule to the B. subtilis enzyme suggests that the substrate interacts with the dimer (Fig. 6 ▶). In the model of Ishii et al. (2003), tRNA binding, although different from our model, also only involve the dimer. So, what is the functional role of a hexameric form? We suggest that the hexamer has a protective role, protecting the highly flexible loop between strand β4 and helix α1, which is used for substrate recognition, from cleavage by a trypsinlike protease. This is supported by the observed electrostatic interactions between neighboring dimers and the arginyl residues of the flexible loop maintaining the hexameric structure and from the crystallization of the triple mutant R68Q-R73Q-R76Q enzyme, which crystallizes as a dimer. However, studies on the molecular weight of E. coli and B. subtilis RNase PH have shown that the enzyme has a tendency to aggregate at higher protein concentrations, and thus the presence of the hexamer could be caused by the high concentration of protein under the crystallization conditions.
Materials and methods
Purification and expression
Expression in the E. coli strain SØ6744 F′lacIq rph from the B. subtilis rph-gene on plasmid pLSH11 and purification of wild-type enzyme will be described elsewhere (L.S. Harlow and K.F. Jensen, in prep.). The R68Q-R73Q-R76Q triple mutant enzyme was prepared as follows: The three point mutations were introduced in the wild-type B. subtilis rph gene on pLSH11 in one step by the overlap extension PCR method (Ho et al. 1989) using mutagenic primers and generating plasmid pLSH15. Strain SØ6744 transformed with plasmid pLSH15 was grown in LB broth medium, supplemented with ampicillin, and induced with IPTG to promote synthesis of B. subtilis RNase PH. The cells were disrupted by ultrasonic treatment in a 12.5 mM MES buffer (pH 6.0; buffer A), and the triple mutant enzyme was purified by a combination of ammonium sulfate precipitation, chromatography on a DEAE ion exchange column in buffer A from which it eluted at a concentration of 0.26 M NaCl, chromatography on a column of hydroxyapatite in buffer A from which it eluted at a concentration of 45 mM potassium phosphate (pH 6), and gel filtration on a Sephacryl S300 column in a 50 mM Tris-HCl buffer (pH 7.6). The pooled fractions of the enzyme were divided in 0.5-mL aliquots of 15.3 mg/mL, which were frozen quickly in dry ice–ethanol and stored at −20°C.
Crystallization and data collection
Two crystal forms, I and II, of native B. subtilis RNase PH have been obtained at 20°C by sitting drop vapor diffusion. The protein concentrations were 12.5 mg/mL and 37.5 mg/mL, respectively, and 2 M (NH4)2SO4 was used as precipitant. For crystal form I, pH was adjusted to 6.0 by adding small amounts of 1.0 M NH4HCO3 to a 3.0 M (NH4)2SO4 stock solution. For crystal form II, pH was adjusted to 8.5 using a 0.1 M Tris-HCl buffer, and 20 mM CdCl2 was present in the precipitant solution. In both cases, the sitting drops composed of 5-μL protein solution and 5-μL precipitant solution were equilibrated against a 1.0-mL reservoir of precipitant solution. Crystals appeared within a week. Both crystal forms belong to the orthorhombic space-group P212121 with unit cell dimensions: a = 51.3 Å, b = 163.6 Å, c = 166.8 Å, and a = 99.8 Å, b = 146.5 Å, c = 152.3 Å for crystal forms I and II, respectively. A third crystal form, III, has been obtained for the triple mutant protein R68Q-R73Q-R76Q. Mutant protein at 15.3 mg/mL was crystallized using a precipitant composed of 1 M Na-citrate (pH 6.2) and 10% PEG400. Sitting drops were composed of 3 μL protein solution, 1 μl 10% β-octyl glycoside, and 3 μL of precipitant and equilibrated against 1 mL of precipitant solution in the reservoir. Crystal form III belongs to the tetragonal space-group P41212 with cell dimensions: a = b = 62.2 Å and c = 203.0 Å. Data collection statistics are summarized in Table 1.
Structure determination
The structure was determined by isomorphous replacement using crystal form II. Heavy-atom derivatives were prepared by soaking with HgCl2. Five different derivative crystals, containing from 2 to 6 Hg sites, were made by soaking in various heavy-atom concentrations. Heavy atoms were refined and initial phases were calculated using the program MLPHARE (Collaborative Computational Project 1994). These phases were subjected to solvent flattening and histogram matching using the program DM (Collaborative Computational Project 1994). In the first electron density map, a Trigonal Planar Pseudo Residue (TPPR) model was constructed using the graphics program TURBO-FRODO (Roussel and Cambillau 1992). This model showed sixfold P32 symmetry that was incorporated as noncrystallographic symmetry averaging in DM. The electron density was much improved and major parts of the amino acid sequence could be assigned to the electron density. The structures of crystal forms I and III were determined by molecular replacement with the program AMoRe (Navaza 1994) using the monomer structure of crystal form II. Similar to crystal form II, crystal form I contains a hexamer in the asymmetric unit. The hexamer is organized as a trimer of dimers. Crystal form III of mutant protein R68Q-R73Q-R76Q contains one molecule per asymmetric unit, but one of the crystallographic twofold axes generates the dimer found as part of the hexamers in crystal forms I and II.
Refinement
All structures were refined with the program CNS (Brünger et al. 1998) using torsion angle dynamics. Rebuilding between rounds of refinement was performed with the graphics program TURBO-FRODO (Roussel and Cambillau 1992) using 3mFobs − 2DFcalc and mFobs − DFcalc electron density maps (Read 1986; Collaborative Computational Project 1994). For crystal forms I and II, sixfold noncrystallographic symmetry restraints were applied during refinement, and electron density maps were accordingly averaged for rebuilding (Kleywegt and Read 1997). In crystal form II, three additional Cd2+ ions were detected in the packing of the nonaveraged map and included in the model. Refinement statistics are given in Table 1.
Acknowledgments
We thank David Friedman, University of Michigan, Ann Arbor, for the plasmid pUB2 bearing the B. subtilis rph gene. Lise Schack is thanked for technical assistance. We are grateful to the staff at EMBL, Hamburg, and at MAXLAB, Lund, for synchrotron beam time and assistance during experiments. This research has been supported by the Danish National Research Foundation.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Abbreviations
RNase, ribonuclease
PNPase, polynucleotide phosphorylase
Tris, 2-amino-2-hydroxymethyl-1,3-propanediol
MES, 2-[N-morpholino]ethanesulfonic acid
PEG, polyethylene glucol
Article published online ahead of print. Article and publication date are at http://www.proteinscience.org/cgi/doi/10.1110/ps.03477004.
References
- Al-Karadaghi, S., Avarsson, A., Garber, M., Zheltonosova, J., and Liljas, A. 1996. The structure of elongation factor G in complex with GDP: Conformational flexibility and nucleotide exchange. Structure 4 555–565. [DOI] [PubMed] [Google Scholar]
- Brünger, A.T., Adams, P.D., Clore, G.M., DeLano, W.L., Gros, P., Grosse-Kunstleve, R.W., Jiang, J.-S., Kuszewski, J., Nilges, M., Pannu, N.S., et al. 1998. Crystallography and NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr. D 54 905–921. [DOI] [PubMed] [Google Scholar]
- Burd, C.G. and Dreyfuss, G. 1994. Conserved structures and diversity of functions of RNA-binding proteins. Science 265 615–621. [DOI] [PubMed] [Google Scholar]
- Bycroft, M., Hubbard, T.J.P., Proctor, M., Freund, S.M.V., and Murzin, A.G. 1997. The solution structure of the S1 RNA binding domain: A member of an ancient nucleic acid-binding fold. Cell 88 235–242. [DOI] [PubMed] [Google Scholar]
- Collaborative Computational Project, Number 4. 1994. The CCP4 suite: Programs for protein crystallography. Acta Crystallogr. D 50 760–763. [DOI] [PubMed] [Google Scholar]
- Craven, M.G., Henner, D.J., Alessi, D., Schauer, A.T., Ost, K.A., Deutscher, M.P., and Friedman, D.I. 1992. Identification of the rph RNase PH. gene of Bacillus subtilis: Evidence for suppression of cold-sensitive mutations in Escherichia coli. J. Bacteriol. 174 4727–4735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deutscher, M.P. 1995. tRNA processing nucleases. In tRNA: Structure, biosynthesis, and function (eds. D. Söll, and U.L. RajBhandary), pp. 51–65. American Society of Microbiol. Press, Washington, DC.
- Esnouf, R.M. 1999. Further additions to Molscript version 1.4, including reading and contouring of electrondensity maps. Acta Crystallogr. D 55 938–940. [DOI] [PubMed] [Google Scholar]
- Godefroy, T. 1970. Kinetics of polymerization and phosphorolysis reactions of Escherichia coli polynucleotide phosphorylase. Evidence for multiple binding of polynucleotide in phosphorolysis. Eur. J. Biochem. 14 222–231. [DOI] [PubMed] [Google Scholar]
- Ho, S.N., Hunt, H.D., Horton, R.M., Pullen, J.K., and Pease, L.R. 1989. Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene 77 51–59. [DOI] [PubMed] [Google Scholar]
- Holm, L. and Sander, C. 1993. Protein structure comparison by alignment of distance matrices. J. Mol. Biol. 233 123–138. [DOI] [PubMed] [Google Scholar]
- Ishii, R., Nureki, O., and Yokoyama, S. 2003. Crystal structure of the tRNA processing enzyme RNase PH from Aquifex aeolicus. J. Biol. Chem. 278 32397–32404. [DOI] [PubMed] [Google Scholar]
- Jensen, K.F., Andersen, J.T., and Poulsen, P. 1992. Overexpression and rapid purification of the orfE/rph gene product, RNase PH of Escherichia coli. J. Biol. Chem. 267 17147–17152. [PubMed] [Google Scholar]
- Kelly, K.O. and Deutscher, M.P. 1992a. The presence of only one of five exoribonucleases is sufficient to support the growth of Escherichia coli. J. Bacteriol. 174 6682–6684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ———. 1992b. Characterization of Escherichia coli RNase PH. J. Biol. Chem. 267 17153–17158. [PubMed] [Google Scholar]
- Kelly, K.O., Reuven, N.B., Li, Z., and Deutscher, M.P. 1992. RNase PH is essential for tRNA processing and viability in RNase-deficient Escherichia coli cells. J. Biol. Chem. 267 16015–16018. [PubMed] [Google Scholar]
- Kleywegt, G.J. and Read, R.J. 1997. Not your average density. Structure 5 1557–1569. [DOI] [PubMed] [Google Scholar]
- Kraulis, P.J. 1991. MOLSCRIPT: A program to produce both detailed and schematic plots of protein structures. J. Appl. Cryst. 24 946–950. [Google Scholar]
- Li, Z. and Deutscher, M.P. 1996. Maturation pathways for E. coli tRNA precursors: A random multienzyme process in vivo. Cell 86 503–512. [DOI] [PubMed] [Google Scholar]
- Littauer, U.Z. and Grunberg-Manago, M. 1998. Polynucleotide phosphorylase. In Encyclopedia of molecular biology (ed. T.E. Creighton), pp. 1911–1918. John Wiley & Sons, Europe.
- Mian, I.S. 1997. Comparative sequence analysis of ribonucleases HII, III, II, PH and D. Nucleic Acid Res. 25 3187–3195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murzin, A.G. 1995. A ribosomal protein module in EF-G and DNA gyrase. Nat. Struct. Biol. 2 25–26. [DOI] [PubMed] [Google Scholar]
- Niranjanakumari, S., Stams, T., Crary, S.M., Christianson, D.W., and Fierke, C.A. 1998. Protein component of the ribozyme ribonuclease P alters substrate recognition by directly contacting precursor tRNA. Proc. Natl. Acad. Sci. 95 15212–15217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nissen, P., Thirup, S., Kjeldgaard, M., and Nyborg, J. 1999. The crystal structure of Cys-tRNACys-EF-Tu-GDPNP reveals general and specific features in the ternary complex and in tRNA. Structure 7 143–156. [DOI] [PubMed] [Google Scholar]
- Ost, K.A. and Deutscher, M.P. 1991. Escherichia coli orfE upstream of pyrE encodes RNase PH. J. Bacteriol. 173 5589–5591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulsen, P., Bonekamp, F., and Jensen, K.F. 1984. Structure of the Escherichia coli pyrE operon and control of pyrE expression by a UTP modulated intercistronic attentuation. EMBO J. 3 1783–1790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Read, R. 1986. Improved Fourier coefficients for maps using phases from a partial structure with errors. Acta Crystallogr. A 42 140–149. [Google Scholar]
- Roussel, A. and Cambillau, C. 1992. TURBO-FRODO. Biographics and AFMB (Architecture et Fonction des Macromolécules Biologiques), Marseille, France.
- Spitzfaden, C., Nicholson, N., Jones, J.J., Guth, S., Lehr, R., Prescott, C.D., Hegg, L.A., and Eggleston, D.S. 2000. The structure of ribonuclease P protein from Staphylococcus aureus reveals a unique binding site for single-stranded RNA. J. Mol. Biol. 295 105–115. [DOI] [PubMed] [Google Scholar]
- Stams, T., Niranjanakumari, S., Fierke, C.A., and Christianson, D.W. 1998. Ribonuclease P protein structure: Evolutionary origins in the translational apparatus. Science 280 752–755. [DOI] [PubMed] [Google Scholar]
- Sulewski, M., Marchese-Ragona, S.P., Johnson, K.A., and Benkovis, S.J. 1989. Mechanism of polynucleotide phosphorylase. Biochemistry 28 5855–5864. [DOI] [PubMed] [Google Scholar]
- Symmons, M.F., Jones, G.H., and Luisi, B.F. 2000. A duplicated fold is the structural basis for polynucleotide phosphorylase catalytic activity, processivity, and regulation. Structure 8 1215–1226. [DOI] [PubMed] [Google Scholar]
- Symmons, M.F., Williams, M.G., Luisi, B.F., Jones, G.H., and Carpousis, A.J. 2002. Running rings around RNA: A superfamily of phosphate-dependent RNases. Trends Biochem. Sci. 27 11–18. [DOI] [PubMed] [Google Scholar]
- Zhou, Z. and Deutscher, M.P. 1997. An essential function for the phosphate-dependent exoribonucleases RNase PH and polynucleotide phosphorylase. J. Bacteriol. 179 4391–4395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuo, Y. and Deutscher, M.P. 2001. Exoribonuclease superfamilies: Structural analysis and phylogenetic distribution. Nucleic Acid Res. 29 1017–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]