

Abstract
Using the tumor suppressor VHL protein as an example, we show that detailed analysis of conservation versus variation pattern in the multiple alignment can be coupled with the genomic pathway/complex conservation analysis to provide a more complete picture of the entire interaction/regulatory network. Results from the present study have allowed us to hypothesize that two additional proteins are involved in the VHL-mediated regulation of angiogenesis. Detailed modeling also has led to a prediction of the possible interaction mode between the known and the proposed parts of the VHL complex. To aid in an analysis of the VHL protein regulation of HIF-1α degradation, an important and only partially understood process that directly influences angiogenesis, we performed a comprehensive search for the orthologs of the VHL as well as for VHL-interacting proteins in all the available eukaryotic genomes. Analysis of a multiple alignment of thus identified VHL orthologs reveals an unusually high degree of conservation of the surface amino acid residues that almost exactly correspond to positions mutated in the VHL disease-associated tumors. In addition, these positions form well-defined clusters in three-dimensional space, and presence or absence of individual clusters correlates with the presence or absence of pathway elements in different genomes. We have also shown that relation trees derived from the multiple sequence alignment, functional surface-mapping, and HIF-1α degradation pathway structure are in complete agreement, linking the functional and structural evolution of the VHL protein and VHL-dependent HIF-1α degradation complex.
Keywords: conservation, VHL, HIF-1α, tumorigenic mutations, evolutionary trace, phylogeny
The analysis of residue conservation in homologs from various species is a well-established research technique that historically was used in combination with evolutionary and, sometimes, structural studies. The main focus of the multiple sequence alignment analysis is usually finding regions of high conservation (Valdar 2002; Frazer et al. 2003). At the same time, a lot of information is hidden in the pattern of change in less- or nonconserved regions, and several attempts have been made to explore such information, especially in the context of substrate binding (Lichtarge et al. 1996a,b, 1997). However, in this paper we go one step further and present a case that such analysis becomes even more powerful and far-reaching when coupled with additional information, such as the presence or absence of the network/pathway elements/protein complex members in various organisms.
Multiple alignment analysis usually focuses on single proteins, disregarding the fact that most proteins are involved in complexes and regulatory networks. Differences in network topologies or protein complexes organization between organisms have a direct influence on the conservation pattern between proteins being part of these networks and, vice versa, the analysis of such patterns can provide information about the networks. Currently, there is no unified theoretical framework that will allow studying the evolution of a single protein and its network simultaneously. The example presented here shows how much such framework is needed as well as what can be learned about the given process when we combine insights from standard multiple alignment analysis, comparative modeling and network/pathway analysis.
The particular system studied here is a VHL protein and the network of proteins involved in the degradation of HIF-1α, a primary regulator of angiogenesis. This network was discovered in the course of a detailed analysis of the von Hippel-Lindau (VHL) disease, a hereditary condition leading to the development of highly vascularized, and often malignant tumors (Kaelin Jr. and Maher 1998). All affected tumor cells express a high level of vascular endothelial growth factor (VEGF) mRNA, which is normally found only in hypoxic cells (Maxwell et al. 2001). VEGF transcription is regulated by the HIF-1 transcriptional activator. Stability of one of its subunits, HIF-1α, is regulated by the oxygen concentration (Semenza 1999). In normal cells under hypoxic conditions HIF-1α expression is high, whereas under normoxic conditions HIF-1α is ubiquitinated and directed for a proteosome-dependent degradation (Ivan and Kaelin Jr. 2001). Ubiquitination of the HIF-1α protein is mediated by the E3 ubiquitin ligase complex, with the VHL protein specifically recognizing oxygen-modified HIF-1α and recruiting it to the E3 ubiquitin ligase complex (Maxwell et al. 1999; Ivan and Kaelin Jr. 2001). Tumorigenic mutations in the VHL protein disable the HIF-1α degradation pathway and allow the tumor cells to mimic the hypoxic conditions despite normal levels of oxygen.
The ubiquitin ligase complex, in addition to the VHL, contains Elongin B, Elongin C, Cullin-2, and Rbx-1 (Lonergan et al. 1998; Iwai et al. 1999; Kamura et al. 1999). Components of the VHL-containing E3 ubiquitin ligase complex have been found in different species, suggesting that the HIF-1α degradation pathway is a part of an ancient and well conserved regulatory pathway.
In addition, there is a second mechanism of deactivation of HIF-1α-activated transcription of VEGF. Numerous studies have demonstrated that VHL binds to HIF-1α transcriptional activator using it as a specific substrate to inhibit HIF-1α–activated transcription of VEGF, thus inhibiting angiogenesis (Mahon et al. 2001). Although transcriptional repression mechanism of HIF-1 inactivation is secondary to the HIF-1 degradation in human cells, it may be a primary mechanism of HIF-1 deactivation in lower species.
The VHL is a 213 amino acid-long protein formed by two major domains: the α-domain, which contains three α-helices, and the β-domain, which consists of seven β-sheets and a single α-helix (Stebbins et al. 1999; Min et al. 2002). Genetic and clinical studies have identified a number of VHL mutations correlated with tumor development (Beroud et al. 1998). These mutations can be mapped to several distinct clusters on the VHL surface. Surface tumor mutant clusters are discussed in details below. For some, but not all, mutation clusters the detailed molecular consequences are known. The results presented in this paper suggest a possible function for the surface residues of the tumor mutant clusters, which currently do not have any assigned function.
The surface VHL residues found mutated in tumors are predominantly involved in interactions between different elements of the HIF-1α degradation complex, and should be conserved if the interaction partners are conserved. By a simple extension of this argument, the presence of clusters with unknown molecular function may suggest that there are still other, as yet unknown, members of the HIF-1α degradation complex and pathway. In this paper we follow this hypothesis, and by a combination of multiple sequence, structure, and genome analyses we find possible candidates for the missing components of the HIF-1α deactivation pathway.
Results
Synergetic, multilevel analysis of protein divergence
In biological systems proteins almost always operate as part of a larger network, with physical interactions between partners in this network being as important for the role of a given protein as active side residues for an enzyme. Yet, even the most sophisticated tools of multiple sequence analysis are based on a single molecule/binding sign paradigm. Here we explore a multilevel multiple sequence alignment analysis, based loosely on the idea of the evolutionary trace (Lichtarge et al. 1996b), but aiming at integrating such analysis with a pathway/network analysis. In this section we describe an outline of this approach (Fig. 1 ▶). First, all the orthologs (or lack thereof) of a protein of interest (protein X) as well as orthologs of other members from the same pathway (protein Y, A, B, and C) are identified in different species. Then, multiple sequence alignment is combined with comparative modeling protein X to identify functionally important patches of surface residues on this protein and tie them to the conservation/absence of pathway elements in various species.
Figure 1.

Combinatorial approach to pathway analysis. Schematic representation of the method’s relationship and pathway reconstruction used in current studies. The atomic-resolution structure of the protein X is known. Using protein X sequence, multiple sequence alignment of the protein X orthologs (right) is performed. Multiple sequence alignment provides necessary information for surface mapping (marked with protein–structure icon). Surface mapping results combined with the multiple sequence alignment and phylogenetic analysis for members of a pathway provide necessary data for a detailed pathway reconstruction (top).
Many proteins would be amendable to such analysis, however, the VHL protein studied here is particularly interesting because a detailed analysis of the functional role of surface was done previously in the context of their correlation to tumorigenesis of VHL syndrome (Beroud et al. 1998) and could be correlated with the multiple alignment/surface analysis. In addition, the analysis of fusion events of VHL lead to a hypothesis about two novel members of the studied pathway. Thus, several different types of information can be integrated in a common sequence conservation/protein surface analysis framework.
Tumor mutant clusters
When all the surface residues that are frequently mutated in the VHL tumors are mapped on the VHL protein structural model, it becomes evident that they are organized in spatially distinct surface clusters. In most cases, each cluster is related to a specific function. The first tumorigenic cluster (further in the text referred to as cluster A) is formed by the surface residues of the α-domain responsible for the interactions between Elongin C and VHL (Fig. 2 ▶; Kibel et al. 1995; Stebbins et al. 1999). This cluster includes the residues T157, L158, K159, C152, Q164, V166, R167, V170, R176, L178, I180, L184, E186, and L188. The residue R167 is the most frequently mutated in the VHL syndrome-associated tumors (Beroud et al. 1998). Several other clusters are located on the β-domain. The second tumor mutant cluster (referred to as cluster B), was implicated in HIF-1α binding, and also in binding to FIH-1 protein, a transcription factor working in concert with the VHL protein (Mahon et al. 2001). This tumor cluster is formed by the second most frequent tumorigenic hotspot Y98 as well as F76, N78, P86, W88, N90, Q96, T100, L101, G106, S111, Y112, H115, and W117 (Fig. 2A ▶). The third tumor mutant cluster (further referred to as cluster C), also located on the β-domain (shown on the Fig. 2B ▶), is represented by the residues R79, S80, R82, L89, D121, Q132, L135, F136, and P138. The role of this surface tumor mutant cluster is not known. Another residue frequently found mutated in tumors is Q145. It lies far from all the tumor mutant clusters discussed so far, and its functional role is not known. Finally, another patch of the tumor mutants is located on the H4 α-helix of the β-domain (Fig. 2B ▶). It is represented by the L198 and R200 and referred to as cluster D. The role that is usually attributed to this surface residue cluster is stabilization of the β-domain tertiary structure.
Figure 2.
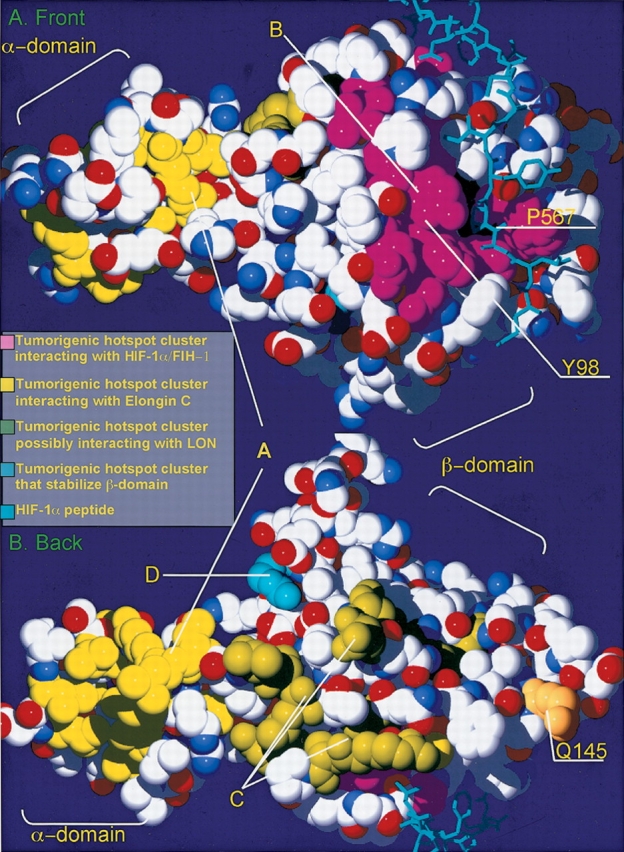
VHL tumor mutant clusters. (A) Front surface; (B) back surface. Structural representation of the human VHL protein. Each tumorigenic hotspot is marked according to the functional location. Residues, which were found to interact with the FIH-1/HIF-1α (tumor mutant cluster B) are marked in purple. The residues that are found to be responsible for Elongin C binding (tumor mutant cluster A) are marked in yellow. The residues that may interact with the LON protease (tumor mutant cluster C) are marked in bright green. The Q145 tumorigenic hotspot that was hypothesized to interact with the M24-like peptidase is marked in orange. Finally, the tumorigenic hotspots that are located within the H4 α-helix of the β-domain, which are thought to be involved in the domain stabilization (tumor mutant cluster D), are marked in blue. The HIF-1α peptide, which is responsible for the interaction with the VHL protein, is marked in bright blue. The Y98 of the VHL protein and the P567 of the HIF-1α are marked with corresponding arrows.
Conservation of the VHL residues found mutated in the VHL-associated tumors
First, by a cascade of PSI-BLAST, FFAS, and organism-specific searches, we confirmed the presence of VHL orthologs in Drosophila melanogaster and Caenorhabditis elegans, and found previously unrecognized orthologs in recently completed genomes of Caenorhabditis briggsae, Xenopus tropicalis, Takifugu rubripes, Anopheles gambiae, and Ciona intestinalis. The VHL homologs from the mouse, rat, and other mammals were excluded from the current analysis, as essentially identical to the human protein. We do not present analysis of the C. intestinalis VHL protein, because the whole genome was not yet analyzed.
The most interesting feature of the VHL tumor mutant hotspots is that their conservation is very high, despite significant full-length protein sequence divergence of the VHL family that varies from 85% identical residues between closely related species, such as humans and the mouse, to about 25% between humans and Drosophila melanogaster (Table 1). The conservation of the tumor hotspots is even higher than the core residues, which usually dominate the list of conserved positions in multiple alignments. Clearly, the evolution rate of the functional surface residues, which are responsible for interaction with other VHL complex partners, is limited, illustrating the evolutionary importance of the VHL complex. At the same time, conservation varies strongly between clusters (see the above section for the discussion and nomenclature of tumorigenic clusters on VHL surface), suggesting that conservation/divergence pattern is dominated by conservation of function of specific clusters.
Table 1.
Conservation of the VHL residues, found in tumors
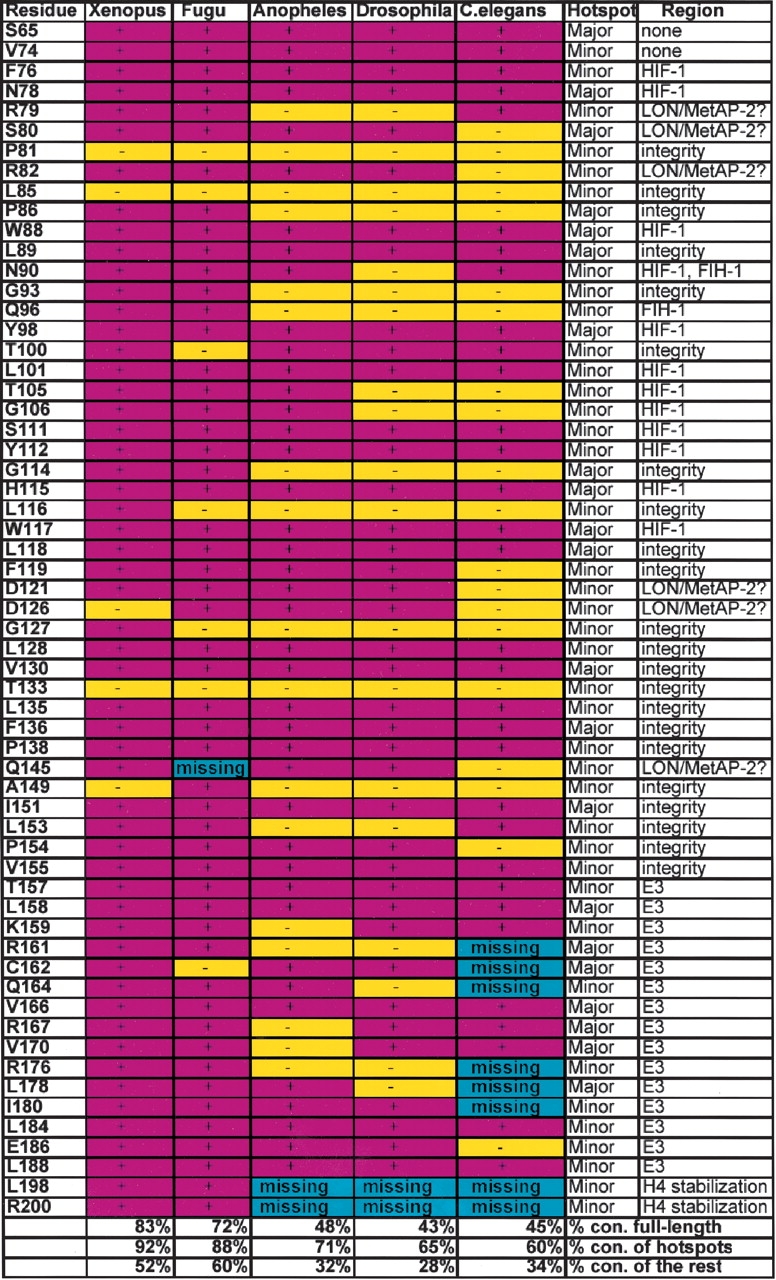
First column, residues in human VHL, which were found in the VHL-associated tumors; second through sixth columns, conservation of the residues in the correspondingly indicated species; seventh column, major or minor tumor mutant hotspot; eighth column possible (?) or proved function of the region, in which the residue is located; bottom of the table, % conservation of the protein in lower species, % conservation of the tumorigenic hotspots in lower species, and % conservation of the rest of the protein in lower species. Conserved residues, purple; not conserved, yellow; missing residues, blue.
The detailed discussion of tumorigenic hotspots is illustrated on the multiple sequence alignment of all the identified VHL proteins (Fig. 3 ▶). Positions that are conserved throughout all the species are mostly coming from residues from the cluster B (F76, N78, W88, L89, Y98, L101, S111, Y112, H115, W117, and L118) (Figs. 2 ▶,3 ▶; Beroud et al. 1998). All residues from this list are responsible for the binding to the HIF-1α and FIH-1 proteins (Fig. 2A ▶).
Figure 3.

Multiple sequence alignment of the VHL orthologs from lower species. Multiple protein sequence alignment of the VHL homologs from Homo sapiens, Xenopus tropicalis, Takifugu rubripes, Anopheles gambiae, Drosophila melanogaster, C. elegans, and C. briggsae. Important tumorigenic hotspots are indicated with arrows.
The tumor mutant cluster A is responsible for binding to the Elongin C, thus tethering other components of the E3 ubiquitin ligase complex to the VHL protein. The most important tumorigenic residues within this cluster are conserved throughout species. However, a large number of tumorigenic residues is missing from the tumor mutant cluster A in C. elegans (R161, C162, Q164, R176, L178, and I180), thus suggesting that this VHL ortholog may not interact with the Elongin C and may have a different mechanism of HIF-1α inactivation (Fig. 3 ▶; Table 1) because the E3 ubiquitin ligase complex is unable to assemble and ubiquitinate the VHL-bound HIF-1α protein.
Q145 is conserved in the frog and the fruit fly, and is not conserved in the mosquito, fugu, and worms. We hypothesize that this residue is responsible for interaction with the MetAP-2, a proposed new member of the pathway (see next section for details on the newly proposed pathway). MetAP-2 is fused to the VHL protein in fugu, thus eliminating the requirement for the MetAP-2-interacting residues. In worms, the mechanism of HIF-1α inactivation may be different from the human mechanism (discussed in Discussion), and does not require the presence of active MetAP-2 ortholog in the VHL complex.
The tumor mutant cluster C without a known binding partner is conserved throughout vertebrates, but is less conserved in the invertebrates (Table 1; Fig. 2B ▶), thus correlating exactly with the presence or absence of the specific LON protease ortholog (see Discussion), naturally leading to a hypothesis that this cluster is essential for either LON binding or LON functional activity (Table 2; Fig. 2B ▶). In the following three sections we will discuss in detail conservation of the tumorigenic VHL hotspots and the presence/absence of different VHL pathway elements in specific species.
Table 2.
VHL-binding protein orthologs in lower species
| Organism | LON ortholog | MetAP-2 | Elongin C | HIF-1α |
| Homo sapiens | present | present | present | full length |
| Xenopus tropicalis | present | present | present | full length |
| Takifugu rubripes | fused to VHL | present | 2 separate polypep. | short |
| Drosophila melanogaster | not present | present | present | short |
| Anopheles gambiae | not present | present | present | short |
| Caenorhabditis briggsae | not present | present | present | short |
| Caenorhabditis elegans | not present | present | present | short |
First column indicates corresponding organisms, in which VHL homolog was found; second column represents presence or absence of the single-domain LON protease orthologs in the corresponding organisms; third column indicates presence or absence of the MetAP-2; fourth column indicates presence of the Elongin C ortholog; and, finally, fifth column indicates presence of the HIF-1α protein and its length.
HIF-1α degradation pathway in xenopus and fugu
The main feature of the vertebrate VHL proteins is nearly complete conservation of the major tumor mutant clusters described above (Fig. 3 ▶). The X. tropicalis VHL protein was found to be 167-amino acids long, lacking 52 N-terminal amino acids, similar to the IVHL translation version. It possesses a 66% identity relative to the human VHL with E value of 6e−52 (Table 1). The conservation of the tumorigenic hotspots is remarkable compared to the rest of the protein showing that all the surface amino acids, associated with the tumors, except P81 and T133, are found to be conserved (Table 1; Fig. 3 ▶).
Recently, sequenced fugu genome provided many examples of pathways and networks that show a greatly simplified structure when compared to their human counterparts, but at the same time, individual components are similar to their human orthologs (Venkatesh et al. 2000; Aparicio et al. 2002). This combination of close homology and simplification makes fugu an ideal model organism to study human processes. We have identified a fugu VHL ortholog, which, in contrast to all other VHL homologs, codes for a multidomain, 1127 amino acid-long protein. In addition to the VHL domain, fugu protein contains two domains with homologies to LON protease and M24-like peptidase, respectively, raising the possibility that both domains participate in the VHL-dependent pathway. Homologs of both LON and M24 metallopeptidase are present in the human genome, as well as in most genomes containing VHL orthologs.
One of the human LON paralogs has mitochondrial localization, binds to oxygen-modified (possibly hydroxylated) proteins and mediates their degradation (Bota and Davies 2002). It appears reasonable to hypothesize that human ortholog of the fugu LON domain, which is fused to the VHL protein in fugu, has a cytoplasmic localization and may also sense the presence of hydroxylated HIF-1α and assist in its degradation under normal oxygen concentration. This particular human LON paralog is not annotated, and is listed as an uncharacterized protein in the NCBI. According to the PFAM (version 7.7b, Washington University of St. Louis, http://pfam.wustl.edu) analysis, most of the eukaryotic LON proteins contain three major domains (Fig. 4 ▶). They contain N-terminal ATP-dependent proteolytic domain, which contains proteolytic active site, ATP-binding site and protein recognition site (PFAM accession no. PF02190). The second domain is ATPase family associated with various cellular activities (AAA) domain (PFAM accession no. PF00004). The third domain is LON C-terminal proteolytic domain (PFAM accession no. PF05362), which also possess a proteolytic activity. Unlike other human LON paralogs as well as other eukaryotic LON domain-containing proteins, the human LON protein (NCBI–NP_057386.1) possesses a single N-terminal proteolytic LON domain, which by itself is fully active in proteolysis (Fig. 4 ▶). We hypothesize that this particular LON homolog is localized in the cytoplasm rather than mitochondria and responsible for binding to the VHL protein and inactivation of the HIF-1 protein.
Figure 4.

Domain analysis of the LON protease. Human cells have several LON protease paralogs. The domain organization of one of them, which is localized in mitochondria, is shown on the top panel. It consists of three domains: N-terminal LON domain (LON-N), which possesses ATP-binding and proteolytic activity and present in all LON proteins; the ATPase family associated with various cellular activities (AAA) domain, which has a chaperone-like function; and the C-terminal LON domain (LON-C), which also has a proteolytic activity. The second one, homologous to the LON protease fused to the VHL protein in fugu (bottom), is shown in the middle panel. Flies and worms possess the LON protease, homologous to the one shown on the top panel, but do not possess the single-domain LON ortholog.
Human M24 metallopeptidase homolog–methionine aminopeptidase-2 (MetAP-2) was described previously as a target for fumagillin, an antiangiogenic drug for the tumor therapy (Liu et al. 1998), opening the possibility that this protein is involved in the HIF-1α degradation pathway. The specific function of human MetAP-2 and mechanism of the fumagillin activity are not known, but its involvement in the angiogenesis regulatory pathways is extremely interesting in the context of the findings presented here.
All the tumorigenic mutant clusters, including the tumor mutant cluster C on the VHL β-domain with an unknown function, are conserved in fugu. Among important tumorigenic mutation sites, only the Q145 residue is missing. In the context of the structure of the fugu VHL protein, it opens up the possibility that Q145 may play a role in interaction with the LON or MetAP-2, and in fugu, this residue is not important, because these three proteins are fused together. Later in the paper we will provide evidence that would suggest that the LON protease binds to a different and distant from the Q145 cluster, thus leaving only MetAP-2 as a legitimate partner for binding to the Q145-containing region.
HIF-1α protein in fugu is significantly shorter than in human and frog, but the VHL-binding peptide (M551–R575), which includes hydroxylated P564, is completely conserved. The only residue not conserved in the peptide is P567 (Fig. 2 ▶).
Another hallmark of the E3 ubiquitin ligase complex in fugu is that the Elongin C is expressed as two different proteins, corresponding to structural domains in the experimentally determined Elongin C structure (Fig. 5 ▶). It has been shown in the crystal structure (Stebbins et al. 1999) that only the domain 2 of the Elongin C is responsible for the interaction with the VHL protein. Therefore, at the same time with the VHL–MetAP-2–LON fusion, we observe a split of the elongin C protein in fugu. It is tempting to speculate that these two domains have different functions. For instance, it is possible that domain 1 of the Elongin C participates in transcriptional elongation, while domain 2 participates in the E3 ubiquitin ligase complex formation. This hypothesis seems reasonable considering a proven role of the Elongin C in transcriptional elongation (Aso et al. 1995) as well as in E3 ubiquitin ligase activity.
Figure 5.

VCB complex in fugu. Ribbon structure of the VHL–Elongin C–Elongin B complex (the structural representation of the complex was designed based on the Protein Data Bank file, described previously [Stebbins et al. 1999]). The α- and β-domains of the VHL protein, domains 1 and 2 of the Elongin C and Elongin B are indicated. In fugu domains 1 and 2, while bearing 93% and 100% identity to the corresponding human domains, are separate proteins according to the computerized prediction. According to the crystal structure of the VCB complex, only domain 2 of the Elongin C plays a role in the VHL binding.
HIF-1α degradation pathway in flies
A similar analysis was performed on D. melanogaster and A. gambiae VHL orthologs. Although anopheles VHL has a significantly higher conservation rate throughout the protein and within the tumor clusters than Drosophila (Fig. 3 ▶; Table 1), they both have similar features, distinct from the vertebrate VHL. In general, the fly’s VHL homologs have a lower conservation rate of the tumorigenic residues than the vertebrates (Table 1). However, the lower conservation rate of the tumorigenic residues does not represent a slow evolutionary progress from the flies to vertebrates, but rather an evolvement of new functions and new members of the HIF-1 inactivation pathway, as illustrated by the fact that some tumor mutant clusters are almost completely conserved, while other clusters are not. One of the distinctions of the fly’s VHL protein from the vertebrate’s protein is the absence of the H4-stabilizing α-helix. Another distinction between vertebrate and inverterbrate VHL is significantly lower conservation of the tumor mutant cluster A in flies (Table 1; Fig. 2 ▶). In the A. gambiae VHL cluster A, K159, R161, R167, V170, and R176 are not conserved. In the D. melanogaster cluster A, R161, Q164, R176, and L178 are not conserved. As it was mentioned before, tumor mutant cluster A is part of the region responsible for interaction with the Elongin C, thus tethering the ubiquitin ligase to the HIF-1–VHL complex. Considering that the Drosophila (NCBI accession no. NP_523794, sequence information—Supplemental Material) and Anopheles (NCBI accession no.EAA05684, sequence information—Supplemental Material) Elongin C proteins possess a 93% and 95% identity to its human counterpart, respectively, we suggest that Elongin C does not evolve together with the α-domain of the VHL protein, but rather either has lower affinity or no affinity for the VHL protein in flies. This fact suggests that invertebrate VHL protein may inactivate HIF-1 by a mechanism distinct from the vertebrate one due to an inability to tether the E3 ubiquitin ligase complex and ubiquitinate HIF-1.
Although the tumorigenic cluster B (HIF-1/FIH-1-binding cluster) is conserved in flies, the cluster C hypothesized in the previous section to be involved in possible LON interaction is not completely conserved (Fig. 2B ▶; Table 1), suggesting that the VHL complex may not bind to the single-domain LON protease. Following on the hypothesis formulated in the previous sections, only extremely distant LON protease homologs are present in Anopheles and Drosophila (Table 2), and they all have a more complex domain structure suggesting that they are involved in other complexes in mitochondria, similar to the ones formed by the human mitochondrial LON (Fig. 4 ▶). Therefore, there is a good indication that the third tumor mutant cluster on the back of the VHL β-domain (Fig. 2B ▶) is important either for interaction with the LON or for its functional activity. In addition, Q145 residue, which is found in the VHL-associated tumors, and is missing from the fugu VHL, is conserved in flies (Table 1). Although the conservation of the Q145 in D. melanogaster is evident from the multiple sequence alignment (Fig. 3 ▶), the conservation of the Q145 VHL residue in A. gambiae is not. However, when local sequence alignment between human and Anopheles VHL homologs is performed, it becomes evident that the Q145 is conserved in the Anopheles (alignment is not shown).
HIF-1α degradation pathway in worms
Next, we analyzed the VHL-dependent HIF-1α degradation pathway in Caenorhabditis elegans. At 30% sequence identity, the C. elegans VHL protein is the most divergent from VHL homologs discussed here. In general, it has a significantly lower degree of conservation of the tumor hotspots than the fly’s VHL (Table 1; Fig. 2 ▶). The largest distinction of the worm VHL protein is divergence of the α-domain. Most of the tumorigenic residues from the tumor mutant cluster A are either missing or not conserved (Table 1). Considering that the worm’s Elongin C has a very high homology to its human counterpart (76% identity, accession no. NP_497405) we can exclude the possibility of the parallel evolution of the Elongin C and the of the cluster A, and suggest that Elongin C does not interact with the VHL protein. Most likely, the mechanism of deactivation of the C. elegans HIF-1 is distinct from the vertebrate mechanism. In addition, worms, similar to flies, still possess a conserved HIF-1α-binding site, but the possible LON-binding site is conserved even less than in flies (Table 1; Fig. 3 ▶). Similar to the fly’s LON, the single-domain LON (Fig. 4 ▶, middle panel) was not found either in C. elegans or C. briggsae. Although the LON homologs are present in worms, they have a different domain composition, similar to the fly’s LON proteins, and probably participate in mitochondrial processes. At the same time, the most prominent tumorigenic hotspot of the VHL β-domain Y98 is conserved in C. elegans (Table 1). Probable elimination of the Elongin C and the whole E3 ubiquitin ligase complex interaction with the VHL due to the absence/divergence of the VHL α-domain suggests that the VHL regulates HIF-1α activity by a mechanism distinct from the proteosome- and LON/MetAP2-dependent degradation (hypothesis described in Discussion).
Phylogenetic analysis of the VHL protein
A phylogenetic tree, built by the nearest joining method from the multiple sequence alignment (Fig. 6A ▶; Nei 1996) broadly follows the canonical phylogenetic tree of eukaryotes.
Figure 6.

Phylogenetic tree of the VHL protein. (A) A phylogenetic tree, derived from the multiple sequence alignment. Reliable bootstrap values are indicated at the branches of the tree. (B) A cladistic tree, derived from the functional analysis of the VHL-dependent HIF-1α degradation system as well as from the surface mapping of the functionally important tumor mutant clusters and their conservation throughout lower species. Instead of the bootstrap values at the branches of the tree, we used functional characteristics and tumor mutant cluster’s conservation as an explanation for one or another branch of the tree or for the distance from a branch. The Homo sapiens VHL protein is marked on the figure as ‘complete.’ The phylogenetic tree in B is not drawn to scale.
At the same time, we can build a cladistic tree (Morrison 1996) using conservation of tumorigenic clusters, which, as discussed in the previous paragraphs, coincides exactly with surface positions of the HIF-1α/FIH-1-, Elongin C-, and possible LON-binding sites (Fig. 6B ▶). Although cladistic trees are seldom built for single proteins, in this case we can use several well-defined features of the VHL proteins. In the following analysis, instead of using the bootstrap values, we have indicated the reason for each branch of the tree and for relative distances from the branches. In this section, C. briggsae was not analyzed, as the entire genome is still not processed. The VHL protein from the C. elegans is separated from the rest of the VHL orthologs because this particular VHL homolog almost completely lacks VHL α-domain cluster A, suggesting a different mechanism of the HIF-1α deactivation, unlike vertebrate VHL homologs, which possess a complete VHL α-domain (Fig. 6B ▶). In addition, the C. elegans VHL homolog tumor mutant cluster C, which was hypothesized to bind to the LON protease, also is not conserved, correlating with the absence of the clear single-domain LON ortholog in worms. The VHL homologs from flies are separated from the vertebrate homologs because the fly VHL lacks conservation of the tumorigenic cluster C as well as partial lack of conservation of the tumorigenic cluster A (Elongin C-binding cluster). The lack of conservation of the tumorigenic cluster C in the fly’s VHL correlates with the absence of the single-domain LON protease ortholog. The Drosophila VHL is distinct from the Anopheles VHL, with Anopheles being closer to the main branch, because Anopheles VHL tumorigenic hotspots exhibit a higher degree of conservation. The fugu VHL is separated from the rest of the vertebrate VHL homologs (from Xenopus and humans) because it has the lower degree of tumor mutant hotspot conservation and possess a fusion between LON protease N-terminal domain, M24-like peptidase and the VHL protein (Fig. 6B ▶). Interestingly, identical cladistic trees can be built separately based on the arguments of missing/present/fused members of the potential VHL complex and presence/absence of the corresponding tumor mutant clusters.
Therefore, the phylogenetic tree based on the multiple sequence alignment and cladistic trees based either on the functional surface mapping or presence/absence of potential complex members, all completely agree (Fig. 6A,B ▶).
Discussion
We presented a detailed analysis of the conservation of the VHL protein tumorigenic hotspots, combined with the analysis of their spatial correlation on the three-dimensional model on the VHL protein and conservation/divergence in various genomes, fusion events in fugu genome, and presence/absence of potential complex members in various genomes. It allowed us to present a novel hypothesis about the existence of additional, previously unknown, participants in the HIF-1α degradation complex. In addition to the potentially important discovery, we are illustrating the need for a new way of studying the protein sequence conservation, by correlating standard multiple alignment analysis with three-dimensional model analysis and whole genome analysis. Strong agreement between sequence alignment-derived and functional surface mapping/whole genome-derived phylogenies clearly illustrates the underlying unity of the cellular processes, which only manifests itself in different observables—which should be, but seldom are, studied together.
The most important information came from the identification of the fusion of the VHL protein to LON protease and M24-like peptidase in fugu. Analysis of genomic fusion events is a very powerful tool to determine possible physical or functional interaction of proteins (Marcotte et al. 1999), and here we show that it can be extended to the identification of unknown members of metabolic or signaling pathways.
The hypothesis put forward in this manuscript suggests that LON and M24-like peptidase take part in the VHL complex responsible for HIF-1α degradation. This would suggest that the VHL/elongin C complex is not only responsible for targeting HIF-1α for the standard ubiquitin protein degradation pathway, but may actively perform first steps in the proteolysis of the hydroxylated HIF-1α. This hypothesis is strengthened by the fact that LON interacts with the proteins hydroxylated in an oxygen-dependent manner (Bota and Davies 2002) and that fumagillin, interacting with MetAP-2, is influencing angiogenesis. The most likely scenario is that the MetAP-2 removes N-terminal methionine from the HIF-1α protein. The methionine removal makes HIF-1 more accessible for proteolytic degradation by LON and for further degradation in proteosomes (Varshavsky 1996). Then LON partially degrades HIF-1, and proteosomes only finalize the degradation process. Our findings also suggest a possible mechanism for the fumagillin antiangiogenic function (Liu et al. 1998). The MetAP-2, as do most of the peptidases, possesses a constituent inhibitor, which blocks the active site from an interaction with the target (in this case, HIF-1α). The fumagillin competitively removes an inhibitor from the MetAP-2 active site without blocking the interaction with HIF-1α. This enhances the interaction between MetAP-2 and HIF-1, which results in a higher rate of HIF-1α degradation, and subsequently a lower VEGF expression. Although the specific of this process are, of course, pure speculation, observations presented in this manuscript make a strong case of the involvement of LON and MetAP-2 in HIF-1 degradation.
A global analysis of the VHL homologs in lower species reveals a number of important features. First, none of the VHL homologs, analyzed in current studies, possess N-terminal 55 amino acids, making it similar to the IVHL transcription version. No significant differences in VHL functionality have been found between IVHL and full-length VHL. It appears reasonable to hypothesize, that the presence of the N-terminal portion is characteristic of the human VHL protein, because even mouse VHL does not possess a homologous N-terminal portion (data not shown).
Mapping of the surface tumor mutant hotspots in lower species has shown that their conservation is significantly higher than the rest of the protein (Table 1). The surface mapping analysis of the von Hippel-Lindau disease-associated tumor mutant clusters as well as genomic analysis of the presence/absence of the VHL-binding orthologs has demonstrated that the evolution of the VHL protein from worms to humans takes place in steps, by adding new functional surface clusters in parallel with the VHL-binding partners, thus adding new function. This evolution leads to a more rapid and efficient regulation of the angiogenesis. It is clear that the HIF-1α–binding site is conserved throughout tested VHL homologs, suggesting that HIF-1 binding is essential for the VHL functionality (Fig. 3 ▶). The hypothesized LON-binding site, which contains the tumor mutant cluster C, is not conserved in the fly’s and worm’s VHL protein. The lack of conservation of the LON-binding site correlates with the absence of a single-domain LON protease in flies and worms, suggesting that LON protease does not participate in HIF-1 degradation. The Elongin C-binding surface within the VHL α-domain is conserved in all of the vertebrates and partially flies, but is not conserved in worms. This leaves no room for E3 ubiquitin ligase binding to the worm VHL, suggesting that the Caenorhabditis VHL protein deactivates HIF-1α by some other means. In humans, the secondary mechanism of the VHL-dependent degradation is VHL-dependent repression of HIF-1–activated transcription using FIH-1 protein as a corepressor (Mahon et al. 2001). VHL binds to the HIF-1α transcriptional activator, using it as a specific substrate for tethering to the HIF-1–activated gene. We suggest that in worms the VHL-dependent transcriptional repression of HIF-1 transcriptional activation of VEGF is a primary mechanism of countering the effect of VEGF activation by HIF-1. With evolution, the transcriptional repression becomes secondary, while VHL-dependent ubiquitination of HIF-1 evolves as a primary mechanism. Even though Elongin C is present in worms despite of the absence of Elongin C-binding site on the worm’s VHL protein, it performs a number of other functions. The most important of them is transcriptional elongation (Aso et al. 1995). It participates in the Elongin SIII complex, and together with Elongin A/B activates transcriptional elongation by the RNA polymerase II.
In conclusion, this data suggests that functional surface residues have a lower rate of evolution than the rest of the surface residues and even lower than the core residues. This seems to be at odds with common wisdom, but it is important to remember that most of that wisdom was developed on studies of single molecule, soluble enzymes.
Materials and methods
Homology search
A homology search was performed using PSI-BLAST on the NCBI BLAST server, (http://www.ncbi.nlm.nih.gov/BLAST/) and FFAS on the Burnham Institute server (http://ffas.ljcrf.edu). D. melanogaster VHL protein: gi:25480297. Anopheles gambiae: gi:21295810. C. elegans protein: gi:3875650. The fugu homologous scaffold was identified using Fugu Blast Server (MRC HGMP-PC, The Fugu Genomics Project, http://fugu.hgmp.mrc.ac.uk/blast/). The VHL-encoding scaffold in fugu is S002894. The corresponding proteins in fugu were predicted using the GENSCAN gene prediction program with the ‘vertebrate’ parameters (MIT server, http://genes.mit.edu/GENSCAN.html). Conserved domains were identified using PFAM. Similarly, fugu Elongin C proteins have been predicted. The VHL-encoding est from Xenopus has been retrieved using the Sanger Institute Xenopus BLAST server, using a TBLASTN subroutine with parameters (The Sanger Institute, UK, http://www.sanger.ac.uk). The Xenopus VHL EST no. AL869123. The Xenopus VHL protein sequence prediction was performed using GENSCAN as described above.
Structural modeling
Structural representation of the VHL protein and tumor hotspot modeling was performed using 1LM8 (Elongin C-Elongin B-Hif-1α) cocrystal structure from the Protein Data Bank (http://www.rcsb.org/pdb/cgi) with the Swiss PDB Viewer.
Tumor hotspot identification
Tumorigenic hotspots were identified using VHL Universal Mutation Database (http://www.umd.necker.fr), described previously (Beroud et al. 1998).
Multiple sequence alignment
Multiple sequence alignment between the VHL protein and its homologs was performed using T-COFFEE software (T-Coffee, version 1.37, Swiss Institute of Bioinformatics). After the first round of T-COFFEE, nonhomologous regions as well as large gaps in sequence alignment were removed, and a second round of alignment was performed. After the third round of the sequence alignment, identical sequences were removed and a third round of T-COFFEE alignment was performed. The resulting output was plotted on using GeneDoc program (GeneDoc, version 1.0, Free Software Foundation).
LON domain organization analysis
LON domain analysis has been performed using PFAM.
Phylogenetic tree generation
A phylogenetic tree was created using Treecon software (Treecon, version 1.3b, University of Konstanz, Belgium). Initial distance estimation was done using Kimura correction. Tree topology was inferred using the neighbor-joining method. Bootstrap analysis was included.
Electronic supplemental material
The supplementary material contains VHL and Elongin C protein sequences from Homo sapiens, Xenopus laevis, Takifugu rubripes, Anopheles gambiae, Drosophila melanogaster, Caenorhabditis briggsae, and Caenorhabditis elegans. It also contains protein sequence of the C. elegans HIF-1α and human LON sequence. The file is MS Word, named “Suppl._VHL_sequences.”
Acknowledgments
The present work was supported by NIH Grant GM60049. We thank Dr. Alex Strongin for useful discussions and Dr. Ana Rojas for technical assistance and discussions.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Supplemental material: see www.proteinscience.org
Article published online ahead of print. Article and publication date are at http://www.proteinscience.org/cgi/doi/10.1110/ps.03454904.
References
- Aparicio, S., Chapman, J., Stupka, E., Putnam, N., Chia, J.M., Dehal, P., Christoffels, A., Rash, S., Hoon, S., Smit, A., et al. 2002. Whole-genome shotgun assembly and analysis of the genome of Fugu rubripes. Science 297 1301–1310. [DOI] [PubMed] [Google Scholar]
- Aso, T., Lane, W.S., Conaway, J.W., and Conaway, R.C. 1995. Elongin (SIII): A multisubunit regulator of elongation by RNA polymerase II. Science 269 1439–1443. [DOI] [PubMed] [Google Scholar]
- Beroud, C., Joly, D., Gallou, C., Staroz, F., Orfanelli, M.T., and Junien, C. 1998. Software and database for the analysis of mutations in the VHL gene. Nucleic Acids Res. 26 256–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bota, D.A. and Davies, K.J. 2002. Lon protease preferentially degrades oxidized mitochondrial aconitase by an ATP-stimulated mechanism. Nat. Cell Biol. 4 674–680. [DOI] [PubMed] [Google Scholar]
- Frazer, K.A., Elnitski, L., Church, D.M., Dubchak, I., and Hardison, R.C. 2003. Cross-species sequence comparisons: A review of methods and available resources. Genome Res. 13 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivan, M. and Kaelin Jr., W.G. 2001. The von Hippel-Lindau tumor suppressor protein. Curr. Opin. Genet. Dev. 11 27–34. [DOI] [PubMed] [Google Scholar]
- Iwai, K., Yamanaka, K., Kamura, T., Minato, N., Conaway, R.C., Conaway, J.W., Klausner, R.D., and Pause, A. 1999. Identification of the von Hippel-lindau tumor-suppressor protein as part of an active E3 ubiquitin ligase complex. Proc. Natl. Acad. Sci. 96 12436–12441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaelin Jr., W.G. and Maher, E.R. 1998. The VHL tumour-suppressor gene paradigm. Trends Genet. 14 423–426. [DOI] [PubMed] [Google Scholar]
- Kamura, T., Koepp, D.M., Conrad, M.N., Skowyra, D., Moreland, R.J., Iliopoulos, O., Lane, W.S., Kaelin Jr., W.G., Elledge, S.J., Conaway, R.C., et al. 1999. Rbx1, a component of the VHL tumor suppressor complex and SCF ubiquitin ligase. Science 284 657–661. [DOI] [PubMed] [Google Scholar]
- Kibel, A., Iliopoulos, O., DeCaprio, J.A., and Kaelin Jr., W.G. 1995. Binding of the von Hippel-Lindau tumor suppressor protein to Elongin B and C. Science 269 1444–1446. [DOI] [PubMed] [Google Scholar]
- Lichtarge, O., Bourne, H.R., and Cohen, F.E. 1996a. Evolutionarily conserved Gαβγ binding surfaces support a model of the G protein–receptor complex. Proc. Natl. Acad. Sci. 93 7507–7511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ———. 1996b. An evolutionary trace method defines binding surfaces common to protein families. J. Mol. Biol. 257 342–358. [DOI] [PubMed] [Google Scholar]
- Lichtarge, O., Yamamoto, K.R., and Cohen, F.E. 1997. Identification of functional surfaces of the zinc binding domains of intracellular receptors. J. Mol. Biol. 274 325–337. [DOI] [PubMed] [Google Scholar]
- Liu, S., Widom, J., Kemp, C.W., Crews, C.M., and Clardy, J. 1998. Structure of human methionine aminopeptidase-2 complexed with fumagillin. Science 282 1324–1327. [DOI] [PubMed] [Google Scholar]
- Lonergan, K.M., Iliopoulos, O., Ohh, M., Kamura, T., Conaway, R.C., Conaway, J.W., and Kaelin Jr., W.G. 1998. Regulation of hypoxia-inducible mRNAs by the von Hippel-Lindau tumor suppressor protein requires binding to complexes containing elongins B/C and Cul2. Mol. Cell Biol. 18 732–741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahon, P.C., Hirota, K., and Semenza, G.L. 2001. FIH-1: A novel protein that interacts with HIF-1α and VHL to mediate repression of HIF-1 transcriptional activity. Genes & Dev. 15 2675–2686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcotte, E.M., Pellegrini, M., Ng, H.L., Rice, D.W., Yeates, T.O., and Eisenberg, D. 1999. Detecting protein function and protein–protein interactions from genome sequences. Science 285 751–753. [DOI] [PubMed] [Google Scholar]
- Maxwell, P.H., Wiesener, M.S., Chang, G.W., Clifford, S.C., Vaux, E.C., Cockman, M.E., Wykoff, C.C., Pugh, C.W., Maher, E.R., and Ratcliffe, P.J. 1999. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature 399 271–275. [DOI] [PubMed] [Google Scholar]
- Maxwell, P.H., Pugh, C.W., and Ratcliffe, P.J. 2001. The pVHL-hIF-1 system. A key mediator of oxygen homeostasis. Adv. Exp. Med. Biol. 502 365–376. [PubMed] [Google Scholar]
- Min, J.H., Yang, H., Ivan, M., Gertler, F., Kaelin Jr., W.G., and Pavletich, N.P. 2002. Structure of an HIF-1α–pVHL complex: Hydroxyproline recognition in signaling. Science 296 1886–1889. [DOI] [PubMed] [Google Scholar]
- Morrison, D.A. 1996. Phylogenetic tree-building. Int. J. Parasitol. 26 589–617. [DOI] [PubMed] [Google Scholar]
- Nei, M. 1996. Phylogenetic analysis in molecular evolutionary genetics. Annu. Rev. Genet. 30 371–403. [DOI] [PubMed] [Google Scholar]
- Semenza, G.L. 1999. Regulation of mammalian O2 homeostasis by hypoxia-inducible factor 1. Annu. Rev. Cell Dev. Biol. 15 551–578. [DOI] [PubMed] [Google Scholar]
- Stebbins, C.E., Kaelin Jr., W.G., and Pavletich, N.P. 1999. Structure of the VHL–ElonginC–ElonginB complex: Implications for VHL tumor suppressor function. Science 284 455–461. [DOI] [PubMed] [Google Scholar]
- Valdar, W.S. 2002. Scoring residue conservation. Proteins 48 227–241. [DOI] [PubMed] [Google Scholar]
- Varshavsky, A. 1996. The N-end rule: Functions, mysteries, uses. Proc. Natl. Acad. Sci. 93 12142–12149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatesh, B., Gilligan, P., and Brenner, S. 2000. Fugu: A compact vertebrate reference genome. FEBS Lett. 476 3–7. [DOI] [PubMed] [Google Scholar]