

Abstract
Hydrophobin SC3 is a protein with special self-association properties that differ depending on whether it is in solution, on an air/water interface or on a solid surface. Its self-association on an air/water interface and solid surface have been extensively characterized. The current study focuses on its self-association in water because this is the starting point for the other two association processes. Size-exclusion chromatography was used to fractionate soluble-state SC3. Real-time multiangular light scattering detection of the eluate indicated that SC3 mainly exists as a dimer in buffer, accompanied with a small amount of monomer, tetramer, and larger aggregates. Dimeric SC3 has very likely an elongated shape, as indicated by the hydrodynamic radius determined by using dynamic light scattering (DLS) and fluorescence anisotropy measurements on dansyl-labeled SC3. Size-exclusion chromatography experiments also indicated that the protein oligomerizes very slowly at low temperature (4°C) but rather rapidly at room temperature. Ionic strength plays an important role in the oligomerization; a short-lived monomeric SC3 species could be observed in pure water. Oligomerization was not affected by low pH but was accelerated by high pH. Fluorescence resonance energy transfer showed that dissociation occurred when the protein concentration was lowered; a large population of oligomers, presumably dimers, dissociate when the protein concentration is <4.5 μg/mL. This value is similar to the critical concentration for SC3 self-assembly. Therefore, dimeric SC3 is indicated to be the building block for both aggregation in solution and self-assembly at hydrophobic/hydrophilic interfaces.
Keywords: hydrophobin SC3, oligomerization, molecular exchange, association/dissociation, structural changes, self-assembly
Hydrophobins are a family of small fungal proteins (±100 amino acids) that have unusual biophysical properties. They are amphipathic and highly surface-active. Because of their amphipathic character, they are able to coat hydrophobic or hydrophilic solid surfaces and reverse the hydropathy character of these surfaces. They are also able to self-assemble at an air/water or water/oil interface, forming a robust, electron-microscopically identifiable monolayer characterized by a rodlet structure. This amphipathic monolayer (~10 nm thick), in fungal aerial hyphae or spores, provides a water-repellent coating, allowing them to emerge from the medium or to disperse aerially (Wösten et al. 1993, 1999; Wösten and Wessels 2000).
Hydrophobins are characterized by eight conserved cysteine residues that are proposed to form four disulfide bridges; otherwise their amino acid sequences are diverse and the length of the N-terminal sequence preceding the first cysteine residue is variable (de Vries et al. 1993; Wessels 1997). Two different types of hydrophobins, class I and class II, have been distinguished based on the differences in their hydropathy patterns and biophysical properties (Wessels 1994). SC3, secreted by Schizophyllum commune, is the best-characterized class I hydrophobin. It is distinguished from other hydrophobins by a long N-terminal sequence preceding the first cysteine residue. There are 16 to 22 mannose residues attached to the 12 threonine residues in this region, which are probably exposed at the hydrophilic side of assembled SC3 and, therefore, determine the surface properties of this side (de Vocht et al. 1998). Circular dichroism spectroscopy (CD) and attenuated total reflection-Fourier transform infrared spectroscopy (ATR-FTIR) revealed that the protein formed a stable intermediate with an α-helical signature upon the binding to a solid hydrophobic surface such as Teflon. This intermediate was able to convert to a structure with a β-sheet signature upon adding detergent and heating the sample. In contrast, no stable α-helical intermediate was seen if SC3 assembled at an air/water interface; instead, a β-sheet structure was observed to form over a period of several hours. SC3 with the α-helical signature appears to be monomeric when bound to a hydrophobic solid surface at low occupancy, as reported by the lack of fluorescence resonance energy transfer (FRET) between populations of fluorescence donor- and acceptor-labeled SC3. Upon heating in the presence of detergent, self-assembly occurred over a period of several minutes, followed by a slow conformational change to a stable structure characterized by a β-sheet signature and the presence of rodlets (Wang et al. 2002).
Although much work has been done on the structural changes and molecular interactions of SC3 bound to a hydrophobic surface, the soluble-state of SC3, which is the starting point of engaging its surface activity, has still to be explored. SC3 had been assumed to be monomeric in solution until our previous FRET studies clearly showed that SC3 dissociates in pure trifluoroacetic acid (TFA) but associates once resuspended in buffer after pure TFA treatment (Wang et al. 2002); however, the details of the association of SC3 in solution are not known. Are there multiple association states? What are their sizes, and under which conditions can they be dissociated? Such questions have been addressed in this article in an effort to understand how subunit interactions might influence the surface activity of SC3 and its self-assembly properties at an air/water interface.
Results
Dimerization of SC3 in buffer
Size-exclusion chromatography (SEC) was used to investigate the oligomerization of soluble-state SC3 in detail. Molecular-weight (MW) markers, consisting of five different proteins (Bio-Rad), were used under the same experimental conditions to calibrate the chromatography column. Figure 1 ▶ shows a typical SEC elution pattern of purified soluble-state SC3 in 50 mM sodium phosphate (pH 7.0). Except for a small portion of large aggregates at the beginning and a broad shoulder between 10 and 18 min, three peaks, indicated as P1, P2, and P3, appeared, with retention times of 19.3, 20.5, and 24.0 min, respectively. Based on the absorption at 215 nm, >80% of the applied protein was eluted during the chromatography, and it could be estimated that P2 contained ~80% of the eluted protein. All three peaks were collected separately, lyophilized, treated with TFA, and analyzed by SDS-PAGE and Western Blot by using an anti-SC3 antibody. The same SC3 band was seen in all three fractions, indicating that these three peaks contained SC3 in different oligomeric states, which, however, could be dissociated by TFA treatment into the monomers (data not shown). To further confirm the mass of the major oligomeric state of SC3, the P2 fraction, we used SEC-MALLS, which combines SEC and multiangle laser light scattering (MALLS). With this technique, the particles can be detected in real-time by the light scattering detectors arranged at 18 different angles around the cell, and the absolute molar masses and distribution can readily be obtained. The result is shown in Figure 2A ▶. Soluble-state SC3 again showed a major peak on SEC-MALLS as it had on normal SEC (dotted line); the molar mass was determined to be ~30 kD, which corresponds to the mass of dimeric SC3 (solid line). The initial area of this peak, the position of which corresponds to the P1 fraction in Figure 1 ▶, has a molar mass of ~70 kD, which is probably tetrameric SC3. The ratio between dimer and tetramer was estimated to be 7 : 3, and the yield of the eluted protein, including the large aggregates at the beginning, was ~70%. The remaining protein either stuck to the column material or was excluded by the column as large aggregates. This experiment therefore confirms the conclusion that SC3 mainly forms a dimer in buffer, but that certain amount of tetramer and larger oligomers are also present.
Figure 1.

Size-exclusion chromatography on hydrophobin SC3. Elution of soluble-state SC3 in 50 mM sodium phosphate (pH 7.0), from TSK250 HPLC column. Retention times are indicated at the top of each peak. A molecular weight marker consisting of five different proteins (Bio-Rad) was used to calibrate the column under the same experimental condition.
Figure 2.

Multiangle light scattering (MALLS) and dynamic light scattering (DLS) on soluble-state SC3. (A) Elution of soluble-state SC3 from a size-exclusion chromatography (SEC) column, which was subsequently detected in-line by a MALLS detector (solid line) and a refractive index detector (dotted line). (B) DLS analysis of soluble-state SC3: soluble-state SC3 in 50 mM sodium phosphate (pH 7.0) before SEC (left) and soluble-state SC3 in 50 mM sodium phosphate (pH 7.0) after SEC (right). Peak P2 was collected and measured immediately.
SC3 in the major peak (P2) was collected and examined further by using DLS (Fig. 2B ▶). First, SC3 prior to chromatography was measured, as described in Materials and Methods. More than two components could be identified after data fitting, indicating that the solution was heterogeneous (Fig. 2B ▶, left). In another experiment, SC3 was subjected to SEC, and peak P2 was collected (the concentration was >100 μg/mL, ensuring a high signal to noise ratio) and subjected to DLS measurement immediately. The resulting data showed a single component upon data fitting, indicating that the solution was relatively homogeneous (Fig. 2B ▶, right). The apparent hydrodynamic radius of the particle in peak P2 was ~3.4 nm, which is larger than that of a dimeric SC3 if it would have a spherical shape (~2.7 nm). This may indicate that the shape of dimeric SC3 in buffer may not be spherical, but rather elongated.
Time-dependent aggregation of dimerized SC3
The stability of dimeric SC3 in buffer has been checked with SEC. Peak P2 was collected and reloaded onto the same column under the same conditions after 2-h incubation at room temperature. Only peak P2 and P3 could be seen, whereas peak P1 and its front shoulders were missing, indicating that a small amount of monomers but no tetramers or higher oligomers formed during the incubation or subsequent chromatography (data not shown). When the same peak P2 was stored overnight at 4°C and then rechromatographed, only peak P2 indicated that dimeric SC3 is very stable at low temperature.
The oligomeric state of SC3 in the P2 fractions was further characterized by fluorescence anisotropy using dansyl-labeled SC3 (dansyl-SC3). Dansyl-SC3 was used in this experiment because the long lifetime of the covalently labeled probe (≅18 nsec), makes it possible to distinguish monomers from multimers on the basis of their rotational correlation times. Dansyl-SC3 also showed three peaks with the same retention times as SC3 on SEC (data not shown). The P2 fractions were collected and subjected to time-resolved fluorescence anisotropy measurement. Three lifetimes—0.8 to 0.9 nsec; 5 to 6 nsec, and 18 to 20 nsec—could be identified after fitting the time-resolved data (Fig. B; Table 1, upper panel). As a control, only one lifetime of 3.3 nsec was determined for the free dansyl group in buffer (Fig. 3A ▶; Table 1). The three different lifetimes observed for dansyl-SC3 might reflect the structural heterogeneity of soluble-state SC3, which had also been indicated by previous fluorescence and CD measurements (Wang et al. 2002). The fluorescence anisotropy decay was then measured on collected P2 fractions of another sample of dansyl-SC3 chromatographed under the same conditions. The data were taken 1 to 2 h after chromatography due to the optimization of the experimental setup. The resulting anisotropy decay can be described by two exponentials, corresponding to rotational correlation times of 1.77 (θ1) and 19.74 (θ2) nsec, respectively (Fig. 3C,D ▶). Table 1, lower panel, lists the correlation times measured for the sample prior to chromatography and for the pooled P2 fractions after 1-, 8-, and 24-h incubation at room temperature. It seems unlikely that θ1 reflects the tumbling of monomeric SC3 because the fraction of θ1 (A1, in Table 1) is 30% to 40%, whereas, as indicated by SEC, only a very low amount of monomeric SC3 (P3) is present. Therefore, θ1 probably reflects the tumbling of a partially restricted dansyl group attached to SC3, whereas θ2 reflects the tumbling of the whole particle. The considerable time-dependent increase of θ2 (19.7, 35.1, and 61.5 nsec for the 1-, 8-, and 24-h samples, respectively) strongly indicates that soluble-state SC3 forms larger oligomers at room temperature. θ1 for dansyl-SC3 prior to chromatography resembled that determined for P2, but θ2 was much larger (64 nsec), most likely due to the larger oligomers that exist in the sample.
Table 1.
Lifetimes and rotational correlation times of dansyl-SC3
| τ1 (nsec) | A1 (%) | τ2 (nsec) | A2 (%) | τ3 (nsec) | A3 (%) | τ (nsec) | |
| Dansyl | 3.29 | 100 | 3.29 | ||||
| Unchromatographed dansyl-SC3 | 0.75 | 28 | 4.15 | 35 | 18.77 | 37 | 8.61 |
| P2 | 0.90 | 22 | 5.50 | 37 | 19.15 | 41 | 10.08 |
| P2 8 h | 1.01 | 33 | 6.37 | 33 | 19.94 | 33 | 9.01 |
| R0 | θ1 (nsec) | A1 (%) | θ2 (nsec) | A2 (%) | θ (nsec) | |
| τ1 through τ3 indicate lifetimes; A1 through A3, fractions of respective lifetimes of the total lifetime; P2, the fraction collected from gel filtration HPLC at retention time of 20.5 min; τ, averaged lifetime; R0, fundamental anisotropy calculated at time 0, according to the extrapolation; θ1 and θ2, rotational correlation times; and θ, averaged rotational correlation time. | ||||||
| Unchromatographed dansyl-SC3 | 0.31 | 1.22 | 41 | 64.09 | 59 | 38.31 |
| P2 | 0.25 | 1.77 | 29 | 19.74 | 71 | 14.53 |
| P2 8 h | 0.35 | 3.47 | 31 | 35.06 | 69 | 25.27 |
| P2 24 h | 0.31 | 3.98 | 28 | 61.45 | 72 | 45.36 |
Figure 3.

Time-resolved fluorescence of soluble-state dansyl-SC3. (A) Experimental and fitted fluorescence lifetime decay for the unreacted dansyl suspended in 50 mM sodium phosphate (pH 7.0), at a concentration of 20 μM. (Top) The smooth line that was superimposed on the raw fluorescence data corresponds to a single exponential fit with a lifetime of 3.29 nsec (see also Table 1). (Bottom) The weighted residuals between the experimental data and the fit. (B) Experimental and fitted fluorescence lifetime decay for 0.5 μM P2-dansyl-SC3 after SEC in 50 mM sodium phosphate (pH 7.0). (Top) The smooth line that was superimposed on the raw fluorescence data corresponds to a three-component exponential fit with lifetimes of 0.9, 5.5, and 19.15 nsec, respectively (see also Table 1). (Bottom) The weighted residuals between the experimental data and the fit. (C) The decay of vertical and horizontal polarization components of chromatographed dansyl-SC3, P2, upon excitation with 350-nm vertically polarized light. Solid line indicates vertical component; dotted line, horizontal component. (D) The corresponding calculated anisotropy curve and the fit for the measurement in C. (Top) The smooth line that was superimposed on the anisotropy curve corresponds to a two-exponential fit with rotational correlation times of 1.77 and 19.74 nsec, respectively (see also Table 1). (Bottom) The weighted residuals between the experimental data and the fit.
The size of the oligomer in the homogeneous sample, peak P2, can be estimated from its θ2, which is ~20 nsec. Depending on the degree of hydration, the calculated rotational correlation time for dimeric dansyl-SC3 varies from 8.8 to 13.2 nsec, assuming that the particles are spherical in shape. The obvious difference between the theoretical and measured rotational correlation time might be caused by a higher degree of hydration, an elongated shape of dimeric SC3 in buffer, or both. The later has also been indicated by the DLS data.
Factors that affect the oligomerization
When TFA-treated SC3 was resuspended in water and the chromatography carried out in pure water instead of 50 mM sodium phosphate (pH 7.0), a completely different elution pattern was observed (cf. Figs. 1 ▶ and 4A ▶). The pH of this sample was 6.0. The presence of a broad peak in Figure 4A ▶, with a retention time lasting from 17 to 22 min, indicates the occurrence of larger oligomers. A sharp peak, with a retention time of ~24 min, however, most likely represents the presence of monomeric SC3. This monomeric SC3, however, was not stable in water. A fraction of Pb, which contained monomeric SC3, was collected and stored for 1 h at 4°C before being loaded onto the same column again equilibrated with water. The pH of sample was checked and determined to be 5.0 to 6.0. Pb disappeared together with the emergence of a peak at the position of dimeric SC3; all monomers appear to have converted to dimers in this case (data not shown). When 50 mM sodium chloride in water (pH −6) instead of pure water was used to repeat the same experiment, the elution pattern was the same as that obtained when using 50 mM sodium phosphate (pH 7.0; Fig. 4B ▶), indicating that increased ionic strength results in oligomerization of SC3. All efforts made to get a pure monomeric form or to keep the monomeric fraction for longer time failed, indicating that SC3 monomers have a high tendency to oligomerize.
Figure 4.

Oligomerization of SC3 under various buffer conditions. (A) The same experiment as Fig. 1 ▶, but pure water was used instead of 50 mM sodium phosphate (pH 7.0) to suspend TFA-treated lyophilized SC3 and to equilibrate the column. The pH of the sample and the column effluent was about 6. (B) The same experiment as Fig. 1 ▶, but 50 mM sodium chloride in water (pH ~6), was used instead of 50 mM sodium phosphate (pH 7.0) to suspend SC3 and equilibrate the column. (C) The same experiment as Fig. 1 ▶, but 50 mM sodium bicarbonate (pH 9.0), was used instead of 50 mM sodium phosphate (pH 7.0) to suspend SC3 and equilibrate the column.
A pH effect on the oligomerization of SC3 was tested by using buffers containing the same salt concentration (50 mM). A buffer with a given pH was used to suspend TFA-treated SC3 and equilibrate the column and to chromatograph SC3. Sodium phosphate (pH 7.0), sodium acetate (pH 4.0), and even 0.1% TFA with pH 1 to 2, yielded primarily the dimeric form of the protein, from which we can conclude that oligomerization of SC3 is not sensitive to low pH. However, the elution pattern totally changed when sodium carbonate-bicarbonate (pH 9.0) was used. The dominant oligomeric form is obviously larger than a tetramer, with a retention time of 17.3 min; a small peak with a retention time ~21 min, probably corresponding to the remainder of the dimeric SC3, could also be observed (Fig. 4C ▶). Therefore, SC3 forms larger oligomers at higher pH, indicating that the sole basic amino acid in SC3, histidine, which is located at the N-terminal domain, may play an important role in keeping SC3 dimeric in solution.
Monomer–dimer–tetramer equilibrium
When P2 fractions collected from the SEC were reloaded onto the same column under the same condition, P1 shown in the first chromatography was no longer present, but the distribution of the last two peaks, P2 and P3, remained unchanged (data not shown). No apparent difference could be observed in the peak distribution whether P2 fractions from the first chromatography were reloaded 1 or 48 h after being pooled and stored at 4°C. Apparently, the equilibrium between SC3 in P1 and P2 is a relatively slow process; however, the equilibrium between SC3 in P2 and P3 is established quickly and remains stable for many hours at 4°C.
SC3 samples at different protein concentrations were studied by SEC, and the transition between the tetramer in P1, the dimer in P2, and the monomer in P3 was monitored by comparing the peak areas (Fig. 5 ▶). After dilution from the same SC3 stock solution, the sample was incubated for ~10 min at room temperature and chromatographed under the same conditions (50 mM sodium phosphate at pH 7.0). After elution, the areas of all three peaks were integrated. When the protein concentration decreased from 1 mg/mL to 20 μg/mL, the ratio between the areas of P1, P2, and P3 varied from 17.9 : 81 : 1.1 to 13.2 : 85.9 : 0.9, indicating that the distribution of the oligomeric forms did not change much in this concentration range. However, upon lowering the concentration from 20 to 3.5 μg/mL, the peak ratios changed from 13.2 : 85.9 : 0.9 to 7.9 : 85.8 : 6.3, demonstrating a substantial increase in the fraction of monomer at the low protein concentration (a few micrograms per milliliter).
Figure 5.

SEC analysis of protein concentration–dependent oligomerization. (A) A 0.2-mL sample of soluble-state SC3 in 50 mM sodium phosphate (pH 7.0), at a concentration of 1 mg/mL was loaded onto the column. The peak areas of P1, P2, and P3 were integrated to be 45.9, 207.8, and 2.8 AU, respectively, yielding the ratio of 17.9 : 81 : 1.1. (B) The same sample as in A but diluted to 20 μg/mL with the same buffer before being loaded onto the column; 0.2-mL sample was loaded. The peak areas of P1, P2, and P3 were integrated to be 0.503, 3.263, and 0.033 AU, respectively, yielding the ratio of 13.2 : 85.9 : 0.9. (C) The same sample as in A but diluted to 3.5 μg/mL with the same buffer before being loaded onto the column; 0.2-mL sample was loaded. The peak areas of P1, P2, and P3 were integrated to be 0.0095, 0.1028, and 0.0075 AU, respectively, yielding the ratio of 7.9 : 85.8 : 6.3.
The concentration-dependent dissociation was quantitatively determined by FRET using a sample containing donor- and acceptor-labeled SC3, which have been completely mixed in the oligomers (for sample preparation, see Materials and Methods). The sample was diluted with buffer stepwise, and after each dilution, the sample was incubated for 10 min at room temperature followed by the measurements of fluorescence. At high concentration, the fluorescence is quenched by the proximity of SC3 molecules labeled with donor or acceptor moieties in the same oligomeric complex. However, stepwise dilution resulted in an increase of donor fluorescence after correction for the fluorescence of the control sample, TFA/dansyl-SC3 alone. Two different concentration dependencies were observed (Fig. 6 ▶). From 80 to 10 μg/mL, the fluorescence increased only slightly as the protein concentration was lowered, whereas at <4.5 μg/mL the fluorescence increased more dramatically. The donor fluorescence increase was obviously caused by the decrease of FRET due to the dissociation of oligomeric SC3. The fact that the protein used in this experiment was not chromatographed might be the cause of the two concentration dependencies. The shallow increase might reflect the dissociation of larger oligomers into smaller ones, which are themselves still quenched by the presence of acceptor-labeled protein within the same oligomer, whereas the steep increase might reflect mainly the monomerization from dimers, which has been indicated by the SEC experiment mentioned above.
Figure 6.

Fluorescence resonance energy transfer (FRET) analysis of protein concentration–dependent oligomerization. A sample of TFA/dansyl-SC3/dabcyl-SC3 was prepared as described in Materials and Methods. Donor fluorescence spectra were measured, and the resulting fluorescence intensities were first corrected for the fluorescence change of the control sample (the same amount of dansyl-SC3 alone) and then corrected for the dilution factors. The arrows point out the concentrations at which dimeric SC3 dissociated dramatically.
Structure of dimeric SC3
The secondary structure of soluble-state SC3 in a buffer has been well characterized by CD and FTIR (de Vocht et al. 1998). However, whether or not different oligomeric forms are structurally different is still questionable. We therefore isolated different oligomeric forms of SC3 in 50 mM sodium phosphate (pH 7.0), followed by immediate CD measurements. We could not see a significant difference between the CD spectra of dimeric SC3 and unchromatographed SC3 (Fig. 7A ▶). Tetrameric SC3 and the sample after overnight incubation at room temperature also had the same CD spectrum (data not shown). All samples showed an ellipticity maximum at 209 nm, whereas a control, performic acid oxidized SC3 in the same buffer, showed a typical unstructured spectrum with a maximum ellipticity at ~200 nm. CD data of monomeric SC3 could not be measured due to the low protein concentration after SEC. The secondary structure elements of dimeric SC3 were then calculated from its CD spectrum based on molar ellipticity, which revealed ~12% α-helix, 42% β-sheet, 18% β-turn, and 28% random coil. This result is consistent with a previous study that used unchromatographed SC3 (de Vocht et al. 1998). Therefore, by this criterion, oligomerization of SC3 in solution does not change the protein structure significantly; structural change only occurs when the protein self-assembles at a hydrophobic/hydrophilic interface. The fact that dimeric SC3 is structured was confirmed by a one-dimensional 1H NMR measurement (Fig. 7B ▶). To increase the signal to noise ratio, SEC-separated, dimeric SC3 was concentrated four times at 4°C, as described in Materials and Methods. Spectral dispersion in the range of 7 to 9 ppm clearly showed that the protein is structured. The same measurement was repeated after storing the sample for 10 d at 4°C and yielded the same spectrum (data not shown). Such stability holds out hope for a successful high-resolution NMR structure determination of dimeric SC3 in the future.
Figure 7.

Structure determination of P2, dimeric SC3. (A) CD spectra of dimeric SC3. The measurement was done at 4°C in the presence of 50 mM sodium phosphate (pH 7.0). Solid line indicates unchromatographed SC3; dotted line, dimeric SC3 collected from the gel filtration column; and dashed line, performic acid oxidized SC3. (B) One-dimensional 1H NMR spectrum of dimeric SC3. The sample in 50 mM sodium phosphate (pH 7.0) after gel filtration was concentrated four times at 4°C. The measurement was done for >15 h at 4°C.
Discussion
Hydrophobin SC3, in a buffered solution, has been shown to exist in different oligomeric states, with the dimer dominant. Diverse biophysical techniques were used in this study in order to characterize this protein. SEC, using commercially available MW markers, identified three oligomeric forms of SC3: a monomer, a dimer, and a tetramer, with the dimer being the dominant form. We then performed SEC-MALLS to determine the absolute molecular mass of the oligomers. The main peak after SEC was a mixture of tetramer and dimer, in a ratio of ~3 : 7, in agreement with the previous SEC experiment. The DLS and time-resolved fluorescence anisotropy measurements on the isolated dimeric form, however, showed a larger hydrodynamic radius than expected for a spherical species. This might indicate that dimeric SC3 is solvated to a higher extent than an average globular protein, that it possesses an elongated shape, or both. Rechromatography experiments showed that dimeric SC3 in 50 mM sodium phosphate (pH 7.0) was relatively stable at low temperature (>2 d) but had a tendency to form larger oligomers at higher temperature, indicating that the oligomerization is probably driven by hydrophobic interactions between monomers. This dynamic aggregation was confirmed with time-resolved fluorescence anisotropy measurements, which showed that the rotational correlation times of dansyl-labeled dimeric SC3 isolated by SEC increased dramatically after some hours of incubation at room temperature.
SC3 oligomerization is relatively independent of pH but is dependent on ionic strength. Significant amount of monomers, along with some oligomers larger than dimer, were observed for SC3 in pure water. This monomeric form, however, is not stable. Rechromatography experiments clearly showed that monomers could convert to dimers completely within a few hours, even at low temperature, indicating that SC3 tends to form stable dimers in solution. Interestingly, the difference of using phosphate buffer or pure water to dissolve hydrophobin SC3 has been noticed previously in an experiment in which the surface activity of SC3 was compared with those of other proteins. It was found that when SC3 was dissolved in water at a concentration of 0.1 mg/mL, the water surface tension decreased from 72 to 32 mJ/m2, whereas the same protein dissolved in phosphate buffer could only decrease the surface tension to 43 mJ/m2 (Van der Vegt et al. 1996). Thus, the monomer–oligomer transition of SC3 seems to play an important role in determining the surface activity.
Despite the relative stability of the aggregates in buffer, the molecules within the aggregate are able to undergo exchange. We have demonstrated this previously by following energy transfer between fluorescence donor- and acceptor-labeled SC3 oligomer populations (Wang et al. 2002). In the current study, the dynamic exchange is visible in the redistribution of SCC3 between monomer and dimer, starting from an isolated dimer population. It is also visible from the concentration dependence of the energy transfer process in Figure 6 ▶, in which a strong decrease in quenching is reported at SC3 concentrations <5 μg/mL. A previous study of SC3 self-assembly using thioflavin T (ThT) fluorescence indicated that a critical concentration of 3.7 μg/mL existed, above which SC3 self-assembled at the air/liquid interface; below this critical concentration, the protein did not assemble (M. L. de Vocht, I. Reviakine, H. A. B. Wösten, A. Brisson, J. G. H. Wessels, and G. T. Robillard, unpubl.). The similarity between these two concentrations, as well as the stability of dimeric SC3, strongly indicates that the dimer is a fundamental building block and that dimerization is the initial event in SC3 self-assembly at an air/water interface and possibly on solid surfaces as well. The fact that, by CD criteria, the secondary structure of dimeric SC3 is identical to its higher aggregated form in solution seems to support such a conclusion, although how the dimer organizes into an assembled form and how it converts to a β-sheet state as a result of assembly is still unknown. The class II hydrophobins, HFBI and HFBII from Trichoderma reesei, were recently characterized to be tetramers in solution by using small-angle X-ray scattering. The same tetramer was proposed to be the building block of the assembled form on a surface, based on the study of two-dimensional crystalline film by using atomic force microscopy (Torkkeli et al. 2002; Paananen et al. 2003). The unit cell in the film was found to have dimensions similar to that of the tetrameric aggregates in solution.
So far there is only one class I hydrophobin, EAS, whose soluble-state structure in solution has been studied systematically (Mackay et al. 2001). Three different isoforms of EAS are distinguishable on reverse-phase HPLC. Two of the three isoforms were present in low amounts and were determined to be unstructured monomer, whereas the dominant isoform could not be studied further due to its propensity to aggregate. The present study shows that SC3 mainly forms structured dimers in solution. Because of its low concentration and high propensity to dimerize, the monomeric form of SC3 could not be characterized, and therefore, we cannot exclude the possibility that the monomeric form of SC3 is also unstructured. According to the modern protein structure–function paradigm, native proteins (or their functional regions) can exist in any of the four thermodynamic states: ordered, molten globule, premolten globule, and random coil. The function of the protein can arise from any of the four conformations and transitions between them (Dunker et al. 2001; Uversky 2002). Accordingly, structured (ordered) oligomeric and possibly unstructured monomeric (premolten globule or random coil) hydrophobin can coexist, and transitions between them can take place. Actually some studies have already shown that self-association of an intrinsically unstructured protein induced folding into a molten globule or ordered structure (Ferre-D’Amare et al. 1993; Uversky et al. 2002). Obviously, such a disorder–order transition benefits proteins in different ways. For hydrophobins, the most important advantage might be preventing aggregation in the cell in the monomeric state and gaining the ability of binding to surfaces in the associated state. Furthermore, the formation of a structured oligomer might make hydrophobins thermodynamically stable and protease resistant in solution.
Stroud et al. (2003) recently proposed a model describing the association process of SC3, which involved three states: a monomer or multimer in solution (U-SC3), solution-assembled SC3 with less-ordered structure (S-SC3), and the interfacially assembled SC3 with a highly ordered and stacked β-sheet structure (I-SC3). S-SC3 formation is a time-dependent self-assembly from a quiescent solution in the absence of an air/water or oil/water interface, whereas I-SC3 formation needs both an air/water interface and sufficient energy, such as vortexing. We have checked the time-dependent aggregation, or assembly, of SC3 by using both SEC and fluorescence anisotropy (Table 1). The aggregates resulting from long incubations without disturbance showed CD spectra similar to the original sample and chromatographed dimeric SC3. This is consistent with the conclusion of Stroud et al. (2003), derived from SDS-PAGE and ThT fluorescence. Furthermore, our study shows that soluble-state SC3 in solution mainly exists as the dimer not the monomer, and thus, the dimer is the principle building block for both solution aggregation and interfacial assembly. The estimation of the fraction of soluble-state SC3 in the various oligomeric forms is also consistent in both studies. In our SEC experiment, dimeric SC3 was estimated to be >70% of the total mass by comparing the peak areas, whereas Stroud et al. found that >48% of the soluble SC3 was monomeric or multimeric (U-SC3) based on density gradient centrifugation and SDS-PAGE. They also reported an inverse soluble-state SC3 (U-SC3) concentration dependence for the solution aggregation (S-SC3), which they were unable to explain but which can be understood in light of the present study. At low protein concentration (<6 μg/mL), monomerization takes place and the resulting unstable monomers rapidly associate into large SDS-insoluble aggregates. In contrast, a stable dimeric form dominates when the protein concentration is high, which prevents or retards the aggregation process. This assumption of course needs to be further investigated.
Based on our experimental data, we propose to extend the three-state model of Stroud et al. (2003) to a four-state model, including a distinct monomer–dimer transition (Fig. 8 ▶). In nature, monomeric SC3 might dimerize immediately after being excreted from fungal hyphae into the aqueous medium. Therefore, apart from the known stabilizing effect of four intramolecular disulfide bonds (de Vocht et al. 2000), dimerization may also play an important role in preventing SC3 being degraded or prematurely assembled. Dimeric SC3 can probably aggregate in solution without largely changing its structure, provided that the solution is not disturbed externally. However, given some external energy (e.g., heating or vortexing) or the presence of some hydrophobic/hydrophilic interfaces (e.g, air bubbles or a Teflon surface), dimeric SC3 assembles into a two-dimensional film with dramatic structural changes, which ends with the so called "β-sheet state." Therefore, dimeric SC3 is the basis for establishing both solution aggregates (S-SC3) and interfacial self-assembly (I-SC3). Whether unstable and possibly unstructured monomeric SC3 can convert directly to S-SC3 and I-SC3 remains to be determined.
Figure 8.

Schematic representation of hydrophobin SC3 solution aggregates or interfacial self-assembly based on its dimeric form.
Materials and methods
Protein concentration determination
Because SC3 lacks aromatic amino acid residues, protein concentration was determined by CD spectroscopy. The exact protein concentration of soluble-state SC3 was determined with total amino acid analysis, from which the mean residue molar ellipticity could be calculated based on the CD signal measured afterward. For each determination, the maximal ellipticity determined at 209 nm was converted to protein concentration according to this mean residue molar ellipticity (de Vocht et al. 1998).
Size-exclusion chromatography
A Bio-Sil TSK-250 gel filtration column (600 × 7.5 mm, Bio-Rad) installed on a ChemStation HPLC system (Hewlett-Packard) was equilibrated with various buffers or water at a flow rate of 1 mL/min at room temperature. Lyophilized SC3 was dissolved in a small amount of TFA, dried with a flow of nitrogen gas, and then suspended in either buffer or pure water to a final concentration of 1 to 3 mg/mL. A 200 μL volume of soluble-state SC3 was injected into the column that had been equilibrated by the same buffer or water, followed by elution at a flow rate of 1 mL/min. The elution of SC3 was monitored at 215 nm. The eluate was collected and stored at 4°C, if necessary. The elution pattern was then analyzed by using the instrument software.
SEC-MALLS
SEC-MALLS was used to determine the molecular mass of soluble-state SC3. A TSK 2000SW column (7.5 × 300 mm, Agilent Technologies) was connected to an HPLC system, which was coupled to an Optilab DSP refractive index (RI) detector and a DAWN 18-angle lighter scattering detector (Wyatt Technology). The eluate from the SEC column could then be detected in terms of both refractive index and light scattering, and the absolute masses and molar mass distributions of particles in the eluate could be obtained in real-time. The whole system was equilibrated at a flow rate of 0.5 mL/min with filtered 50 mM sodium phosphate (pH 7.0), for >4 d before use, in order to remove all the large particles and dust from the system. Soluble-state SC3 at 23 mg/mL in the same buffer was centrifuged at 14,000 rpm for 10 min at 4°C, and the supernatant was injected into the column. The experiment was run for 45 min at the same flow rate as used for equilibration.
The data analysis was done by using Wyatt software, which follows the theory of classical light scattering:
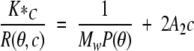 |
where R(θ,c) is the excess Rayleigh ratio of the solution as a function of scattering angle θ and concentration c. It is directly proportional to the intensity of the scattered light in excess of the light scattered by the pure solvent. c is the solute (SC3) concentration, Mw is the weight-averaged solute molar mass, and A2 is the second virial coefficient in the virial expansion of the osmotic pressure. K* is the constant 4p2(dn/dc)2n02/Naλ 04. dn/dc is the refractive index increment and was set to be 0.15, which is an average value for proteins, Na is Avogadro’s number, n0 is the index of refraction of the solvent, and λ0 is the vacuum wavelength of the laser. P(θ) describes the angular dependence of the scattered light and can be related to the root mean square radius.
Dynamic light scattering
Measurements were performed at 4°C by using a DynaPro instrument (Protein Solutions Inc.) equipped with a thermostated cell. This instrument uses light from an Nd:YAG laser lasing at 883 nm to measure the fluctuation in intensity of light scattered by molecules, which is then analyzed by using single-photon-counting electronics hardware. Under the assumption of Brownian motion and a hard-sphere model for the particle, the diffusion coefficient can be converted to the hydrodynamic radius (Rh) by using the Stokes-Einstein equation:
Rh = kBT/6πηDT, where kB is Boltzmann’s constant, T is the absolute temperature, and η is the viscosity.
The estimated MW is based on the assumption that the protein particles are approximate spheres. The MW was calculated by using an algorithm MW = (1.549Rh)2.426, which is based on linear calibration curves (log Rh versus log MW) constructed by using 18 different proteins. In practice, Rh and MW were calculated by using the software Dynapro V.4, which uses the above equations and autocorrelation coefficients. Soluble-state SC3 in 50 mM sodium phosphate (pH 7.0) with a concentration of 1 mg/mL was centrifuged at 14,000 rpm for 10 min at 4°C. Supernatant with a volume of >20 μL was added to the cuvette, and the measurement was started immediately to collect 10 to 20 data points. The theoretical two-exponential autocorrelation function (bimodal analysis) was used to fit the data. In the case of quantitatively determining the MW of SC3 in solution, only a highly homogeneous protein solution after purification with gel filtration was used. For a typical Rh of SC3 of ~3.4 nm, only those measurements with standard deviations in calculated hydrodynamic radius (Rh) of <0.2 nm were considered.
Specific labeling of hydrophobin SC3
The specific labeling of SC3 with amine reactive fluorescence dyes was performed as described previously (Wang et al. 2002). Briefly, to obtain soluble-state SC3, an amount of lyophilized SC3 was weighed, treated with chilled pure TFA, and then dried with a stream of nitrogen gas. Buffer containing 0.1M NaHCO3 (pH 7.2, for dansyl labeling) or 0.1M NaHCO3 (pH 8.3) plus 0.1% Empigen (for dabcyl labeling) was added to the dried material to a concentration of ~5 mg/mL. Concentrated fluorescence dye in dimethylformamide, either succinimidyl ester of 6-((5-dimethylaminonaphthalene-1-sulfonyl)amino)hexanoic acid (dansyl) or succinimidyl ester of 4-(4-(dimethylamino)azo) benzoic acid (dabcyl; Molecular Probes), was added slowly to the stirred solution of soluble-state SC3 until the molar ratio of fluorescence dye to SC3 reached 20 : 1; the volume added was <10% of the total volume. The reaction mixture was stirred slowly overnight in the dark and then centrifuged at 10,000g for 15 min. Subsequently, 0.5 mL supernatant was applied immediately to water-equilibrated desalting PD-10 column, followed by elution with water. The stoichiometry of labeling with dansyl was determined by MALDI-TOF mass spectrometry; 50% to 60% of SC3 was labeled. The CD spectrum showed that labeling occurred without gross conformational changes. In the case of dabcyl SC3, the stoichiometry of the reaction was based on the absorbance of dabcyl at 453 nm using an extinction coefficient of 32,000M−1cm−1. The labeling yield was 90% to 100%, assuming that one dabcyl molecule coupled to one SC3 molecule. The CD spectra were again similar to those of the unlabeled protein.
Steady-state fluorescence and time-resolved fluorescence measurements
A SPF-500C spectrofluorometer (SLM Aminco) with a 300-W Xenon lamp type 300 UV was used to measure the steady-state fluorescence of dansyl-labeled protein. Soluble-state dansyl-SC3 was prepared in 10 mM sodium phosphate (pH 7.0). The fluorescence spectra were then recorded with an excitation wavelength of 340 nm and an emission wavelength ranging from 400 to 650 nm. The resulting spectra were corrected for buffer and instrumental distortions.
For time-resolved fluorescence measurements, polarized fluorescence decay curves were measured by the time-correlated single photon counting technique. The laser system consisted of a Verdi-5W laser that pumped the mode-locked Mira Ti:Sapphire laser, followed by a pulse picker and a harmonics generator delivering subpicosecond pulses. The vertically polarized excitation provided an excitation wavelength of 350 nm with a pulse frequency of 1.9 MHz. Fluorescence emission was measured from 400 to 700 nm, using a WG360 cutoff filter (Schott). For fluorescence anisotropy decay measurements, the emitted light passed through a polarizer and was recorded with a Hamamatsu streak camera model C5680 equipped with a Cromex 250IS imaging spectrograph. Because the polarizer could be oriented both horizontally and vertically, the intensity of parallel Ih(t) and perpendicular Iv(t) components could be measured separately. For fluorescence lifetime measurements, the emitted light was recorded directly as I(t) without the polarizer. Fluorescence intensities, either polarized or not polarized, were then recorded in a time range of 50 nsec at 20°. To determine the time-dependent fluorescence anisotropy, r(t), the equation r = (Ih − GIv)/(Ih + 2G Iv) was used, where G is the grating factor that corrects for wavelength-dependent distortion of the polarizing system. It was determined by comparing the fluorescence intensities using horizontally polarized excitation light, with the polarizer in both the horizontal and vertical direction. The data, I(t) or r(t), were then fit to a sum of exponential decays by using a nonlinear least-squares algorithm, Y(t) = ∑αiexp(−t/xi), in which Y(t) is the fluorescence intensity or anisotropy at time t, and αi is the amplitude of the ith component xi such that ∑iαi = 1. xi can be lifetime τi or rotational correlation time θi. The adequacy of the fit was judged by inspection of the plots of weighted residuals, which should be randomly distributed for a satisfactory fit. The average fluorescence lifetime (τ) was calculated according to τ = ∑αiτi/∑αi, which is similar to that used to calculate the average rotational correlation time θ. The correlation time of the Brownian rotation of a protein (θ) depends on the hydrated volume (V) and the shape of the protein, as well as on the temperature (T) and viscosity (η) of the solution as given by the Stokes-Einstein equation (spherical particle): θ = ηV/KT = M(ν + h) η/RT, where K is the Boltzmann constant, M is the molecular mass of the protein, ν is the partial specific volume (milliliters per gram), and h the amount of hydration (milliliters per gram) of the rotating particle. Therefore, the rotational correlation time, θ, of monomeric SC3, with MW of 14 kD, can be calculated to be 4.37 (hydration is 0), 5.5 (hydration is 0.2), or 6.63 (hydration is 0.4).
Sample preparation for FRET
Based on the assumption that SC3 molecules associate in solution but dissociate in TFA, two different ways to prepare the samples for FRET measurements were used, as described previously (Wang et al. 2002). In the present study, only the second method was used. Briefly, the same amount of dansyl (energy donor)- and dabcyl (energy acceptor)-labeled protein was used. After mixing of the two species, the solution was freeze-dried, treated with TFA, dried with nitrogen gas, and finally dissolved in buffer to a total protein concentration of 10 μg/mL. Because dabcyl does not fluoresce, FRET can only be determined by the fluorescence quenching of the donor. The fluorescence intensity of donor was measured as described in Steady-State Fluorescence and Time-Resolved Fluorescence Measurements.
Circular dichroism spectroscopy
CD spectra of SC3 were recorded over the wavelength range from 190 to 250 nm on an Aviv 62A DS CD spectrometer, using a 1-mm quartz cuvette. The temperature was kept at 4°C, and the sample compartment was continuously flushed with nitrogen gas. The final spectra were obtained by averaging six scans, using a bandwidth of 1 nm, a stepwidth of 1 nm, and a 5-sec averaging per point. The spectra were then corrected for the background signal by using a reference solution without the protein.
Soluble-state SC3 and the chromatographed samples were prepared as mentioned. To prepare performic acid oxidized SC3, lyophilized SC3 was treated with TFA, dried with a flow of nitrogen gas, and then suspended in formic acid. The sample was incubated on ice for 5 min, followed by adding freshly made performic acid (a 1 : 10 mixture of hydrogen peroxide and formic acid incubated for 2 h at room temperature) to a final concentration of 10%. After 15-min incubation on ice, SC3 was separated from performic acid by elution through a PD-10 column, which was equilibrated with water. Fractions containing SC3 were pooled and lyophilized.
One-dimensional 1H NMR
Soluble-state SC3 in 50 mM sodium phosphate (pH 7.0) was chromatographed, and the peak P2 was collected and concentrated by using a Centricon centrifugal filter YM-10 (Millipore; MW cut-off, 10,000). The centrifugation was performed for 1 h at 4°C at 5000g. Subsequently, D2O was added to the concentrated sample to a final volume of 8%; the final protein concentration was ~1 mg/mL. One-dimensional NMR data, with water suppression, were recorded for 15 h at 4°C on a Varian Unity INOVA 600 NMR spectrometer. In total, 23,856 scans were acquired and averaged.
Acknowledgments
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.03367304.
References
- de Vocht, M.L., Scholtmeijer, K., van der Vegte, E.W., de Vries, O.M.H., Sonveaux, N., Wosten, H.A.B., Ruysschaert, J.M., Hadziioannou, G., Wessels, J.G.H., and Robillard, G.T. 1998. Structural characterization of the hydrophobin SC3, as a monomer and after self-assembly at hydrophobic/hydrophilic interfaces. Biophys. J. 74 2059–2068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Vocht, M.L., Reviakine, I.R., Wosten, H.A.B., Brisson, A., Wessels, J.G.H., and Robillard, G.T. 2000. Structural and functional role of the disulfide bridges in the hydrophobin SC3. J. Biol. Chem. 276 28428–28432. [DOI] [PubMed] [Google Scholar]
- de Vries, O.M.H., Fekkes, M.P., Wosten, H.A.B., and Wessels, J.G.H. 1993. Insoluble hydrophobin complexes in the walls of Schizophyllum commune and other filamentous fungi. Arch. Microbiol. 159 330–335. [Google Scholar]
- Dunker, A.K., Lawson, J.D., Brown, C.J., Williams, R.M., Romero, R., Oh, J.S., Oldfield, C.J., Campen, A.M., Ratliff, C.M., Hipps, K.W., et al. 2001. Intrinsically disordered protein. J. Mol. Graph. Model. 19 26–59. [DOI] [PubMed] [Google Scholar]
- Ferre-D’Amare, A.R., Prendergast, G.C., Ziff, E.B., and Burley, S.K. 1993. Recognition by max of its cognate DNA through a dimeric b/HLH/Z domain. Nature 363 38–45. [DOI] [PubMed] [Google Scholar]
- Mackay, J.P., Matthews, J.M., Winefield, R.D., Mackay, L.G., Haverkamp, R.G., and Templeton, M.D. 2001. The hydrophobin EAS is largely unstructured in solution and functions by forming amyloid-like structures. Structure 9 83–91. [DOI] [PubMed] [Google Scholar]
- Paananen, A., Vuorimaa, E., Torkkeli, M., Penttila, M., Kauranen, M., Ikkala, O., Lemmetyinen, H., Serimaa, R., and Linder, M.B. 2003. Structural hierarchy in molecular films of two class II hydrophobins. Biochemistry 42 5253–5258. [DOI] [PubMed] [Google Scholar]
- Stroud, P.A., Goodwin, J.S., Butko, P., Cannon, G.C., and McCormick, C.L. 2003. Experimental evidence for multiple assembled states of SC3 from Schizophyllum commune. Biomacromolecules 4 956–967. [DOI] [PubMed] [Google Scholar]
- Torkkeli, M., Serimaa, R., Ikkala, O., and Linder, M. 2002. Aggregation and self-assembly of hydrophobins from Trichoderma ressei: Low resolution structural models. Biophys. J. 83 2240–2247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uversky, V.N. 2002. Natively unfolded proteins: A point where biology waits for physics. Protein Sci. 11 739–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uversky, V.N., Permyakov, S.E., Zagranichny, V.E., Rodionov, I.L., Fink, A.L., Cherskaya, A.M., Wasserman, L.A., and Permyakov, E.A. 2002. Effect of zinc and temperature on the conformation of the γ-subunit of retinal phosphodiesterase: A natively unfolded protein. J. Proteome Res. 1 149–159. [DOI] [PubMed] [Google Scholar]
- Van der Vegt, W., van der Mei, H.C., Wösten, H.A.B., Wessels, J.G.H., and Busscher, H.J. 1996. A comparison of the surface activity of the fungal hydrophobin SC3p with those of other proteins. Biophys. Chem. 57 253–260. [DOI] [PubMed] [Google Scholar]
- Wang, X., de Vocht, M.L., de Jonge, J., Poolman, B., and Robillard, G.T. 2002. Structural changes and molecular interactions of hydrophobin SC3 in solution and on a hydrophobic surface. Protein Sci. 11 1172–1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wessels, J.G.H. 1994. Developmental regulation of fungal cell wall formation. Ann. Rev. Phytopathol. 32 413–437. [Google Scholar]
- ———. 1997. Hydrophobins: Proteins that change the nature of a fungal surface. Adv. Microb. Physiol. 38 1–45. [DOI] [PubMed] [Google Scholar]
- Wösten, H.A.B. and Wessels, J.G.H. 2000. Surface-active proteins enable microbial aerial hyphae to grow in the air. Microbiology 146 767–773. [DOI] [PubMed] [Google Scholar]
- Wösten, H.A.B., de Vries, O.M.H., and Wessels, J.G.H. 1993. Interfacial self-assembly of a fungal hydrophobin into a hydrophobic rodlet layer. Plant Cell 5 1567– 1574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wösten, H.A.B., van Wetter, M.-A., Lugones, L.G., van der Mei, H.C., Busscher, H.J., and Wessels, J.G.H. 1999. How a fungus escapes the water to grow into the air. Curr. Biol. 9 85–88. [DOI] [PubMed] [Google Scholar]