

Abstract
Expression of recombinant proteins as fusions to the eukaryotic protein ubiquitin has been found to significantly increase the yield of unstable or poorly expressed proteins. The benefit of this technique is further enhanced by the availability of naturally occurring deubiquitylating enzymes, which remove ubiquitin from the fusion product. However, the versatility of the system has been constrained due to the lack of a robust, easily purified deubiquitylating enzyme. Here we report the development of an efficient expression system, utilizing the ubiquitin fusion technique, which allows convenient high yield and easy purification of authentic protein. An Escherichia coli vector (pHUE) was constructed for the expression of proteins as histidine-tagged ubiquitin fusions, and a histidine-tagged deubiquitylating enzyme to cleave these fusions was expressed and purified. The expression system was tested using several proteins varying in size and complexity. These results indicate that this procedure will be suitable for the expression and rapid purification of a broad range of proteins and peptides, and should be amenable to high-throughput applications.
Keywords: protein purification, ubiquitin-fusion, deubiquitylating enzyme, affinity purification
The expression of a cloned gene to isolate large quantities of its protein product demands a highly efficient expression system in which protein can be purified to homogeneity, especially for crystallographic and therapeutic purposes. Unfortunately, high-level expression of biologically active recombinant proteins, especially those from eukaryotes, is often difficult to achieve. In response to this need for efficient expression, several systems have emerged that involve fusing the gene of interest downstream of a second gene to produce a fusion protein (Uhlen and Moks 1990). This strategy generally gives reliably high protein yields and can also allow simple purification methods due to the affinity of certain fusion partners for a particular ligand (Baker 1996). A major drawback of this approach is the covalent linkage of the two proteins, where the presence of the fusion partner may prevent or interfere with subsequent use of the desired protein. To overcome this problem a protease recognition site can be constructed between the two fused proteins; however, this involves altering the N terminus of the desired product, resulting in the expression of an unauthentic protein (Butt et al. 1989). Furthermore, cleavage of the fusion protein is rarely complete, causing a reduction in protein yield, and it may also occur nonspecifically within the fused protein (Baker 1996).
A fusion partner that has been used for some years is ubiquitin (Ub). This small eukaryotic protein provides two benefits. First, like other fusion partners, it offers a natural yield enhancement, and, second and uniquely, the Ub moiety can be removed by highly specific proteases known as deubiquitylating enzymes (DUBs) that do not cleave non-specific sequences and do not leave additional amino acids at the N terminus of the protein of interest (Baker 1996; Hondred et al. 1999). This cleavage occurs precisely after the final glycine residue at the carboxyl terminal of Ub irrespective of the amino acid immediately following, with the sole exception of proline, which is cleaved inefficiently (Bachmair et al. 1986). To date, the main drawbacks of the Ub fusion technique have been no simple affinity purification for Ub and no readily available deubiquitylating enzyme. Most DUBs that have been isolated from various species have been relatively large enzymes and difficulties have been encountered with expressing and purifying large quantities, along with problems in finding a stable DUB with general activity against a range of fusion proteins (Varshavsky 2000).
We have developed an efficient Escherichia coli-based expression system where the protein of interest is expressed as a fusion to poly-histidine-tagged Ub, enabling a simple one-step purification of the fusion protein by immobilized metal affinity chromatography (IMAC). We have also engineered a mouse DUB, Usp2, to provide a minimal catalytically active deubiquitylating domain and expressed and purified this as poly-histidine-tagged protein. The tagged protease allows the in vitro cleavage of Ub from the desired protein as well as its selective removal from the cleavage reaction, along with the cleaved Ub, any uncleaved fusion protein, and any copurified contaminants, leaving the desired protein as the only soluble product. This system was found to be very effective and applicable to the expression of a broad range of proteins and peptides, and should be useful for high-throughput applications.
Results and Discussion
Using pHUE for the expression and purification of ubiquitin fusion proteins
The pHUE vector was constructed for the expression of His-tagged ubiquitin fusion proteins by modifying pET15b (Novagen). It contains the inducible T7 RNA polymerase promoter, a histidine tag at the 5′ end of a Ub open reading frame and an extended polylinker (Fig. 1A ▶). When the presence of extra residues at the N terminus of the protein cannot be tolerated, a precise fusion to Ub can be generated using the SacII site (Fig. 1B ▶), which has been engineered into the 3′ end of Ub (see Baker et al. 1994 for details). The ligated DNA fragment must encode Gly 75-Gly 76 residues of Ub, which are essential for cleavage (Ecker et al. 1989). This can be achieved by PCR amplifying the gene of interest using a primer with the 5′ extension dCTC-CGC-GGT-GGT, encoding Leu 73, Arg 74, Gly 75, and Gly 76 and containing the SacII site (Baker et al. 1994; Baker 1996).
Figure 1.

The Histidine-tagged Ubiquitin Expression vector, pHUE. (A) Plasmid map of pHUE showing the ubiquitin (Ub) coding region (black box), the T7 polymerase promoter (black triangle), and other regions (shaded boxes). Arrows indicate the direction of transcription. Restriction enzyme recognition sites within the multiple cloning site (MCS) are listed and other useful recognition sites in the vector backbone are also shown (unique, except BglII); locations are given relative to the start codon upstream of the his-tag, ATG = 1. (His)6, poly histidine tag; Ampr, β-lactamase gene; ori, colE1 origin of replication; lacI, lacI repressor gene. (B) DNA and encoded protein sequence of the 5′ and 3′ end of the ubiquitin coding region showing the engineered SacII site (underlined) within codons Leu 73, Arg 74, and Gly 75, and the 3′ polylinker. Restriction sites and protein translation are given above and under the DNA sequence, respectively.
To test the efficiency of this system for both high-level expression and easy purification, several pHUE constructs were made using a number of different genes. They included (1) SUMO, a small Ub-like protein; (2) human Pi class glutathione S-transferase, with an amino terminal methionine reside (M-GSTP1); (3) human Pi class glutathione S-transferase, with an amino terminal proline reside (P-GSTP1); (4) human glutathione synthetase (GSH-S); and (5) lacZ, encoding the E. coli protein β-galactosidase (βgal). These were selected to represent a cross section of proteins, for their range in size and complexity. The two GSTP1 proteins differ only in their N-terminal residue and were chosen to investigate the cleavage efficiency of our chosen DUB against a Ub-proline bond, which has been an observed limitation of most ubiquitin-specific proteases (Gilchrist et al. 1997). The system was also used to synthesize peptides as Ub fusions. Protein expression from the empty pHUE vector (no inserted DNA fragment) produces Ub fused to a 34-residue (3.4 kD) peptide translated from the polylinker, with presumably no ordered secondary structure. Fusions were also constructed to a 28-residue (3.0 kD) peptide containing the nuclear localization signal (NLS) of SV40 large T antigen (T-ag; residues 111–135; Hubner et al. 1999) and with a 39-residue (3.8 kD) peptide containing an antigenic determinant of chicken ovalbumin (residues 328–340; Gautam et al. 1992).
Induced E. coli cells containing the pHUE constructs were analyzed for the synthesis of Ub fusion proteins by SDS-PAGE. Each Ub fusion protein was detected as an abundant band on Coomassie blue-stained SDS gels, reflecting a high level of protein expression regardless of protein size (achieved from nonoptimized cell culture; Fig. 2 ▶; data not shown). A comparison of the total and soluble protein fractions indicated most of the Ub fusions were expressed as completely soluble proteins (data not shown). Two exceptions were Ub–GSH-S and Ub–β-gal.
Figure 2.
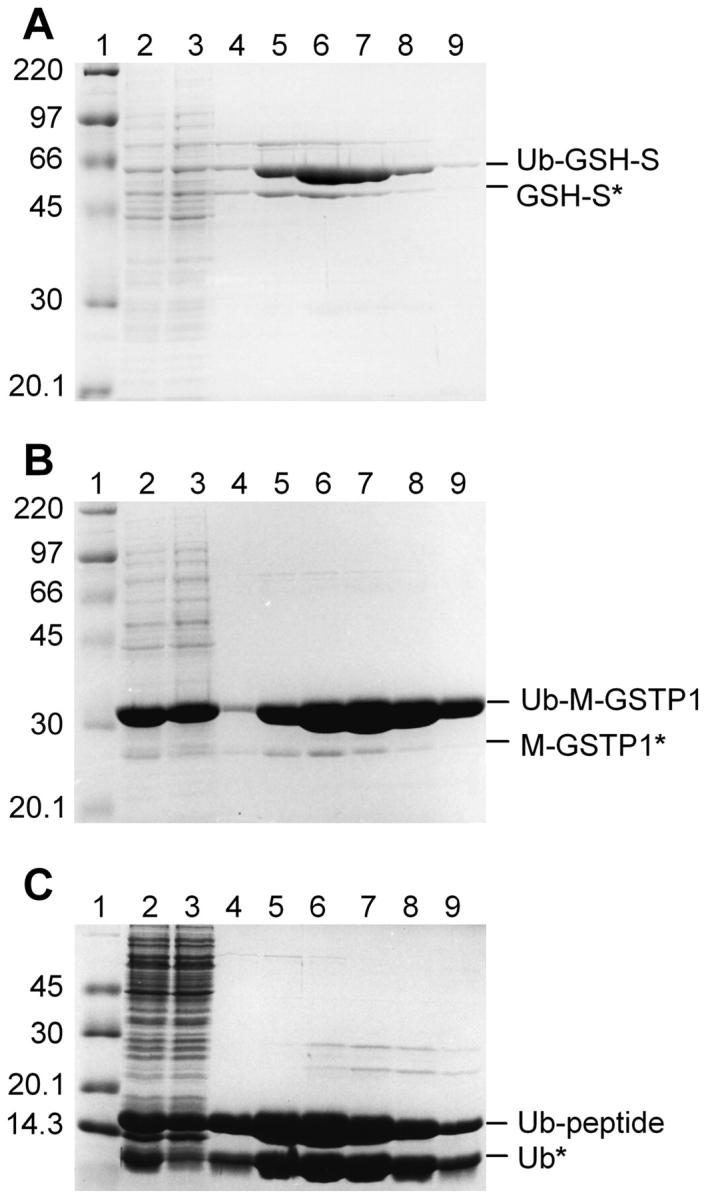
Purification of His-tagged Ub fusion proteins. Ub fusion proteins were isolated from crude E. coli extract by Ni-affinity chromatography under native conditions. Samples from sequential steps in the purification were resolved by 10% SDS-PAGE and stained with Coomassie blue. (A) Ub–GSH-S; (B) Ub–M-GST; (C) Ub-peptide. Molecular mass marker (lane 1); crude E. coli extract (lane 2); unbound proteins (lane 3); and elutions with 50 mM (lane 4), 100 mM (lane 5), 150 mM (lane 6), 200 mM (lane 7), or 250 mM (lanes 8,9) imidazole. Proteins migrating at the expected molecular mass of the Ub fusion are indicated on the right. Proteins with an asterisk represent apparent cleavage by an unknown E. coli protease (see text).
Each of the His-tagged Ub fusion proteins was successfully purified from crude E. coli extracts by nickel-affinity chromatography under native conditions (Table 1). A substantial amount of protein was recovered despite a large amount lost in the flow-through by failing to bind to the Ni-NTA agarose beads (Fig. 2 ▶), which was due to overloading. This protein loss could be overcome by increasing the volume of Ni-NTA agarose, and thus protein yields could easily double those shown. This is supported by recent protein expression from the pHUE/M-GSTP1 construct, which yielded 53 mg/L (data not shown). Only two proteins, Ub–GSH-S and Ub–β-gal, failed to give a high recovery, which was due to their reduced solubility. In both cases, half of the expressed protein was in an insoluble form and thus unable to be purified under the native conditions used. The production of insoluble protein is a commonly encountered problem with the overexpression of heterologous proteins and under current estimates only 15%–20% of human genes expressed in E. coli produce soluble protein (Stevens 2000). Although fusion to Ub generally leads to increased protein solubility (Baker 1996; Varshavsky 2000), it does not guarantee a completely soluble product (Welch et al. 1995). Although not performed here, the poly-histidine tag allows the purification of insoluble proteins under denaturing conditions, which can be followed by protein refolding, thus retaining high protein yields.
Table 1.
Yield of soluble protein for each of the Ub-fusion proteins expressed from pHUE
| Purified protein | Size (kD)a | Structureb | Yield (mg/L)c |
| His6Ub–peptide | 14.1 | Polypeptide | 14.30 |
| His6Ub–T-ag-NLS | 13.7 | Polypeptide | 13.30 |
| His6Ub–Ova | 14.5 | Polypeptide | 17.55 |
| His6Ub–SUMO | 24.5 | Monomer | 22.83 |
| His6Ub–M-GSTP1 | 34.0 | Dimer | 25.56 |
| His6Ub–P-GSTP1 | 33.9 | Dimer | 26.85 |
| His6Ub–GSH-S | 63.1 | Dimer | 3.58 |
| His6Ub–β-gal | 129.2 | Tetramer | 3.30 |
a Size of intact fusion protein. His6Ub portion contributes 10.7 kD.
b Expected subunit composition in solution.
c Yield given as purified soluble protein in milligrams per liter of Escherichia coli culture.
Each purified Ub fusion protein, except Ub–P-GSTP1, copurified with smaller amounts of a protein with the apparent molecular mass of cleaved product (Fig. 2 ▶; data not shown). To identify these extra proteins, bands from the Ub–SUMO and Ub–M-GSTP1 samples were excised from an SDS-polyacrylamide gel and the N terminus was sequenced by Edman degradation. Both sequences were a precise match to the expected sequence after Ub cleavage (see below), although no DUB was present. These results indicate that the Ub fusions are being accurately cleaved in E. coli at the peptide bond between Ub and the fused protein. This result is surprising given that E. coli lacks Ub, DUBs, and all enzymes of the Ub pathway (Tobias and Varshavsky 1991). This processing was not seen with the Ub-P-GSTP1 fusion, which indicates, like most proteases, this unknown processing activity has difficulty in cleaving a Ub–proline bond. Furthermore, this activity must be extremely inefficient, as intact Ub fusions were isolated from E. coli in large quantities. Jonnalagadda et al. (1987) also observed E. coli processing of Ub fusions and offered a number of explanations, which were not addressed in this study.
Differences in the amount of E. coli processing was noted among the Ub fusions, with the most cleaved protein present in the Ub–SUMO and Ub–β-gal, and all Ub-peptide purified fractions (Fig. 2 ▶; data not shown). For Ub–β-gal, a large percentage of the free β-gal protein would come from the endogenous E. coli lacZ gene. For Ub-peptides and Ub–SUMO this could be explained by greater accessibility to the cleavage site due to the presence of amino acids derived from the multiple cloning site, unlike the Ub–GSTP1 and Ub–GSH-S fusion proteins, which consist of two highly structured proteins. Copurification of the E. coli cleaved products is most likely due to the cleaved passenger protein forming dimers or tetramers with other His-tagged subunits or, in the case of SUMO, which is a monomer, aggregating with the intact fusion.
Usp2-cc expression, purification, and optimization
The catalytic core of the Usp2–45 open reading frame (Gousseva and Baker 2003) was cloned into pET15b (Novagen) for expression as a His-tagged protein. The expressed enzyme (termed Usp2-cc) was detectable as a predominant band on a Coomassie blue-stained SDS-PAGE gels (Fig. 3A ▶) and the majority remained in the soluble fraction (data not shown). It was purified from crude E. coli extracts by nickel-affinity chromatography under native conditions at a final yield of ~20 mg per liter of E. coli culture with ~95% purity, as determined by densitometry.
Figure 3.

Purification of His-tagged Usp2-cc and time course cleavage assay. (A) Purification of His6Usp2-cc by Ni-affinity chromatography under native conditions. Samples from sequential steps in the purification were resolved by 10% SDS-PAGE and stained with Coomassie blue. (Lane 1) Marker (masses shown at left); (lane 2) crude E. coli extract; (lane 3) unbound proteins; (lane 4) elution with 150 mM imidazole. The protein migrating with the expected molecular mass of poly-his-tagged Usp2-cc is indicated on the right. (B) Purified Usp2-cc was assayed against the Ub-M-GSTP1 test substrate over a 60-min time course at a 1 : 100 molar ratio. Samples were taken at 0, 5, 10, 15, 20, 30, 40, 50, and 60 min (shown above lanes), then resolved by 10% SDS-PAGE and Coomassie blue staining. The position of Ub–M-GSTP1 and the two cleaved products, M-GSTP1 and Ub, are indicated on the right. (C) Densitometry was used to quantify the bands from panel B, and plotted as percent uncleaved substrate remaining (on a natural log scale) over time. Values were normalized for background pixelation and loading errors. The initial rate was found to be 0.4 μg Ub–M-GSTP1 cleaved per minute at this 1 : 100 enzyme : substrate ratio. This equates to a rate of 6.3 mg Ub–M-GSTP1 cleaved per minute per milligram enzyme.
Various Ub time-course cleavage assays were performed and quantified by densitometry to optimize Usp2-cc activity (Fig. 3 ▶; data not shown). For each assay the substrate, temperature, and reducing agent remained fixed whereas the pH or NaCl concentration was varied. Usp2-cc displayed maximum activity at pH 8.5 (tested range 5.5 to 9.5) but retained >80% activity between pH 7.5 and 9.0 (data not shown). Whereas Usp2-cc showed maximum activity at 0 mM NaCl, >90% activity was displayed at NaCl concentrations between 0 and 300 mM (data not shown). Thus the enzyme is quite versatile, and cleavage conditions can be chosen to suit individual substrates.
Quantifying the cleavage efficiency of Usp2-cc
The efficiency of Usp2-cc against each of the Ub fusions was assayed by incubating each substrate with Usp2-cc at a 1 : 100 enzyme to substrate molar ratio for 60 min. Samples were resolved by SDS-PAGE, stained with Coomassie blue, and quantified by densitometry. His-tagged Usp2-cc was able to cleave all the test Ub fusion proteins to varying extents, with ~75% Ub–M-GSTP1, 65% Ub–SUMO, 50%–60% for all three Ub–peptides, 24% Ub–β-gal, and 15% Ub-GSHS cleaved. Less than 5% of Ub–P-GSTP1 was cleaved, consistent with known limitations of DUBs against the Ub-Pro bond (Gilchrist et al. 1997). Notably, other pro-teases such as enterokinase and factor Xa are also inhibited when their recognition sequence is followed by proline (Stevens 2000).
Complete cleavage of all test fusions was easily achieved by increasing the enzyme concentration to a 1 : 10 molar ratio, or by overnight incubation at 16°C (data not shown). This is not always the case for other proteases, where steric hindrance is a common cause of ineffective processing of fusion proteins (Kapust and Waugh 2000). This is possibly due to differences in substrate recognition. Both genetic and structural studies reveal that cleavage by DUBs involves the recognition of other regions of the complete Ub structure in addition to its C terminus (Johnsson and Varshavsky 1994; Johnston et al. 1999; Hu et al. 2002), whereas many other proteases only recognize a specific sequence of several amino acids. When a passenger protein obstructs the site of cleavage, the fusion protein may quickly disperse from the protease active site. However, for DUBs, the fusion protein may be retained for an extended duration due to the recognition of the Ub moiety, which may allow adequate time for structural fluctuations of the passenger protein to expose the cleavage site, allowing proteolysis to occur.
Our observations show that Usp2-cc is capable of cleaving a broad range of Ub fusion proteins independent of their size or complexity, making this enzyme highly versatile and well suited for many applications requiring the production of authentic protein. In addition to the examples reported here, we have recently used this system to express recombinant human Kappa-class glutathione transferase (Robinson et al. 2004) and the human intracellular chloride channel regulatory protein CLIC-2 (Board et al. 2004).
Purification, N-terminal sequencing, and activity of cleaved proteins
The strategy of this expression system includes a final purification step using IMAC to isolate the cleaved product from the cleavage reaction. To investigate the effectiveness of this method, each Ub fusion protein (excluding Ub–P-GSTP1) was incubated with Usp2-cc for a shortened time period to intentionally achieve partial digestion, and we used a high enzyme concentration, enabling both to be observed by SDS-PAGE to monitor their removal from the digest. After cleavage, Ni-NTA agarose (20–50 μL bed volume) and NaCl (300 mM) were added to the solution to bind the His-tagged Usp2-cc, His-tagged Ub, the uncleaved His-tagged fusion protein, and any copurified contaminants. After binding for 30 min, the mixture was centrifuged for 1–2 min and the supernatant collected for analysis by SDS-PAGE. This procedure was found to work extremely well, recovering almost all of the cleaved protein while successfully removing all other proteins present in the digest (Fig. 4A ▶; data not shown). The N terminus of the cleaved and purified M-GSTP1 was sequenced by Edman degradation to confirm precise cleavage by Usp2-cc, returning a sequence identical to the expected amino acid sequence (Fig. 4B ▶). As the test peptides were too small to be visualized by SDS-PAGE and Coomassie blue staining, Edman degradation N-terminal sequencing was performed on protein absorbed onto polyvinylidene difluoride membrane immersed in the supernatant. Again the results gave unequivocal sequences identical to the expected amino acid sequence, confirming the peptide’s presence, purity, and accurate cleavage by Usp2-cc (Fig. 4B ▶). N-terminal sequencing of cleaved and purified GSH-S was also attempted; however, no reliable sequence data was obtained.
Figure 4.

Purification and N-terminal sequencing of cleaved proteins. (A) The Ub fusion proteins named above the gels were cleaved with Usp2-cc, then purified from the reaction mix by nickel-affinity chromatography. For each substrate, samples were taken before cleavage (lane 1), after cleavage (lane 2), and the supernatant after purification (lane 3). Proteins were resolved by SDS-PAGE and Coomassie blue staining in a 10% Tricine gel. Protein positions are indicated on the right. Black arrowheads indicate contaminating proteins. Numbered arrows show protein bands excised from the gel for N-terminal sequencing in panel B. (B) N-terminal sequencing of Usp2-cc cleaved and apparent E. coli-cleaved products. (Upper) The expected site of Usp2-cc cleavage is shown (↓). (Lower) Sample numbers correspond to the protein band excised from the SDS-polyacrylamide gel shown in panel A or data not shown. (*) Supernatant after purification of Ub-peptide cleavage reaction (panel A, lane 3, Ub-peptide gel). (#) Following HPLC purification of cleavage reaction supernatant.
To examine whether proteins expressed as poly-histidine-tagged Ub fusions retain enzyme activity before and after cleavage by Usp2-cc, the specific activity of Ub-M-GSTP1 and cleaved M-GSTP1 were determined using GSH and 1-chloro-2,4-dinitrobenzene as substrates (Habig et al. 1974). For GSTP1 as a fusion to Ub, the specific activity was found to be 82.0 ± 12.48 μmole/min/mg, and for cleaved GSTP1 the specific activity was 88.1 ± 7.2 μmole/ min/mg. These figures fall into the range of published specific activity values for purified recombinant GSTP1 (64–128 μmole/min/mg; Baker et al. 1994 and references therein), suggesting that proteins purified via the poly-His-tagged Ub fusion approach and subsequently cleaved in vitro by Usp2-cc retain full enzyme activity. Furthermore, these comparable activities indicate that the presence of the poly-histidine-tagged Ub at the N terminus of GSTP1 does not interfere with its biological activity, and thus is assumed not to interfere with its folding. Similarly, for those examples that have been studied (Baker et al. 1994; Gali and Board 1995), the bound Ub did not inhibit enzyme activity, and thus for some applications the fusion protein may be left intact, reducing the number of experimental steps.
We also assayed the functionality of the SV40 large T antigen (T-ag) NLS-containing peptide produced as a Ub fusion in an ELISA assay to measure its binding to an Importin α/β heterodimer. The ovalbumin peptide and his-tagged Ub (His6-Ub) were used as negative controls. Data are presented as either without (Fig. 5A ▶) or with (Fig. 5B ▶) readings from the His6-Ub control subtracted before curve fitting. A synthetic T-ag NLS peptide was used as a positive control (Fig. 5C ▶). The Importin α/βheterodimer bound very tightly to the T-ag-NLS peptide with a Kd of 3.0 ± 1.0 nM (Fig. 5A ▶), which is comparable to a Kd of 3.4 ± 0.1 nM for the synthetic T-ag NLS peptide (Fig. 5C ▶) and to published values of 3.0 nM (Hubner et al. 1999). Binding to the control ovalbumin peptide or his-tagged Ub was far weaker (Fig. 5A ▶). Correction of the values from Figure 5A ▶ by subtracting the His6-Ub control gave a better curve fit (correlation coefficient of 0.989 versus 0.93) and a Kd of 1.7 ± 0.2 nM (Fig. 5B ▶). Thus, peptides produced by this system retain full activity, at least in protein binding assays.
Figure 5.

Peptides produced as ubiquitin fusions retain activity. An ELISA-based binding assay was performed using microtiter plates coated with the protein or peptide indicated, and the change in absorbance (405 nm) plotted against concentration of Importin α/β heterodimer used. Apparent dissociation constants (Kd) were determined following curve-fitting (see Materials and Methods). (A) Data not corrected for His6-Ub binding; (B) data corrected for His6-Ub binding; (C) data obtained from a synthetic T-ag NLS peptide.
Conclusions
We have developed an E. coli-based recombinant protein expression system, which delivers high-level expression, includes an easy means of purification, and allows the production of a variety of proteins and peptides with authentic N termini for a range of downstream applications. The steps involved in the expression system include (1) cloning the gene of interest into His-tagged Ub expression vector (pHUE); (2) expressing the fusion protein and purifying by IMAC; (3) cleaving the fusion with the purified His-tagged Usp2-cc; and (4) passing the cleavage reaction back over IMAC to remove the His-tagged Ub, His-tagged DUB, any uncleaved His-tagged fusion, and any copurifying contaminants, leaving the protein of interest as the only soluble product. Although we have not performed a totally exhaustive survey, we have tested the system with six different proteins and three unrelated peptides, with excellent results. This system exceeds the numerous expression systems currently available by meeting many of the demands of heterologous gene expression with minimal experimental steps in a convenient, low-cost manner, and would be readily amenable to high-throughput applications. The relatively small size of ubiquitin also makes this system attractive for metabolic labeling of proteins for applications such as NMR.
While this manuscript was in preparation, Wang et al. (2003) published a similar expression system that used biotin-tagged ubiquitin and a biotin-tagged chicken DUB.
Materials and methods
Construction of plasmids
The histidine-tagged ubiquitin expression vector (pHUE) was constructed using pET15b (Novagen) as a backbone plasmid. The EcoRI, ClaI, and HindIII sites in the backbone were destroyed by EcoRI/HindIII digestion followed by blunt ending with Klenow and self-ligation. The resulting vector was then digested with BamHI and ligated with a double-stranded oligonucleotide encoding an extended polylinker (5′-GATCCGAATTCGAGCTCGGT ACCGTCGACGCGGCCGCAAGCTTA-3′; 3′-GCTTAAGCTC GAGCCATGGCAGCTGCGCCGGCGTTCGAATCTAG-5′) resulting in pET15b.ep. To generate pHUE, pET15b.ep was digested with NdeI and BamHI, and ligated with a DNA insert encoding a Ub open reading frame amplified from a human adrenal gland UBA52 cDNA clone (Baker and Board 1991). The PCR primers used were 5′-GCACATATGCAGATCTTTGTGAAGAC-3′ and 5′-AATGGATCCACCGCGGAGGCGCAAC-3′.
Each of the genes cloned into pHUE was obtained by digestion of a previously constructed plasmid containing a cDNA clone. SUMO, 250-bp SacI/HindIII fragment of pRB580 (R.T. Baker, unpubl.); M-GSTP1, 646-bp SacII/HindIII fragment of pRB307 (Baker et al. 1994); P-GSTP1, 643-bp SacII/HindIII fragment of pRB481 (Gilchrist et al. 1997); GSH-S, 1425-bp SacII/HindIII fragment of pHUG (P.G. Board, unpubl.); lacZ, 3800-bp BamHI/ HindIII fragment of pLL (Baker and Varshavsky 1995); SV40-TAg, 83-bp BamHI-EcoRI fragment from pPR28 (Hubner et al. 1999); and chicken ovalbumin peptide, 80-bp XmaI-SpeI fragment from pGEM3T/Ovalbumin (Gautam et al. 1992).
The Usp2-cc open reading frame was obtained by PCR amplification of a mouse Usp2–45 cDNA from IMAGE clone 1922050 (AY255637; Gousseva and Baker 2003). The PCR primers used were 5′-CGTGGATCCTCTGCTCACCAAAGCCAAGAATTC-3′ and 5′-TCCGGATCCTTACATACGGGAGGGTGGACTG-3′. The PCR product was digested with BamHI producing a 1281-bp fragment, which was ligated into the BamHI site of pET15b in the correct orientation, resulting in pHUsp2-cc.
Expression and purification of recombinant proteins
Overnight cultures in E. coli strain BL21(DE3) were subcultured 1 : 120 into 400 mL Luria broth containing ampicillin and grown to a late exponential phase at 37°C. Protein expression was induced by adding isopropyl-1-thio-β-D-galactopyranoside (IPTG) to a final concentration of 0.4 mM, with a further 4–6 h growth. The harvested cells were resuspended in 20 mL of buffer A (50 mM Na2HPO4/NaH2PO4 at pH 7.4, 300 mM NaCl, 12 mM imidazole, 20 mM β-mercaptoethanol [β-ME], 30% glycerol) or buffer 2A for expression of Usp-cc (50 mM Na2HPO4/NaH2PO4 at pH 7.4, 300 mM NaCl, 20 mM imidazole, 20 mM β-ME, 30% glycerol), plus 1 mM Phenylmethylsulphonyl fluoride. Cells were then frozen at −70°C. His-tagged recombinant proteins were purified by nickel-affinity chromatography using batch mode under native conditions based on the QIAexpress protocol (Qiagen). To the thawed cells, ~12 mg of lysozyme were added and incubated on ice for 5–12 min. The cells were then lysed by sonication (3 × 1 min bursts at 0°C) and the soluble protein fraction recovered by centrifugation at 4°C (15 min at 15,300g). To the supernatant, 0.75–1.5 mL of a 50% slurry of nickel-nitrilotriacetic acid (Ni-NTA) agarose in buffer A was added, then placed on a rotary wheel at 4°C for 1 h. The lysate/Ni-NTA mixture was centrifuged (5 min at ~550g) and the supernatant collected as flow-through for SDS-PAGE analysis. The remaining Ni-NTA agarose pellet was washed 4–6 times in 50 mL buffer 2A. The poly-His-tagged protein was eluted from the Ni-NTA resin in 1 mL fractions with buffer A containing 50–250 mM imidazole. Chosen fractions were pooled and dialyzed for 8–16 h at 4°C.
For small-scale IMAC used to purify proteins after cleavage with Usp2-cc, the Ni-NTA agarose beads were washed in 50 mM Na2HPO4/NaH2PO4 (pH 8.0), 300 mM NaCl, and the buffer removed to leave a bed of “dry” beads. The completed cleavage reaction was made 300 mM NaCl to inhibit nonspecific ionic interactions with the Ni-NTA resin, added to the beads and mixed on a rotary wheel at 4°C for 30 min to allow binding. The cleaved target protein was recovered by centrifuging the protein/Ni-NTA solution and collecting the supernatant.
Ubiquitin cleavage activity assay
Purified proteins (Usp2-cc and Ub fusions) were dialyzed against 12–50 mM Na2HPO4/NaH2PO4 (pH 7.4–8.0), 0–300 mM NaCl, 1–2 mM β-ME, 0%–30% glycerol, and the protein concentration was determined by a Bradford microprotein assay (Pierce). The deubiquitylating activity of Usp2-cc was assayed against different Ub fusion proteins by incubating both enzyme and substrate at 37°C for various time periods under different conditions, including enzyme/substrate molar ratio, NaCl concentration, and pH. β-ME (2 mM) was always used as the reducing agent. All reactions were terminated by adding SDS-PAGE sample buffer and analyzed by SDS-PAGE, Coomassie staining, and densitometry.
Densitometry and analysis of ubiquitin cleavage activity
Densitometry was used to quantify the amount of cleaved protein by calculating the loss of the intact Ub fusion protein after a cleavage assay with Usp2-cc, compared to an uncleaved sample. Image files of Coomassie blue-stained polyacrylamide gels were created with Fuji Image Reader Las1200 LiteV1.12 film; and were opened in Image Gauge V3.46 for densitometry analysis. Boxes of equal size were drawn around each protein band and quantified as the pixel intensity per square millimeter minus a background box value. For gels that resolved the intact plus both cleaved proteins, these values were normalized for loading errors. The data were then plotted as the amount of substrate cleaved against the parameter under study (e.g., incubation time, pH, or NaCl concentration).
Functional assays
GST activity was determined using GSH and 1-chloro-2,4-dinitro-benzene as substrates (Habig et al. 1974). The ELISA was performed essentially as described by Hubner et al. (1999). Ninety-six-well microtiter plates were coated with ubiquitin-fused peptides and incubated with increasing concentrations of Importin α/Importin β heterodimer (both as GST-fusion proteins). Detection was performed using goat anti-GST primary antibody (Pharmacia) and alkaline phosphatase-coupled rabbit anti-goat secondary antibody with p-nitrophenyl phosphate as substrate and absorbance read at 405 nm using a plate reader (Molecular Devices), with values corrected by subtracting absorbance both at 0 min, and in wells without added importins, and where indicated, in wells coated with his-tagged ubiquitin. Data were fitted to the formula, B(x) = Bmax (1 − e kx), where x is the concentration of importins, as previously described (Hubner et al. 1999).
Accession numbers
Human adrenal gland UBA52 ubiquitin cDNA: X56998. Mouse Usp2–45 cDNA: AY255637.
Acknowledgments
We thank Anand Gautam for the chicken Ovalbumin plasmid and Peter Milburn for HPLC and protein sequencing. A.-M.C. was supported in part by a JCSMR Medical Sciences Honours Scholarship.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.04618904.
References
- Bachmair, A., Finley, D., and Varshavsky, A. 1986. In vivo half-life of a protein is a function of its amino-terminal residue. Science 234 179–186. [DOI] [PubMed] [Google Scholar]
- Baker, R.T. 1996. Protein expression using ubiquitin fusion and cleavage. Curr. Opin. Biotechnol. 7 541–546. [DOI] [PubMed] [Google Scholar]
- Baker, R.T. and Board, P.G. 1991. The human ubiquitin-52 amino acid fusion protein gene shares several structural features with mammalian ribosomal protein genes. Nucleic Acids Res. 19 1035–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker, R.T. and Varshavsky, A. 1995. Yeast N-terminal amidase. A new enzyme and component of the N-end rule pathway. J. Biol. Chem. 270 12065–12074. [DOI] [PubMed] [Google Scholar]
- Baker, R.T., Smith, S.A., Marano, R., McKee, J., and Board, P.G. 1994. Protein expression using cotranslational fusion and cleavage of ubiquitin. Mutagenesis of the glutathione-binding site of human Pi class glutathione S-transferase. J. Biol. Chem. 269 25381–25386. [PubMed] [Google Scholar]
- Board, P.G., Coggan, M., Watson, S., Gage, P.W., and Dulhunty, A.F. 2004. Clic-2 modulates cardiac ryanodine receptor Ca2+ release channels. Int. J. Biochem. Cell Biol. (in press). [DOI] [PubMed]
- Butt, T.R., Jonnalagadda, S., Monia, B.P., Sternberg, E.J., Marsh, J.A., Stadel, J.M., Ecker, D.J., and Crooke, S.T. 1989. Ubiquitin fusion augments the yield of cloned gene products in Escherichia coli. Proc. Natl. Acad. Sci. 86 2540–2544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ecker, D.J., Stadel, J.M., Butt, T.R., Marsh, J.A., Monia, B.P., Powers, D.A., Gorman, J.A., Clark, P.E., Warren, F., Shatzman, A., et al. 1989. Increasing gene expression in yeast by fusion to ubiquitin. J. Biol. Chem. 264 7715–7719. [PubMed] [Google Scholar]
- Gali, R.R. and Board, P.G. 1995. Sequencing and expression of a cDNA for human glutathione synthetase. Biochem. J. 310 353–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautam, A.M., Pearson, C.I., Sinha, A.A., Smilek, D.E., Steinman, L., and McDevitt, H.O. 1992. Inhibition of experimental autoimmune encephalo-myelitis by a nonimmunogenic non-self peptide that binds to I-Au. J. Immunol. 148 3049–3054. [PubMed] [Google Scholar]
- Gilchrist, C.A., Gray, D.A., and Baker, R.T. 1997. A ubiquitin-specific protease that efficiently cleaves the ubiquitin-proline bond. J. Biol. Chem. 272 32280–32285. [DOI] [PubMed] [Google Scholar]
- Gousseva, N. and Baker, R.T. 2003. Gene structure, alternate splicing, tissue distribution, cellular localization, and developmental expression pattern of mouse deubiquitinating enzyme isoforms Usp2–45 and Usp2–69. Gene Expr. 11 163–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habig, W.H., Pabst, M.J., and Jakoby, W.B. 1974. Glutathione S-transferases. The first enzymatic step in mercapturic acid formation. J. Biol. Chem. 249 7130–7139. [PubMed] [Google Scholar]
- Hondred, D., Walker, J.M., Mathews, D.E., and Vierstra, R.D. 1999. Use of ubiquitin fusions to augment protein expression in transgenic plants. Plant Physiol. 119 713–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu, M., Li, P., Li, M., Li, W., Yao, T., Wu, J.W., Gu, W., Cohen, R.E., and Shi, Y. 2002. Crystal structure of a UBP-family deubiquitinating enzyme in isolation and in complex with ubiquitin aldehyde. Cell 111 1041–1054. [DOI] [PubMed] [Google Scholar]
- Hubner, S., Smith, H.M., Hu, W., Chan, C.K., Rihs, H.P., Paschal, B.M., Raikhel, N.V., and Jans, D.A. 1999. Plant importin α binds nuclear localization sequences with high affinity and can mediate nuclear import independent of importin β. J. Biol. Chem. 274 22610–22617. [DOI] [PubMed] [Google Scholar]
- Johnsson, N. and Varshavsky, A. 1994. Split ubiquitin as a sensor of protein interactions in vivo. Proc. Natl. Acad. Sci. 91 10340–10344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston, S.C., Riddle, S.M., Cohen, R.E., and Hill, C.P. 1999. Structural basis for the specificity of ubiquitin C-terminal hydrolases. EMBO J. 18 3877–3887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonnalagadda, S., Butt, T.R., Marsh, J., Sternberg, E.J., Mirabelli, C.K., Ecker, D.J., and Crooke, S.T. 1987. Expression and accurate processing of yeast pentaubiquitin in Escherichia coli. J. Biol. Chem. 262 17750–17756. [PubMed] [Google Scholar]
- Kapust, R.B. and Waugh, D.S. 2000. Controlled intracellular processing of fusion proteins by TEV protease. Protein Expr. Purif. 19 312–318. [DOI] [PubMed] [Google Scholar]
- Robinson, A., Huttley, G.A., Booth, H.S., and Board, P.G. 2004. Modelling and bioinformatics studies of the human κ class glutathione transferase predict a novel third glutathione transferase family with homology to prokaryotic 2-hydroxychromene-2-carboxylate (HCCA) isomerases. Biochem. J. (in press). [DOI] [PMC free article] [PubMed]
- Stevens, R.C. 2000. Design of high-throughput methods of protein production for structural biology. Structure Fold. Des. 8 R177–185. [DOI] [PubMed] [Google Scholar]
- Tobias, J.W. and Varshavsky, A. 1991. Cloning and functional analysis of the ubiquitin-specific protease gene UBP1 of Saccharomyces cerevisiae. J. Biol. Chem. 266 12021–12028. [PubMed] [Google Scholar]
- Uhlen, M. and Moks, T. 1990. Gene fusions for purpose of expression: An introduction. Methods Enzymol. 185 129–143. [DOI] [PubMed] [Google Scholar]
- Varshavsky, A. 2000. Ubiquitin fusion technique and its descendants. Methods Enzymol. 327 578–593. [DOI] [PubMed] [Google Scholar]
- Wang, T., Evdokimov, E., Yiadom, K., Yan, Z., Chock, P.B., and Yang, D.C. 2003. Biotinubiquitin tagging of mammalian proteins in Escherichia coli. Protein Expr. Purif. 30 140–149. [DOI] [PubMed] [Google Scholar]
- Welch, A.R., Holman, C.M., Browner, M.F., Gehring, M.R., Kan, C.C., and Van Wart, H.E. 1995. Purification of human matrilysin produced in Escherichia coli and characterization using a new optimized fluorogenic peptide substrate. Arch. Biochem. Biophys. 324 59–64. [DOI] [PubMed] [Google Scholar]