

Abstract
The crystal structure of fosfomycin resistance protein FosA from transposon Tn2921 has been established at a resolution of 2.5 Å. The protein crystallized without bound Mn(II) and K+, ions crucial for efficient catalysis, providing a structure of the apo enzyme. The protein maintains the three-dimensional domain-swapped arrangement of the paired βαβββ-motifs observed in the genomically encoded homologous enzyme from Pseudomonas aeruginosa (PA1129). The basic architecture of the active site is also maintained, despite the absence of the catalytically essential Mn(II). However, the absence of K+, which has been shown to enhance enzymatic activity, appears to contribute to conformational heterogeneity in the K+-binding loops.
Keywords: fosfomycin, fosfomycin resistance protein FosA, antibiotic resistance, X-ray crystallography
Antimicrobial resistance of pathogenic microorganisms has become increasingly widespread during the past decades. Many of the previously known “miracle” drugs are no longer effective against bacteria commonly encountered in the clinic (Walsh 2000). The mechanisms of resistance that bacteria use to evade the effect of antibiotics fall into three categories, including (1) removal of the antibiotic through membrane pumps, (2) the accumulation of mutations that decrease uptake or the affinity of the target for the antibiotic, and (3) the destruction or sequestration of the antibiotic by enzymes or resistance proteins. An understanding of the principles and specific mechanisms that pathogens use to evade drugs is important in providing a basis for the rational design of new and improved therapeutic agents.
Fosfomycin [(1R,2S)-epoxypropylphosphonic acid] is an effective antibiotic against both gram-positive and gram-negative bacteria. It has been most successfully used for the treatment of lower urinary tract infections because of its excellent stability and broad spectrum of activity. The bactericidal activity of the drug is exerted through inhibition of the enzyme UDP-(N-acetyl)glucosamine-3-enolpyruvyl transferase, MurA, which catalyzes the first committed step in cell wall biosynthesis (Kahan et al. 1974; Marquardt et al. 1994).
Two general types of thiol transferase enzymes (FosA and FosB) that confer resistance to fosfomycin have been identified in the genomes of microorganisms. All of these enzymes belong to the vicinal oxygen chelate (VOC) superfamily of enzymes (Armstrong 2000). The enzymes share substantial sequence identities, between 23% and 40%. The enzymes that have been characterized are functional as homodimers that require divalent cations for activity. However, they differ in their preferred divalent metal ion and thiol substrate. FosA is a Mn2+/K+-dependent GSH transferase (Bernat et al. 1997,1999), whereas FosB is a Mg2+-dependent L-cysteine transferase (Cao et al. 2001) with no monovalent cation requirement.
The most extensively studied family, FosA, catalyzes the addition of GSH to fosfomycin (Scheme 1 ▶). The protein has been characterized from two sources. The plasmid-encoded protein from the transposon Tn2921 has been extensively characterized from a mechanistic standpoint and exhibits exceptionally high catalytic activity (Bernat et al. 1997,Bernat et al. 1998,Bernat et al. 1999). More recently, a version of FosA (or PA1129) was identified in the genome of the opportunistic pathogen Pseudomonas aeruginosa. The PA1129 protein exhibits a more modest catalytic activity but confers robust resistance to fosfomycin in the biological context of Escherichia coli (Rife et al. 2002). The high-resolution X-ray crystal structure of PA1129 in complex with Mn2+, K+, and fosfomycin was recently reported (Rife et al. 2002). The structure revealed that the active site of the enzyme is composed of paired βαβββ-motifs that form the metalion binding cavity. The two subunits have a three-dimensional domain-swapped arrangement so that each subunit contributes one βαβββ-motif to each active site. The manganese ion appears to act as an electrophilic catalyst in the addition of GSH.
Scheme 1.

Despite the availability of a high-resolution structure of the PA1129 enzyme, a number of questions remain to be answered concerning the structure and function of the FosA proteins. For example, What are the principal differences in the structures of the plasmid and genomically encoded enzymes? Why is the plasmid-encoded protein so much more efficient? Does the plasmid-encoded protein have a domain-swapped arrangement? What is the influence of the monovalent and divalent cations on the structure of the proteins? As a part of our program to define a basis for microbial resistance to fosfomycin, we report here a 2.5-Å resolution crystal structure of the plasmid-encoded FosA from transposon Tn2921. The protein crystallized as the apo enzyme with no bound monovalent or divalent cation. Thus, the structure helps define the influence of the essential metal ions on the structure of the protein.
Results and Discussion
The plasmid-encoded FosA and PA1129 share 60% sequence identity (Fig. 1 ▶). As is expected from such a high level of sequence identity, the basic molecular structure of the plasmid-encoded FosA is very similar to that of PA1129. Indeed, both proteins form very similar homodimers with the structural fold found in all other known members of the VOC superfamily of metalloenzymes (Fig. 2 ▶). Each monomer is essentially two tandem βαβββdomains (referred to as the N-terminal domain and the C-terminal domain) connected by a flexible linker (residues 52–65). One notable difference between the two structures is that the linker in the Tn2921 FosA is three residues longer than the corresponding region in PA1129 and contains a turn of helix. The monomers are arranged in the homodimer with three-dimensional domain swapping so that both subunits participate in the formation of each U-shaped Mn(II) binding pocket.
Figure 1.

Sequence alignment between FosA and PA1129. The image was generated using the program ESPript (Gouet et al. 1999).
Figure 2.
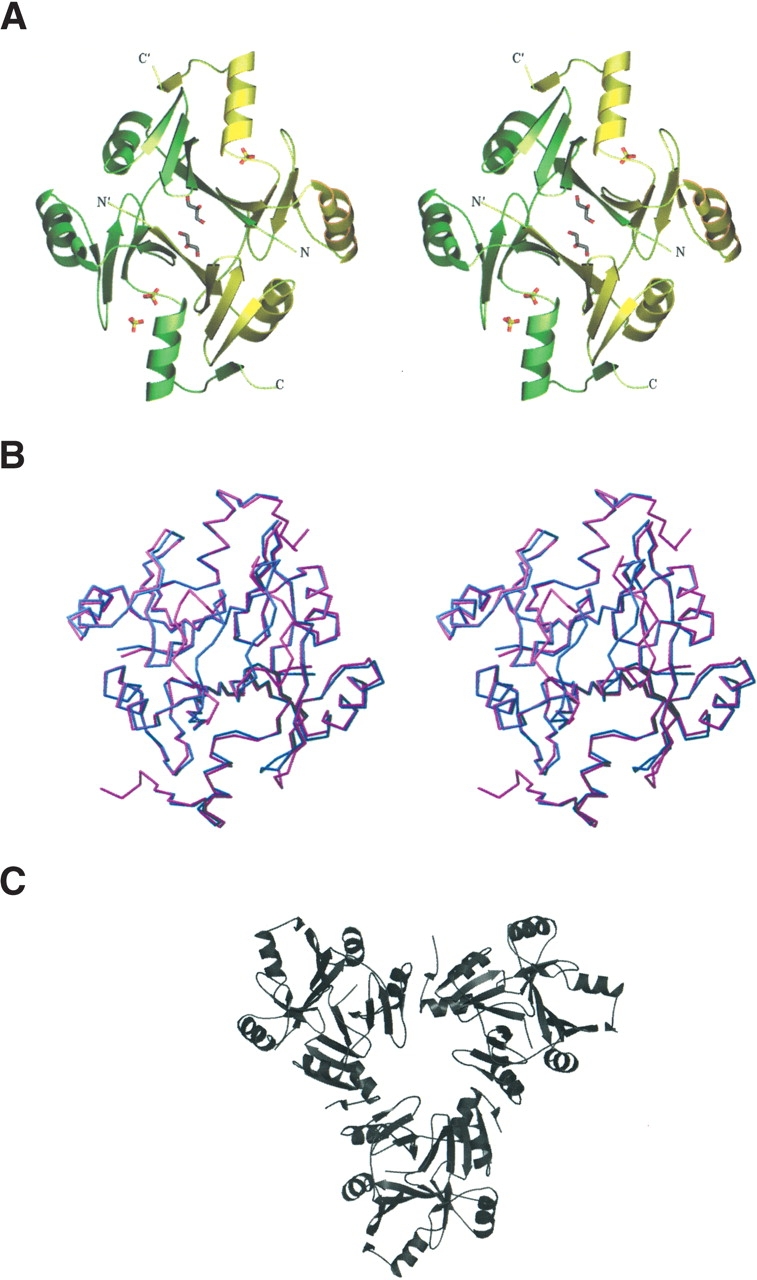
View of the FosA dimer. (A) A stereo view of FosA dimer along a twofold NCS symmetry axis with bound sulfate ions and glycerol molecules. Monomers A and B are shown in green and yellow, respectively. (B) A stereo view of superposition of FosA (magenta) and PA1129 (blue) dimers. (C) A trimer of dimers as observed in the asymmetric unit of the FosA structure. Pictures 2–4 were made by using SETOR (Evans 1993).
In the context of crystal packing, the FosA dimers pack with an almost perfect threefold rotation axis (Fig. 2C ▶) to form a hexameric (trimer of dimers) asymmetric unit. Intermolecular packing contacts between the hexamers in the unit cell are sparse, so that large solvent channels permeate the relatively loosely packed crystal. The Matthews coefficient is 3.9 Å3/Da, a value that is much higher than that expected for a compactly folded protein of this size (~2.2 Å3/Da; Matthews 1968).
Superposition of the FosA and PA1129 dimers gives a very good fit for all of the secondary structure elements (Fig. 2B ▶). The RMSD calculated over 178 equivalent CA atoms is 0.58 Å when the flexible linker and the K+ loop residues are omitted from the calculations. Significant differences between FosA and PA1129 are observed in the vicinity of several loops. The first major difference is in the linker between the tandem βαβββ-motifs, which contains three more amino acid residues than does the same region in the homologous PA1129. In PA1129, the loops from two monomers make close packing contacts in the context of the functional dimer. In FosA, the same loops are wide open and provide access to the dimer interface (Fig. 2A,B ▶). In the crystal structure, the cavity so formed is filled with ordered water and glycerol (present in the crystallization solution).
The K+ loops (residues 93–99) appear to be particularly flexible in Tn2921 FosA and display different conformations in all six monomers of the hexameric packing unit in the crystal structure. Only the K+ loop of molecule B is in essentially the same conformation as that observed in the PA1129 structure. All of the K+ loops have higher-than-average B-factors (1.8 times higher than the rest of the protein). The most significantly disordered K+ loop of molecule F might be described as an “inverted” conformation when compared with the same loop in molecule B (Fig. 3 ▶). Superposition of these two particular K+ loops shows the largest deviation in CA positions between single monomers in the structure: 7.7 Å for E98(B) and E98(F). As stated earlier, crystals of FosA protein have remarkably large solvent channels, and consequently any conformational differences induced by packing contacts are minimal. However, the conformations of the K+ loops in the B and F molecules may be a result of the packing of FosA hexamers in the crystal lattice. In contrast, the structures of the loops from the other four monomers in the hexamer are not influenced by crystal packing contacts. The multiple observed conformations of the K+ loops of molecules A, C, D, and E fill intermediate positions between the two extreme conformations (Fig. 3 ▶) of the B and F molecules. The relatively increased flexibility of the K+ loops may be due to the absence of potassium ions, but it is somewhat surprising that ammonium ions, which are present in high concentration in the crystallization solution, do not appear bound in the loops. Ammonium is known to activate the enzyme to essentially the same extent as K+ (Bernat et al. 1999), and yet no density that might be attributed to an ammonium ion (or water molecule) is apparent in the K+ loop regions.
Figure 3.

A stereo view of superposition of K+ loops from six independent molecules of metal-free (FosA, light gray) and metal-bound (PA1129, dark gray) enzymes. The K+ loops in the metal-free enzyme adopt a variety of conformations in the six monomers of the asymmetric unit. The side chain of glutamate 98 is included in the loops that represent the extremes of the spectrum of conformations.
Despite the fact that the crystallization experiments were performed in the presence of MnCl2 and fosfomycin, electron density that might be attributed to these species also does not appear in the structure. Nevertheless, the active-site geometry in FosA is very similar to that described for PA1129 (Fig. 4 ▶). The absence of Mn(II) in the metal site may be due to the relatively low pH (pH 6.5) of the buffer in the crystallization experiments, in which one or more of the histidine ligands may be protonated. Interestingly, electron density consistent with a sulfate ion is observed in the active site near the position where the phosphonyl group of fosfomycin is located in the PA1129 structure (Rife et al. 2002). Thus, even in the absence of the essential mono- and divalent cations, the vestige of an anion-binding site is apparent in the enzyme active site. There is also an additional sulfate-binding site in some of the molecules in the vicinity of the potassium-binding loop. It is interesting to note that the secondary sulfate-binding site is positioned in the place of side chain of E95 in PA1129 (Fig. 4 ▶; equivalent residue E98 in FosA).
Figure 4.

(A) The active site cavity of subunit A of FosA with bound sulfate ions. Superimposed active site residues, Mn(II), fosfomycin, and K+ loop from PA1129 FosA are shown in pink. (B) Close-up stereo view of the active site.
The structure of the Tn2921-encoded FosA has several implications for the function of the enzyme. The more extended loops that connect the βαβββ-motifs and more open structure near the dimer interface suggest that this enzyme may be more flexible than PA1129, a factor that would favor rapid release of product. Although the Mn(II) ion is crucial for substrate binding and catalysis, in the absence of divalent cation there remains at least a vestige of an anion-binding site to accommodate the phosphonyl group of the substrate. The side chain of R122 is likely to be directly involved in substrate binding.
It is surprising that even in the presence of high concentrations of ammonium ion, a good K+ mimetic (Bernat et al. 1999), none of the K+ loops show any indication of being occupied by NH4+. It may be the case that K+ loop conformation and occupancy requires that Mn(II) or fosfo-mycin be bound in the active site. The acquisition of Mn(II) by the protein has been shown to be a kinetically complex process that might suggest significant conformational heterogeneity in the apoenzyme (Bernat and Armstrong 2001). The crystal structure of the apoenzyme indicates that this heterogeneity does not involve the metal binding site directly. However, it is possible that the kinetic complexity relates to conformational heterogeneity in the K+ loops nearby.
Conclusions
The structure of the apoenzyme of Tn2921 FosA clearly indicates that the divalent cation-binding site is preformed by the paired βαβββ-motifs and is probably enforced by the three-dimensional domain-swapped dimeric structure. In contrast, the neighboring monovalent cation-binding loop is more flexible and remains unoccupied even in the presence of high concentrations of NH4+. These observations suggest that the K+ ion, which is rather loosely bound (Kd = 6.2 mM), may be recruited to the active site only after Mn(II) and fosfomycin are bound.
Materials and methods
Crystallization
FosA was expressed and purified as previously reported (Bernat et al. 1997). Crystals of the enzyme were obtained using the hanging drop vapor-diffusion method by mixing equal volumes of protein at 12 mg/mL, 0.6 mM manganese (II) chloride, 0.6 mM fosfomycin, and the reservoir solution (1.6 M ammonium sulfate, 100 mM sodium citrate at pH 6.5, 5% glycerol) at 22° C. The crystals grew in 5–7 d and belonged to tetragonal space group I422 with a = 208.597 Å, c = 136.358 Å.
Data collection, structure solution, and refinement
Diffraction data were collected at 113 K from a single frozen crystal on an ADSC Quantum 4 CCD area detector at Argonne National Laboratory (APS) BIOCARS beamline 14-D (λ = 0.9797 Å). The images were processed and scaled using DENZO and SCALEPACK (Otwinowski and Minor 1997). Data collection and data processing statistics are summarized in Table 1.
Table 1.
Data collection and refinement statistics
| Wavelength (Å) | 0.9797 |
| Resolution (Å) | 12–2.5 |
| Temperature (K) | 113 |
| Space group | I422 |
| Cell dimensions | |
| a (Å) | 208.597 |
| c (Å) | 136.358 |
| No. of molecules per asymmetric unit | 6 |
| No. of unique reflections | 51,262 |
| Rsym (%)a,b | 8.2 (44.2) |
| Completeness (%) | 98.9 (90.3) |
| Redundancies | 7.3 |
| I/σ (I) | 20.5 (2.7) |
| Refinement statistics | |
| Resolution range | 12–2.5 |
| No. of reflections used in refinement | 46,761 |
| σ cutoff used in refinement | 0 |
| R/Rfree (%)c | 18.32/23.04 |
| Number of refined atoms | |
| Protein | 6613 |
| Heterogen atoms | 92 |
| Water | 348 |
| Average B-factors (Å2) | |
| Protein | 30.5 |
| Sulphate anions | 41.9 |
| Glycerol | 33.3 |
| Water | 46.4 |
| R.m.s. deviations | |
| Bonds (Å) | 0.016 |
| Angles (°) | 1.672 |
a Values in parentheses are for the highest-resolution shell.
bRsym = ∑|Ii - 〈Ii〉|/∑Ii, where Ii is the intensity of the ith observation and 〈Ii〉is the mean intensity of the reflection.
cR = ∑||Fo | - ||Fc||/∑|Fo|, where Fo and Fc are the observed and calculated structure factors amplitudes. Rfree is calculated by using 3.0% of reflections omitted from the refinement.
We initially assumed that there were four dimers in the asymmetric unit, as such packing would give a Matthews coefficient of 2.9 Å/Da, and a solvent content of 57.2% (Matthews 1968), values consistent with well-ordered protein crystals of relatively small proteins. The structure was solved by molecular replacement procedure as implemented in AMORE (CCP4 1994). A dimer of PA1129 (PDB accession code 1LQK; Rife et al. 2002) was used as the search model. The cross-rotation functions, calculated either with a dimer or a monomer model, however, did not show any peaks with a reassuring signal-to-noise ratio, presumably a result of the high number of protein molecules in the asymmetric unit and moderate sequence identity between two proteins. In addition, no relationship among the continuum of peaks that would be consistent with four dimers in the asymmetric unit was apparent. Nevertheless, the first peak of the cross-rotation function calculated with a dimer model (Table 2) was assumed to be correct and the corresponding translation function solution was fixed to search for additional dimers in the asymmetric unit. Only two additional dimers could be located in the asymmetric unit to give a total of three. This solution corresponds to a solvent content of 67.9% and a Matthews coefficient of 3.9 Å3/Da. The correlation coefficient and R-factor for this solution were 31.0 and 54.4, respectively.
Table 2.
Cross-rotation function peaks
| Peak | CCa | Rfb |
| 1 | 9.5 | 56.5 |
| 2 | 9.2 | 56.7 |
| 3 | 9.1 | 56.6 |
| 4 | 9.0 | 56.7 |
| 5 | 8.9 | 56.8 |
| 6 | 8.8 | 56.7 |
| 7 | 8.7 | 56.7 |
| 8 | 8.6 | 56.8 |
| 9 | 8.6 | 56.8 |
| 10 | 8.6 | 56.8 |
a (CC) Correlation coefficient (mean CC and σ are 8.44 and 2.5, respectively).
b (Rf) R factor of cross-rotation function.
Refinement of the structure was performed with REFMAC (CCP4 1994). No sigma cutoff was applied to the data. A bulk solvent correction as well as NCS restraints between all six molecules were applied. However, the flexible parts of the protein molecules were released from the NCS restraints. Bound sulfate ions and glycerol molecules were identified according to high peaks in difference Fourier maps. The final model consists of residues 1–140, 1–139, 1–138, 1–137, 1–138, and 1–138 for protein molecules A, B, C, D, E, and F, respectively, 10 sulfate ions, 7 glycerol molecules, and 348 water molecules. The molecules display good stereochemistry with 91.9% of nonglycine residues falling in the most favored regions of a Ramachandran plot and none in disallowed regions.
Data deposition
The atomic coordinates have been deposited to the Protein Data Bank with the accession code 1NPB.
Acknowledgments
This research was supported by NIH grants R01 AI42756, P30 ES00267, T32 GM08320, and R01 GM55420, as well as by the Louisiana Governor’s Biotechnology Initiative. Use of the Advanced Photon Source was supported by the U.S. Department of Energy, Basic Energy Sciences, Office of Science, under Contract No. W-31–109-Eng-38. Use of the BioCARS Sector 14 was supported by the NIH, National Center for Research Resources, under grant no. RR07707.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Abbreviations
FosA, fosfomycin resistance protein from transposon Tn2921
PA1129, fosfomycin resistance protein from PA1129 gene from P. aeruginosa
GSH, glutathioneK+ loop, potassium binding loop
RMSD, root-mean-square deviation
NCS, noncrystallographic symmetry
VOC, vicinal chelate superfamily of enzymes
Article published ahead of print. Article and publication date are at http://www.proteinscience.org/cgi/doi/10.1110/ps.03585004.
References
- Armstrong, R.N. 2000. Mechanistic diversity in a metalloenzyme superfamily. Biochemistry 39 13625–13632. [DOI] [PubMed] [Google Scholar]
- Bernat, B.A. and Armstrong, R.N. 2001. Elementary steps in the acquisition of Mn2+ by the fosfomycin resistance protein (FosA). Biochemistry 40 12712–12718. [DOI] [PubMed] [Google Scholar]
- Bernat, B.A., Laughlin, L.T., and Armstrong, R.N. 1997. Fosfomycin resistance protein (FosA) is a manganese metalloglutathione transferase related to glyoxalase I and the extradiol dioxygenases. Biochemistry 36 3050–3055. [DOI] [PubMed] [Google Scholar]
- ———. 1998. Regiochemical and stereochemical course of the reaction catalyzed by the fosfomycin resistance protein, FosA. J. Org. Chem. 63 3778–3780. [Google Scholar]
- ———. 1999. Elucidation of a monovalent cation dependence and characterization of the divalent cation binding site of the fosfomycin resistance protein, FosA. Biochemistry 387462–7469. [DOI] [PubMed] [Google Scholar]
- Cao, M., Bernat, B.A., Wang, Z., Armstrong, R.N., and Helmann, J.D. 2001. FosB, a cysteine-dependent fosfomycin resistance protein under the control of αW, an extracytoplasmic function αfactor in Bacillus subtilis. J. Bacteriol. 183 2380–2383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CCP4 (Collaborative Computational Project No. 4). 1994. The CCP4 suite: Programs for protein crystallography. Acta Crystallogr. D 50 760–763. [DOI] [PubMed] [Google Scholar]
- Evans, S.V. 1993. SETOR: Hardware lighted three-dimensional solid model representations of macromolecules. J. Mol. Graph. 11 134–138. [DOI] [PubMed] [Google Scholar]
- Gouet, P., Courcelle, E., Stuart, D.I., and Metoz, F. 1999. ESPript: Multiple sequence alignments in PostScript. Bioinformatics 15 305–308. [DOI] [PubMed] [Google Scholar]
- Kahan, F.M., Kahan, J.S., Cassidy, P.J., and Kroop, H. 1974. The mechanism of action of fosfomycin (phosphonomycin). Ann. N.Y. Acad. Sci. 235 364–385. [DOI] [PubMed] [Google Scholar]
- Marquardt, J.L., Brown, E.D., Lane, W.S., Haley, T.M., Ichskawa, Y., Wong, C.-H., and Walsh, C.T. 1994. Kinetics, stoichiometry, and identification of the reactive thiolate in the inactivation of UDP-GlcNAc enolpyruvoyl transferase by the antibiotic fosfomycin. Biochemistry 33 10646–10651. [DOI] [PubMed] [Google Scholar]
- Matthews, B.W. 1968. Solvent content of protein crystals. J. Mol. Biol. 33 491–497. [DOI] [PubMed] [Google Scholar]
- Otwinowski, Z. and Minor, W. 1997. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276 307–326. [DOI] [PubMed] [Google Scholar]
- Rife, C.L., Pharris, R.E., Newcomer, M.E., and Armstrong, R.N. 2002. Crystal structure of a genomically encoded fosfomycin resistance protein (FosA) at 1.19 Å resolution by MAD phasing off L-III edge of Tl+. J. Am. Chem. Soc. 124 11001–11003. [DOI] [PubMed] [Google Scholar]
- Walsh, C. 2000. Molecular mechanisms that confer antibacterial resistance. Nature 406 775–781. [DOI] [PubMed] [Google Scholar]