

Abstract
The kinetics of disulfide-coupled folding and unfolding of four circularly permuted forms of bovine pancreatic trypsin inhibitor (BPTI) were studied and compared with previously published results for both wild-type BPTI and a cyclized form. Each of the permuted proteins was found to be less stable than either the wild-type or circular proteins, by 3–8 kcal/mole. These stability differences were used to estimate effective concentrations of the chain termini in the native proteins, which were 1 mM for the wild-type protein and 2.5 to 4000 M for the permuted forms. The circular permutations increased the rates of unfolding and caused a variety of effects on the kinetics of refolding. For two of the proteins, the rates of a direct disulfide-formation pathway were dramatically increased, making this process as fast or faster than the competing disulfide rearrangement mechanism that predominates in the folding of the wild-type protein. These two permutations break the covalent connectivity among the β-strands of the native protein, and removal of these constraints appears to facilitate direct formation and reduction of nearby disulfides that are buried in the folded structure. The effects on folding kinetics and mechanism do not appear to be correlated with relative contact order, a measure of overall topological complexity. These observations are consistent with the results of other recent experimental and computational studies suggesting that circular permutation may generally influence folding mechanisms by favoring or disfavoring specific interactions that promote alternative pathways, rather than through effects on the overall topology of the native protein.
Keywords: bovine pancreatic trypsin inhibitor, protein folding, circular permutation, effective concentration
The covalent connectivity of a protein’s polypeptide backbone is a fundamental feature of its structure, and defines the topological relationships among the numerous interactions that stabilize the folded conformation. In recent years, much attention has been focused on the possible role of the topology of native interactions in determining folding mechanisms and kinetics. The folding rates of single domain proteins, especially those that fold without the detectable accumulation of intermediates, have been found to be correlated with simple parameters that describe the relative prevalence of short- and long-range interactions in the native protein. These parameters include the relative contact order (RCO; Plaxco et al. 1998), long-range order (Gromiha and Selvaraj 2001) or fraction of local interactions (Mirny and Shakhnovich 2001). These correlations and other results suggest that the rate-determining step in folding involves the formation of a transition-state ensemble in which the polypeptide has an overall topology similar to that of the native protein (Itzhaki et al. 1995; Sosnick et al. 1996; Goldenberg 1999; Fersht 2000; Bulaj and Goldenberg 2001b; Paci et al. 2002; Makarov and Plaxco 2003). It is not yet clear, however, how formation of such a transition state might be specified by the amino acid sequence of a protein. Just as analyses of protein variants with amino acid replacements have elucidated the roles of individual residues in determining protein stability and folding mechanisms, variants in which the chain connectivity is altered may aid in deducing the possible relationship between native-state topology and the folding process.
One approach to modifying protein topology is circular permutation of the amino acid sequence. For many proteins, the N and C termini are in relatively close proximity in the native structure, making it feasible to connect the termini to one another, either directly with a peptide bond or via a short linker sequence, thereby generating a circular molecule. If the backbone is then cleaved at another site, the result is a circularly permuted sequence that is compatible, in principle, with the native structure of the natural protein. This procedure was first applied to bovine pancreatic trypsin inhibitor (BPTI) using direct chemical and enzymatic modification of the protein (Goldenberg and Creighton 1983). Subsequently, genetic engineering techniques have been used to generate circularly permuted forms of several other proteins, including phosphoribosyl anthranilate isomerase (Luger et al. 1989), dihydrofolate reductase (Buchwalder et al. 1992; Iwakura et al. 2000), bacteriophage T4 lysozyme (Zhang et al. 1993), ribonuclease T1 (Mullins et al. 1994), aspartate transcarbamoylase (Yang and Schachman 1993), the α-spectrin SH3 domain (Viguera et al. 1996), the Escherichia coli DsbA protein (Henneke et al. 1999), and ribosomal protein S6 (Lindberg et al. 2001, 2002; Miller et al. 2002). Pairs of naturally occurring proteins related by circular permutation have also been found by analysis of protein sequences and three-dimensional structures, although there is some uncertainty about the prevalence of natural permuted proteins and the genetic mechanisms by which they might arise during evolution (Lindqvist and Schneider 1997; Jung and Lee 2001; Uliel et al. 2001).
There is also relatively little known about how circular permutation affects protein folding mechanisms. Although permutation often destabilizes native proteins, most of the variants described so far are able to fold into structures similar to those of the natural proteins. Systematic studies of large collections of permuted forms of dihydrofolate reductase (Iwakura et al. 2000) and the DsbA protein (Henneke et al. 1999) indicate that these proteins are able to fold following disruption of their sequences at many sites, but have also identified regions at which connectivity of the chain is essential. When it does not prevent folding altogether, circular permutation appears to cause only modest changes in folding rate, although it may cause changes in the transition state for folding (Viguera et al. 1996; Lindberg et al. 2002; Miller et al. 2002).
Studies of disulfide-bonded proteins offer yet another view of the emergence of the native topology during folding. Many proteins in this class, including BPTI, ribonuclease A, and hen lysozyme, can be unfolded by reducing their disulfides and then refolded under conditions where folding and disulfide formation are thermodynamically and kinetically linked to one another. Provided that folding takes place under conditions where disulfide formation is freely reversible, measuring the rates and equilibria for disulfide formation provides information about the tendency of different segments of the chain to be brought together at various stages of folding (Creighton 1992b). Such studies have revealed that there is relatively little specificity in the initial disulfide formation steps, but subsequently, the cooperative influence of noncovalent interactions can lead to preferential accumulation of specific intermediates, often, although not always, containing native disulfides (Creighton 1974,Creighton 1975,Creighton 1992b; Weissman and Kim 1991; Goldenberg 1992; van den Berg et al. 1999; Wedemeyer et al. 2000).
In the present study, we have used disulfide formation to monitor the folding of four circularly permuted forms of BPTI, thus combining the two approaches described above. The folding and unfolding kinetics of these proteins were compared with those previously measured for both wild-type BPTI and a circular form, making it possible to distinguish the effects of linking together the natural N and C termini from those arising from “cleaving” the backbone at other sites, a distinction that has not been feasible in most other studies of permuted proteins. The results indicate that the four peptide bonds in the natural protein are much more stabilizing than is the artificial bond used to circularize BPTI. The kinetic effects of cleaving the circular protein differ significantly among the permuted proteins, although all show significantly enhanced rates of unfolding. For two of the proteins, circular permutation significantly increases the contribution of a direct disulfide formation mechanism, relative to the contribution of the rearrangement pathway that dominates the folding of the wild-type protein. The results of this and other studies suggest that circular permutation can alter the observed mechanism of folding by shifting the relative contributions of alternative pathways, but that these changes may generally reflect the effects of the modifications on specific interactions, rather than changes in global topology.
Results and Discussion
Design and initial characterization of circularly permuted proteins
The sites of circular permutation in the variants used for this study are illustrated in Figure 1 ▶. The N and C termini of wild-type BPTI are in close proximity to one another in the native structure, and the terminal residues can be linked together in a peptide bond by treating the native protein with a water-soluble carbodiimide (Goldenberg and Creighton 1983). More recently, a circular variant with a slightly altered sequence was constructed using the the method of native chemical ligation (Botos et al. 2001). High-resolution NMR spectroscopy and X-ray crystallography indicate that the cyclizing peptide bond is accommodated with minimal perturbation of the native structure (Chazin et al. 1985; Botos et al. 2001). The four sites at which new N and C termini were introduced were chosen to separate the major elements of regular secondary structure and are all located near the surface of the folded protein. The proteins are designated cp16, cp27, cp41, and cp46, with the numbers indicating the sequence number of the new N-terminal residue. In the cp16 variant, the cleavage site corresponds to the peptide bond that is normally poised for hydrolysis when wild-type BPTI binds to trypsin.
Figure 1.
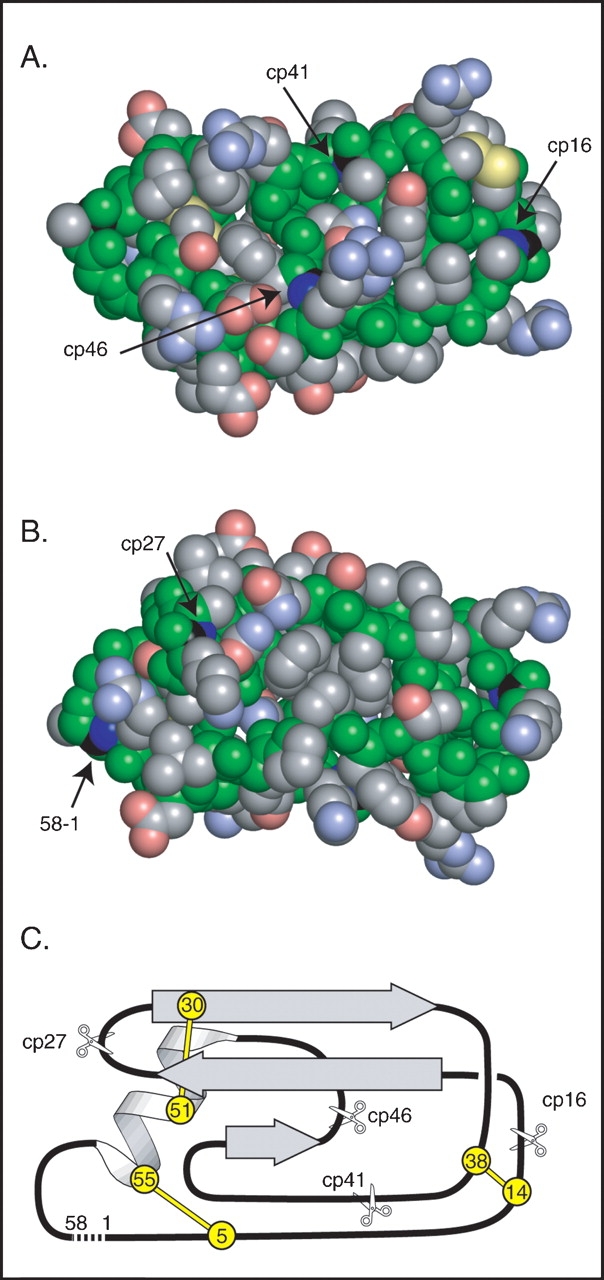
Design of circularly permuted BPTI variants. (A) Space-filling representation of a model of circular BPTI indicating the sites of the natural N and C termini and the sites of cleavage in the circularly permuted proteins. Backbone atoms are colored green, and those of side chains are colored gray, blue, red, or yellow to indicate C, N, O, or S atoms, respectively. The C and N atoms of the peptide bonds broken to generate the termini in the natural and circularly permuted protons are colored black and dark blue. The model of circular BPTI was constructed as described in Materials and Methods. (B) Space-filling model, as in A, but rotated 180°C about the horizontal axis to reveal the sites of the termini in the natural protein and cp27 variant. (C) Schematic representation of the topology and location of secondary structure elements in native BPTI. The locations and residue numbers of the six disulfide bonded Cys residues are indicated by circles, and the sites of cleavage in the circularly permuted proteins are indicated with scissors. A broken line connects the N and C termini of the natural sequence. The orientation in C is roughly equivalent to that in B.
Circularly permuted proteins were produced in E. coli as fusions to a polypeptide derived from the E. coli Trp operon. To facilitate cleavage of the fusion proteins, a Met residue was introduced between the E. coli and BPTI sequences, and the one Met residue in the natural BPTI sequence (at position 52) was replaced with Arg. Arg is commonly found in this position in natural BPTI homologs (Pritchard and Dufton 1999), and previous studies indicate that the M52R replacement causes little or no change in folding kinetics (Darby and Creighton 1993). The polypeptides accumulated in E. coli in an insoluble form. After being solubilized in 6 M GuHCl, the fusion proteins were cleaved with cyanogen bromide, purified in their reduced and unfolded state, and then folded in vitro in the presence of oxidized glutathione.
The protease inhibitor activities of the permuted proteins were measured by incubating the native proteins with bovine trypsin and then measuring the concentration of free trypsin in a spectrophotometric assay (Fig. 2 ▶). After 20 min of incubation, the wild-type protein and the the cp27, cp41, and cp46 variants all inhibited trypsin stoichiometrically at μM concentrations. The cp16 variant, however, produced only incomplete inhibition after 20 min, but inhibited stoichiometrically after an incubation of 120 min, consistent with previous results for natural BPTI after cleavage of the Lys15–Ala16 peptide bond (Goldenberg and Creighton 1983).
Figure 2.

Trypsin inhibitor activity of cpBPTI variants. The native BPTI variants at the indicated total concentrations were incubated with bovine trypsin, at a concentration of 3.4 μM, for either 20 or 120 min, as indicated. The residual trypsin activity was determined spectrophotometrically using a chromogenic substrate, benzoyl-l-arginine 4-nitroanilide, and monitoring the absorbance at 405 nm.
In Figure 3 ▶ are shown the amide and aromatic side-chain regions of one-dimensional 1H-NMR spectra of wild-type BPTI and the permuted variants. The spectra of the four mutant proteins were all very similar to that of the wild-type protein, and all displayed the chemical shift dispersion characteristic of a well-folded protein. A particularly striking diagnostic of the wild-type BPTI spectrum is the large downfield chemical shift of the amide proton of Tyr23, at approximately 10.5 ppm. This proton is positioned near the side chains of Phe22 and Phe45, and the downfield shift is reduced if either of these residues is replaced with with Leu, suggesting that it arises from ring-current effects induced by the aromatic side chains (Zhang and Goldenberg 1997a). This resonance was clearly detectable in all four of the circularly permuted proteins, indicating that the Phe22 and Phe45 side chains were properly positioned in the native proteins, including the cp46 protein, in which Phe45 was no longer linked to residue 46. The resonances from one of the two ɛ-protons of Tyr 23 was also readily identifiable in the spectra of the permuted proteins. This side chain forms part of the interface between the α-helix and β-sheet, and the presence of the resonance at approximately 6.4 ppm provides further evidence that the overall fold is preserved in the circularly permuted proteins.
Figure 3.
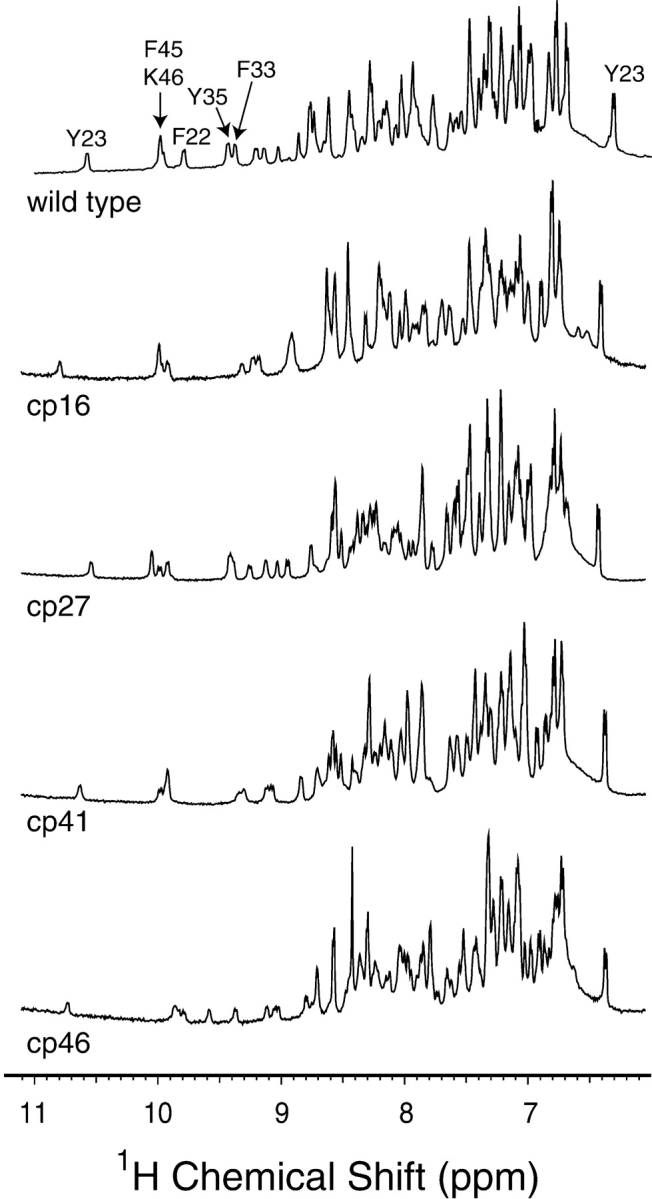
1H-NMR spectra of cpBPTI variants. Spectra were collected at a proton resonance frequency of 600 MHz, and at pH 4.7 and room temperature as described previously (Zhang and Goldenberg 1997a), and the spectrum for the wild-type protein is taken from that publication. The well-resolved amide proton resonances and that of the ɛ proton of Tyr23 were identified from the published assignments (Wagner et al. 1987a,b).
Kinetic and thermodynamic analysis of unfolding and refolding
The kinetics of disulfide-coupled unfolding and refolding of the circularly permuted proteins were analyzed using methods previously applied to the wild-type protein and variants with single amino acid replacements (Creighton and Goldenberg 1984; Goldenberg et al. 1989; Zhang and Goldenberg 1997b; Bulaj and Goldenberg 2001a). As in the previous studies, the reaction kinetics were studied at 25°C (pH 8.7) in the absence of any denaturants, with the folding or unfolding reactions favored by the relative concentrations of the oxidized and reduced forms of dithiothreitol (DTT SS and DTTSH SH, respectively).
Progress of the reactions was monitored by nondenaturing gel electrophoresis, as illustrated in Figure 4 ▶. Prior to electrophoresis, samples withdrawn from the reaction mixtures at various times were reacted with iodoacetate to irreversibly block any free thiols. Reaction with the negatively charged alkylating agent decreases the net positive charge of the proteins, so that molecules with fewer disulfides migrate more slowly during electrophoresis. The bands containing intermediates with one or two disulfide bonds were tentatively identified by comparing their electrophoretic mobilities with those of the well-characterized intermediates in the folding of the wild-type protein. These identities were confirmed by the kinetic behaviors of the various species, on the assumption that one-disulfide intermediates would appear prior to two-disulfide species during folding and in the reverse order during reductive unfolding.
Figure 4.
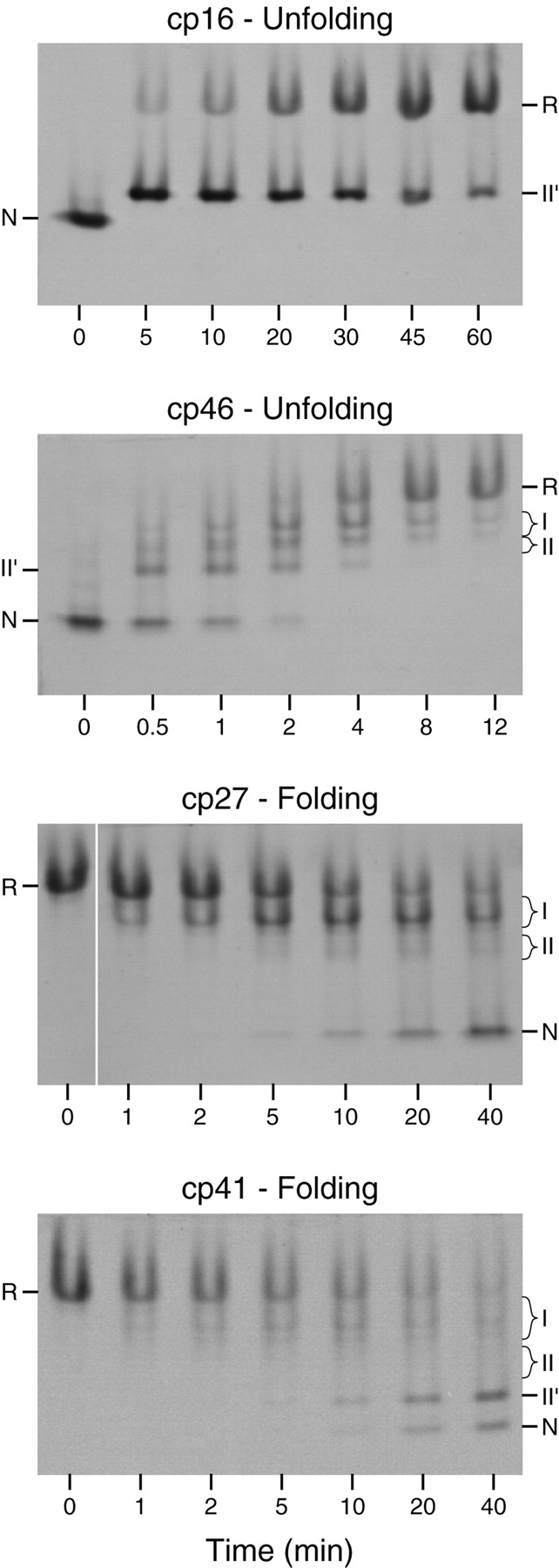
Electrophoretic analysis of unfolding and folding kinetics of cpBPTI variants. The unfolding experiments shown here were performed by incubating the native proteins in the presence of 20 mM (for cp16) or 0.2 mM (for cp46) reduced dithiothreitol (DTTSH SH) at pH 8.7, 25°C. Folding reactions were carried out under the same conditions except that the reactions were initiated by mixing the fully reduced proteins with 40 mM oxidized dithiothreitol (DTTSS). At the indicated times, aliquots of the reactions were reacted with iodoacetic acid and subjected to gel electrophoresis. The gels were stained with Coomassie blue. For the cp27 variant, all of the samples shown were electrophoresed on the same slab gel, but the image of the lane containing the initial reduced protein has been moved to facilitate comparison with the early time points.
As observed previously for wild-type BPTI and various mutants, one two-disulfide intermediate was readily detectable as the initial product of reducing the native protein during unfolding experiments. For the three cpBPTI variants other than cp46, the initial reduction intermediate had an electrophoretic mobility approximately 85% that of the native protein, as observed previously for the [30–51,5–55] intermediate in the folding of the wild-type protein. This species lacks the 14–38 disulfide and has a native-like conformation (Naderi et al. 1991; Beeser et al. 1998). For the cp46 variant, the initial reduction intermediate had a somewhat lower electrophoretic mobility, about 70% that of the native protein, suggesting that this species was at least partially unfolded. Because the other two disulfides are completely buried in the native structure, it is likely that 14–38 is the first disulfide to be reduced in the native circularly permuted proteins, as it is in the wild-type protein and all of the mutant proteins that have previously been studied. This is particularly true for the cp16 and cp41 variants, for which the rates of reducing a second disulfide were approximately 1000-fold slower than that for reducing the 14–38 disulfide of the wild-type protein (below). The [5–55,30–51] intermediate is often designated NSHSH to indicate its native-like conformation. Because, however, the disulfide bonds in the corresponding intermediates for the cpBPTI variants were not identified directly, and the cp46 intermediate did not appear to be native-like, the initial unfolding intermediates are identified here simply as II′.
Other two disulfide intermediates were detectable during folding or unfolding of the various proteins, but the concentrations of these species, relative to that of II′, varied at different times in the reactions or in experiments carried out at different concentrations of DTTSS or DTT SHSH. These observations indicate that there are at least two kinetically distinct populations of two-disulfide intermediates and that the following scheme represents the minimal mechanism necessary to account for the kinetic data: In this scheme, R and N represent the fully reduced and native proteins, respectively, and I and II are populations of intermediates with one or two disulfide bonds. The solid lines represent direct disulfide formation and reduction steps, with observed rates proportional to the concentration of DTTSS or DTTSHSH, and the dashed lines represent intramolecular rearrangements with rates independent of the thioldisulfide reagent concentrations. The same scheme has previously been used for the analysis of the wild-type protein and BPTI mutants with amino acid replacements.
For each of the variants, multiple folding reactions were carried out in the presence of different concentrations of DTTSS, as well as in the presence or absence of added DTTSH SH. Similarly, unfolding reactions were carried out with different concentrations of DTTSHSH, with and without added DTTSS. The resulting kinetic data were analyzed by comparing the observed time-dependent changes in concentrations of the native, reduced, and intermediate species with those predicted by numerical simulations based on Scheme 1 ▶. The rate constants in the scheme were manually adjusted until all of the data for each variant could be accounted for by a single set of parameters for that protein. Examples of the unfolding and refolding data are shown in Figures 5 ▶ and 6 ▶.
Scheme 1.

Figure 5.
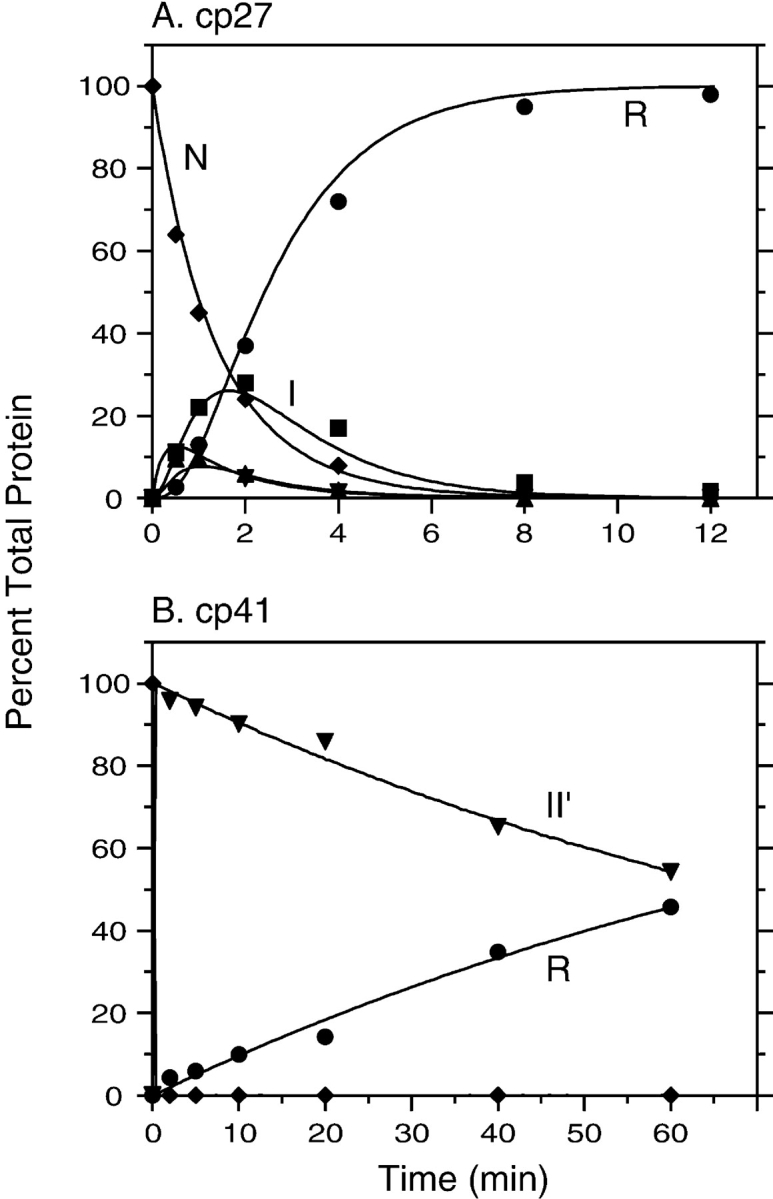
Kinetics of reductive unfolding of cpBPTI variants. (A) The native form of cp27 BPTI was incubated in the presence of 0.5 mM DTTSH SH at pH 8.7, 25°C. At the indicated times, aliquots of the reaction were withdrawn, reacted with iodoacetate, and subjected to nondenaturing gel electrophoresis, as illustrated in Fig. 4 ▶. (B) Unfolding kinetics of cp41 BPTI were measured as described above, except that the DTTSHSH concentration was 10 mM. In each panel, the filled symbols represent the relative intensities of the bands identified with the various species defined in Scheme 1 ▶: R (circles), I (squares), II (triangles), II′ (down triangles), and N (diamonds). The curves represent the values calculated by numerical integration of the kinetic scheme using the rate constants listed in Table 1.
Figure 6.
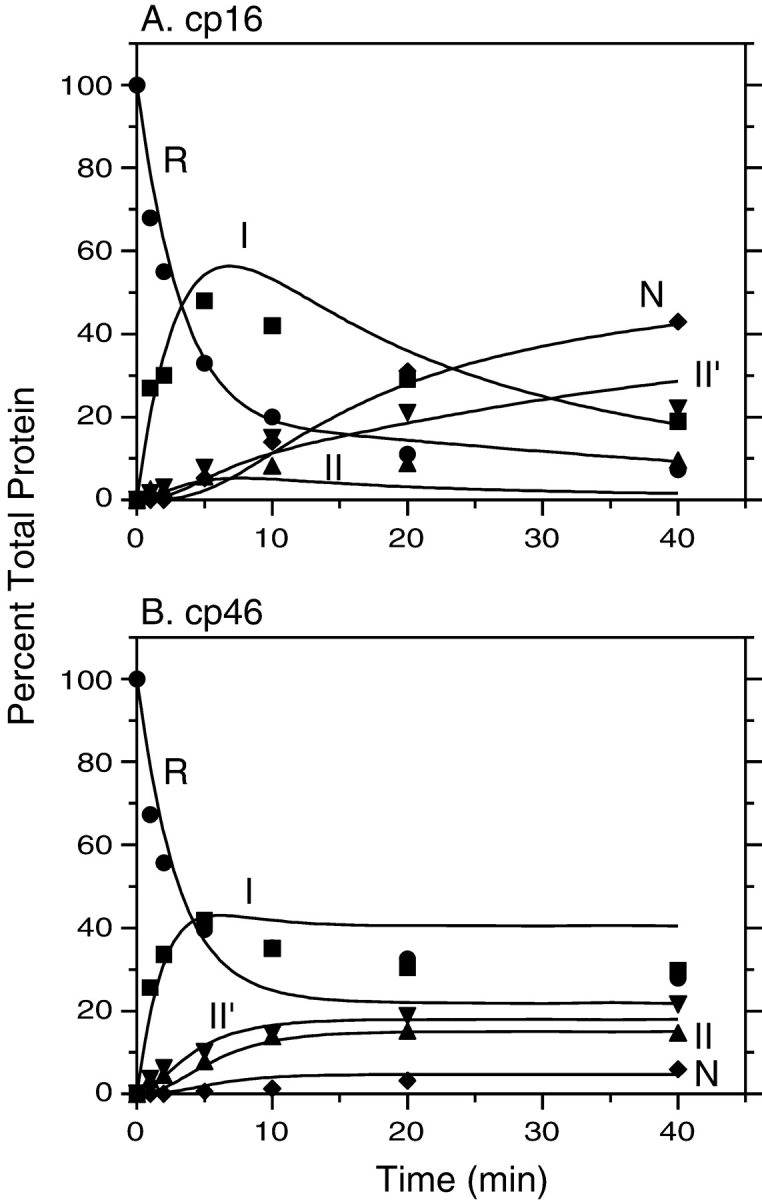
Kinetics of disulfide-coupled folding of cp16 (A) and cp46 (B) BPTI. The fully reduced proteins were incubated in the presence of 80 mM DTTSS at pH 8.7, 25°C, and progress of the reactions was monitored by gel electrophoresis as described in the legends of Figs. 4 ▶ and 5 ▶. The filled symbols represent the relative intensities of the bands identified with the various species defined in Scheme 1 ▶: R (circles), I (squares), II (triangles), II′ (down triangles), and N (diamonds). The curves represent the values calculated by numerical integration of the kinetic scheme using the rate constants listed in Table 1.
As indicated in Scheme 1 ▶, the model used to analyze the kinetic data includes two pathways by which II′ can be formed during folding or reduced during unfolding. In unfolding, this species can undergo either direct reduction to generate a one-disulfide intermediate or intramolecular rearrangements that generate other two-disulfide species, which are then reduced further. The contributions of the two pathways were estimated from the rates of disappearance of II′ during unfolding reactions in the presence of different DTTSH SH concentrations. For each of the cpBPTI variants, the kinetics of reduction could be accounted for only by a model that included both a DTTSHSH-concentration independent step, that is, intramolecular rearrangement, and a step with a rate proportional to the DTTSHSH concentration. These results also constrained the relative roles of direct disulfide formation and intramolecular rearrangement in the formation of II′, because the rate constants for the six steps making up the cycle composed of I, II, and II′ were required to satisfy the principle of microscopic reversibility.
The rate constants derived from this analysis are listed in Table 1, along with those previously published for the wild-type and circular proteins. For comparison, the table also includes rate constants previously determined for four mutants with amino acid replacements that destabilize the native protein to roughly the same extent as do the circular permutations. The rate constants are estimated to be reliable within a factor of 2, based on attempts to adjust their values while maintaining consistency with the experimental data.
Table 1.
Rate constants for individual steps in the folding and unfolding of BPTI variants
| R ↔ I | I ↔ II | I ↔ II′ | II ↔ II′ | II′ ↔ N | ||||||
| Variant | k1 (sec−1M−1) | k−1 (sec−1M−1) | k2 (sec−1M−1) | k−2 (sec−1M−1) | kdir (sec−1M−1 | k−dir (sec−1M−1) | krearr (sec−1) | k−rearr (sec−1) | k3 (sec−1M−1) | k−3 (sec−1M−1) |
| Wild typea | 0.028 | 17 | 0.05 | 240 | 1.4 × 10−5 | 1.1 × 10−4 | 5.0 × 10−3 | 8.0 × 10−6 | 5.7 | 16 |
| cBPTIb | 0.04 | 32 | 0.13 | 100 | nd | nd | 1.0 × 10−3 | 1.6 × 10−5 | 5 | 50 |
| cp16 | 0.05 | 30 | 0.015 | 100 | 2.6 × 10−4 | 1.7 × 10−2 | 1.0 × 10−2 | 1.0 × 10−4 | 0.06 | 50 |
| cp27 | 0.05 | 40 | 0.028 | 80 | 1.6 × 10−3 | 7.5 × 101 | 1.8 × 10−3 | 3.0 × 10−2 | 2 | 25 |
| cp41 | 0.05 | 50 | 0.05 | 100 | 7.0 × 10−5 | 7.0 × 10−3 | 2.0 × 10−3 | 1.0 × 10−4 | 0.1 | 125 |
| cp46 | 0.05 | 60 | 0.005 | 30 | 3.0 × 10−2 | 1.5 × 102 | 6.0 × 10−3 | 5.0 × 10−3 | 0.02 | 17 |
| F22Lc | 0.018 | 45 | 0.015 | 250 | 4.7 × 10−5 | 0.7 | 1.0 × 10−2 | 9.0 × 10−3 | 2.5 | 80 |
| Y35Ic | 0.02 | 25 | 0.013 | 250 | 6.4 × 10−5 | 4.0 × 10−2 | 1.0 × 10−2 | 3.5 × 10−4 | 0.13 | 900 |
| N43Ga | 0.05 | 25 | 0.07 | 140 | 4.2 × 10−4 | 1.2 | 7.0 × 10−3 | 1.0 × 10−2 | 4 | 140 |
| F45Lc | 0.022 | 35 | 0.01 | 150 | 1.8 × 10−4 | 8 | 1.0 × 10−2 | 3.0 × 10−2 | 1 | 60 |
a From Bulaj and Goldenberg (2001a).
b From Goldenberg and Creighton (1984). The rate constants for direct formation and reduction of II′ were not determined in this study of the circular
protein, but the value of kdir was estimated to be less than 2 × 10−3 sec−1M−1.
c From Zhang and Goldenberg (1997b).
Because the forward and reverse rate constants were all determined under the same solution conditions, they can be used to calculate the relative stabilities of the species making up the pathway for each protein. For the analysis below, it is convenient to compare the stabilities of the native circular protein and its folding intermediates with those of the corresponding species for a linear form, where “linear” in this context refers to the presence of N and C termini, irrespective of the disulfides that are present. These values are listed in Table 2, and were calculated according to:
Table 2.
Stability changes of native BPTI and folding intermediates, relative to circular BPTI
| Variant | I (kcal/mole) | II (kcal/mole) | II′ (kcal/mole) | N (kcal/mole) |
| Wild-type | −0.1 | 0.9 | −0.4 | −1.2 |
| cp16 | −0.2 | 1.1 | 0.8 | 3.4 |
| cp27 | 0.0 | 0.8 | 4.9 | 5.0 |
| cp41 | 0.1 | 0.7 | 1.4 | 4.2 |
| cp47 | 0.2 | 1.5 | 3.8 | 7.8 |
Calculated using equation 1 and the rate constants of Table 1. A positive value indicates that the indicated linear form is less stable than the corresponding form of circular BPTI. For I, the equilibrium constants were calculated as K = k1/k−1; for II, K = (k1k2)/(k−1k−2); for II′, K = (k1k2krearr)/(k−1k−2k−rearr); for N, K = (k1k2krearrk3)/(k−1k−2k−rearrk−3).
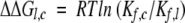 |
(1) |
where Kf,c and Kf,l are the equilibrium constants for forming the species of interest from the fully reduced protein, for the circular and linear proteins respectively; R is the gas constant, and T the temperature. Using this definition, a positive value of ΔΔGl,c indicates that the linear form is less stable than the circular form. The equilibrium constants were calculated as the product of the forward rate constants for the steps making up the path from R to the species, divided by the product of the reverse rate constants. As discussed in Materials and Methods, the uncertainties in stabilities determined by this procedure are estimated to be about 1 kcal/mole.
The thermodynamic and kinetic differences among the topologically modified proteins, and possible structural interpretations of these effects, are discussed in detail below.
Thermodynamic consequences of circular permutation: Effective concentrations of the chain termini in wild-type and circularly permuted BPTI
The previous study of circular BPTI revealed that the peptide bond introduced between the natural N and C termini had little effect on the stability of the native protein, as reflected in Table 2 by the small increase in stability of the wild-type protein relative to cBPTI. In contrast, each of the circularly permuted proteins was measurably less stable than either the wild-type protein or cBPTI, by as much as 9 kcal/mole. To interpret these changes in stability, we consider the effects of forming or hydrolyzing a peptide bond at different sites in both the native and unfolded proteins. The analysis is facilitated by a thermodynamic cycle composed of the unfolded and native states of the linear and circular forms of BPTI:
In this scheme, Ul and Uc are the unfolded forms of the linear and circular proteins, and Nl and Nc are the corresponding native forms. Kf,l and Kf,c are the equilibrium constants for folding, as defined above, and Ksyn,U and Ksyn,N are the equilibrium constants for synthesizing a peptide bond between the terminal residues in the unfolded or native forms of the linear protein. For the disulfide-coupled folding reaction considered here, U refers to the fully reduced and unfolded state. Thermodynamic linkage requires that the four equilibrium constants be related to one another according to:
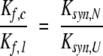 |
(2) |
Thus, folding is favored by cyclization (or other types of cross links) to the extent that forming the new bond is more favorable in the native protein than in the unfolded state (Schellman 1955; Johnson et al. 1978; Goldenberg 1985; Lin and Kim 1989; Matsumura et al. 1989).
For purposes of comparing different intramolecular reactions, it is convenient to express the equilibrium constants for the bond synthesis reactions as effective concentrations, Ceff , defined so that:
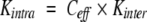 |
(3) |
where Kintra is the equilibrium constant for the intramolecular reaction and Kinter is the equilibrium constant for a chemically equivalent intermolecular reaction (Creighton 1983). Expressed in terms of effective concentrations, equation 3 can be rewritten as:
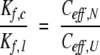 |
(4) |
For most systems involving the formation or hydrolysis of peptide bonds, the equilibrium constants cannot be measured directly, and only the ratio of equilibrium constants (or effective concentrations) can be derived from experimental data. In addition, most studies of circularly permuted proteins have relied on the wild-type protein as a reference, rather than a circular form, so that the effects of two reactions, forming a new peptide bond between the termini and opening the chain at another site, are not easily disentangled. For BPTI, however, two special circumstances allow a more complete analysis. First, because native BPTI interacts very specifically with trypsin and slowly undergoes hydrolysis, the equilibrium constant for synthesis of the Lys15–Ala16 peptide bond can be determined directly. At the pH used for the folding studies (8.7), the apparent equilibrium constant for peptide synthesis, Ksyn,N is estimated to be 0.28 (Estell et al. 1980; Siekmann et al. 1988). Under similar conditions, the apparent intermolecular equilibrium constant for peptide synthesis has been estimated to be 0.11 M−1 (Homandberg et al. 1978; Deschrevel et al. 2003). From equation 4, the effective concentration of the carboxyl group of Lys15 and the amino group of Ala16 in the cleaved native protein is 2.5 M.
The second special feature of the BPTI system is that the folding thermodynamics of the circular form have been measured (Goldenberg and Creighton 1984). With the relative stabilities of the various forms defined as in equation 1 and Table 2, the ratio of effective concentrations in the native and unfolded forms can be expressed as:
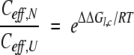 |
(5) |
From this relationship, Ceff,U can be calculated for the termini in unfolded cp16, provided that the value of Ceff,N for Lys15 and Ala16 in cp16 is assumed to be the same as in the selectively cleaved wild-type protein. With this assumption, Ceff,U for the termini of cp16 is estimated to be 7.5 mM. If Ceff,U is further assumed to have the same value in the other linear proteins, then the stability data for these proteins can be used to calculate Ceff,N for the termini in the wild-type protein and the other circularly permuted forms (Table 3).
Table 3.
Calculated effective concentrations of chain termini
| Variant | Ceff,II′ | Ceff,N |
| Wild type | 3.7 × 10−3 | 1.0 × 10−3 |
| cp16 | 3.0 × 10−2 | 2.5 |
| cp27 | 2.9 × 101 | 3.6 × 101 |
| cp41 | 7.6 × 10−2 | 9.5 |
| cp46 | 4.6 | 3.9 × 103 |
Ceff,N for the cp16 variant was calculated from equation 3, using the published equilibrium constants for enzymatic hydrolysis of the Lys15–Ala16 peptide bond of wild-type BPTI (Estell et al. 1980; Siekmann et al. 1988) and of a dipeptide (Homandberg et al. 1978; Deschereverl et al. 2003). The other values in the table were calculated from equation 5 and the destabilizations listed in Table 2, assuming Ceff,U = 7.5 mM.
In the native proteins, the lowest effective concentration, by far, is that for the termini of the natural protein, 1 × 10−1 M. The termini of wild-type BPTI are quite flexible, as indicated by both X-ray crystallographic temperature factors (Wlodawer et al. 1987) and NMR relaxation experiments (Beeser et al. 1997), and a recent theoretical analysis by Zhou suggests that the failure of the crosslink to stabilize BPTI may be largely due to the reduction of conformational entropy in the native protein (Zhou 2003). It is also possible that the new bond introduces steric strain in the folded protein. The higher values of Ceff,N for each of the circularly permuted proteins indicates that the peptide bonds in the natural protein are much more readily accommodated in the native structure.
Of the permuted proteins examined, the two with cleavage sites in the trypsin binding region, cp16 and cp41, displayed lower values of Ceff,N than the two cleaved at the opposite end of the folded protein. NMR relaxation experiments have shown that residues in this region undergo motions on the microsecond to millisecond time scale in the native wild-type protein (Szyperski et al. 1993; Millet et al. 2000), and some amino acid replacements in this region greatly extend the segments of the chain participating in these motions (Beeser et al. 1997, 1998; Hanson et al. 2003). It thus seems likely that the termini generated by cleavage of either the Lys15–Ala16 or Ala40–Lys41 peptide bonds could be quite flexible, accounting for their relatively low effective concentrations. Conversely, the very high value of Ceff,N for cp46 BPTI, 3.9 × 103 M, suggests that the termini in this protein are held in close proximity to one another by the cooperative effects of other interactions in this region of the protein. Indeed, Phe45 and its neighbors participate in a large number of long-range interactions in the native wild-type protein (see Fig. 1c ▶ of Oas and Kim 1988).
Buczek et al. (2002) have measured the effects of cleaving individual peptide bonds on the thermal stability of BPTI (with the disulfides intact), and have used these data to estimate the equilibrium constants for breaking the bonds in the native protein, employing arguments similar to those outlined above. These authors utilized a series of nine BPTI variants in which Met was used to replace individual residues throughout the sequence, allowing site-specific cleavage with cyanogen bromide. When expressed as effective concentrations, the stabilities of the cyanogen bromide-generated termini range from 1.1 × 103 to 3.5 × 107 M, considerably higher than most of the values reported here. At present, the reasons for this discrepancy are not clear, although it may reflect differences in solution conditions (pH 4.3 versus pH 8.7), covalent chemistry (generation of a homoserine residue during CNBr cleavage), or the nature of the unfolding reactions studied. Both studies, however, demonstrate the high energetic cost of breaking the backbone connectivity and the large influence of local structural factors.
Table 3 also includes effective concentrations for the termini in the II′ forms of the linear proteins. For the wild-type protein there is only a fourfold difference in the effective concentrations in the native and II′ forms, consistent with other results indicating that the effects of reducing the 14–38 disulfide are restricted to the immediate vicinity of this bond (Naderi et al. 1991; Beeser et al. 1998), which is located at the opposite end of the protein from the natural termini. The termini of the cp27 variant are also distant from the 14–38 disulfide, and have very similar effective concentrations in the native and II′ forms. On the other hand, the termini of the cp16 and cp41 proteins are relatively close to Cys14 and Cys38, and their effective concentrations are approximately 100-fold lower in the II′ forms than in the native proteins, suggesting that the termini of these proteins become less constrained when the nearby disulfide is reduced.
The effective concentration of the termini in cp46 BPTI also decreased upon reduction of a disulfide in the native protein, by nearly 1000-fold. This variant was also notable for the relatively low electrophoretic mobility of the II′ form (Fig. 4 ▶) and the low rate of forming the third disulfide (Table 1). Because the three disulfides in this variant were reduced much more rapidly than in the wild-type protein, it is difficult to rule out the possibility that the 30–51 or 5–55 disulfide may have been reduced at a rate comparable to that for the 14–38 disulfide. Whatever disulfides it contains, however, it appears that the II′ form of cp46 BPTI is significantly less structured than the native protein.
Effects of circular permutation on the kinetics of folding and disulfide formation
Under the conditions used for the kinetic experiments described here, with DTTSS employed as the oxidant, the observed rates of disulfide formation are expected to be proportional to the rate of the intramolecular step in which one protein thiol reacts with the mixed disulfide transiently formed between the disulfide reagent and another Cys sulfur atom (Creighton and Goldenberg 1984; Creighton 1992a). As a consequence, the observed rates can be a sensitive probe of the conformational properties of the polypeptide chain at different stages of folding.
Neither linking the normal termini of BPTI in a peptide bond nor the subsequent cleavage of the chain at other sites significantly altered the overall rate of the initial step in the folding pathway, formation of the population of one-disulfide intermediates from the fully reduced protein. Previous studies of the wild-type protein have shown that the observed rate reflects the sum of rates for forming the 15 possible one-disulfide species, although the rate of forming one, [14–38], represents approximately 20% of the rate at pH 8.7 (Dadlez and Kim 1996; Bulaj and Goldenberg 1999). Although circular permutation may have altered some of the rate constants for forming individual species, it appears that any large effects compensated one another. This observation is consistent with previous results indicating that any structure present in the unfolded protein plays only a small role in determining the overall rate of forming the one-disulfide intermediates (Creighton 1977; Bulaj and Goldenberg 1999; Zdanowski and Dadlez 1999).
Changes in the native topology of BPTI had somewhat larger effects on the rate of forming II, the population of two-disulfide intermediates other than II′. The previous study of cBPTI showed that linkage of the termini increased this rate by about threefold. This effect appeared to be eliminated in the circularly permuted proteins, for which this rate ranged from 0.005 (for cp46 BPTI) to 0.05 sec−1 M−1, the value observed for the wild-type protein. The very low value observed for the cp46 variant suggests that separation of residues 45 and 46 significantly destabilizes the productive intermediates in the one-disulfide population, as has been seen for some destabilizing amino acid replacements, including Y23L and F45L (Zhang and Goldenberg 1997b).
Two of the variants, cp27 and cp46, displayed greatly enhanced rates for directly forming II′ from the one disulfide intermediates (Kdir). For the wild-type protein, this reaction is approximately 3000-fold slower than the rate of forming II (primarily [30–51,14–38]), and this preference results in the great majority of molecules forming II′ via intramolecular rearrangements. For the cp46 variant, the rate of direct formation of II′ was actually greater than that for forming II, and the preference for the direct pathway was only 20-fold for cp27 BPTI. In contrast to the large effects on Kdir, the rate constants for forming II′ from II via intra-molecular rearrangement, krearr′, were nearly the same in the wild-type and modified proteins. These results suggest that the preference for the rearrangement mechanism observed for the wild-type protein may reflect, in part, topological constraints on the emerging structure, as discussed further below.
The final disulfide-formation step in the pathway was decreased significantly by three of the circular permutations, cp16, cp41, and cp46. The low rates observed for these variants suggests that in the absence of the final disulfide these proteins are significantly less ordered than the wild-type protein, at least in the region of the missing disulfide.
Topological effects on the major transition states for folding and unfolding
During the folding and unfolding of wild-type BPTI, the slowest steps are associated with the formation and reduction of II′ by either the direct or indirect pathways. Our previous studies of BPTI variants have revealed a correlation between the stability of II′ and the rate constants for either the direct reduction of this species or its intramolecular rearrangement (Zhang and Goldenberg 1997b; Bulaj and Goldenberg 2001a). These correlations are illustrated in Figure 7 ▶, where the rate constants, expressed as RTlnk, are plotted as a function of the destabilization of II′ relative to the fully reduced protein. The figure includes data both for variants with amino acid replacements (open circles) and for the topologically modified proteins (filled circles) described here. Lines fit to the data for the replacement mutants have correlation coefficients and slopes close to unity (Fig. 7 ▶ legend). As we have argued previously, these results are most readily accounted for by a model in which II′ must undergo extensive unfolding for its disulfides to be either reduced or rearranged. This interpretation is consistent with the native-like structure of the II′ form of the wild-type protein, in which the two disulfides are buried. Among the replacement mutants, the most notable deviations from this pattern are those with replacements of Asn43, for which both rates are 10–100-fold greater than predicted by the correlation observed with the other mutants (Bulaj and Goldenberg 2001a). The side chain of Asn43 participates in a network of hydrogen bonds within the β-sheet of the native protein, and removal of these hydrogen bonds may facilitate alternative folding and unfolding mechanisms that involve a less extensively unfolded transition state.
Figure 7.
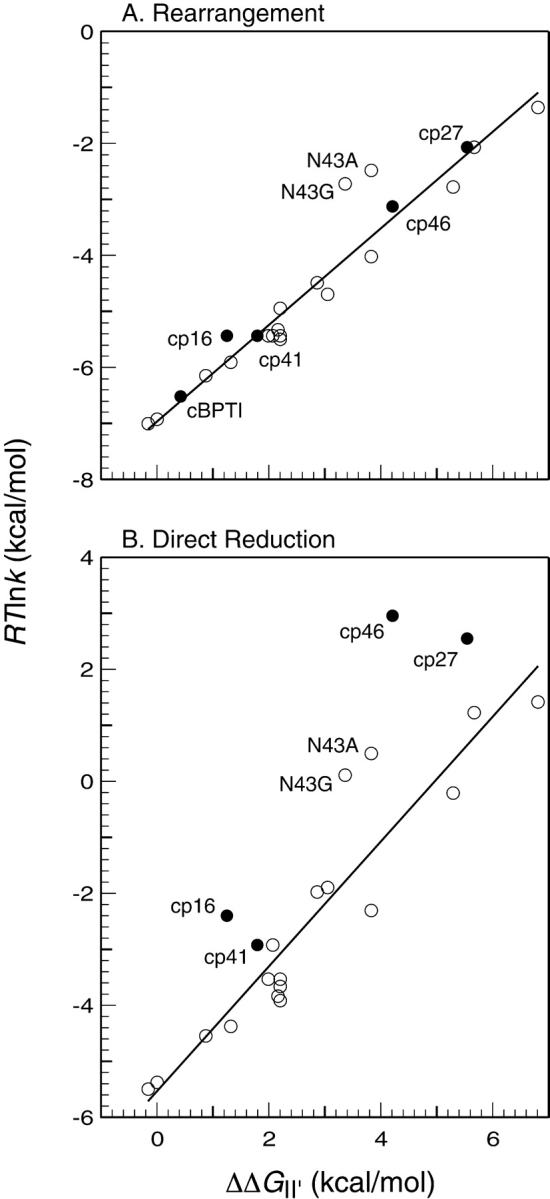
Correlations between the rates of (A) intramolecular rearrangement or (B) direct reduction of II′ and the destabilization of this species with respect to the fully reduced and unfolded protein. The rate constants are expressed as RTln(k), which is related to the free energy difference between II ′ and the corresponding transition state by a constant of addition. The filled symbols represent the rate constants for the circular and circularly permuted proteins, and the open symbols represent the previously Zhang and Goldenberg 1997b; Bulaj and Goldenberg 2001a). The lines shown were fit to the data for the replacement mutants by the method of least squares. For k−rearr′, R2 = 0.95 and the slope is 0.86; for k−dir′, R2 = 0.93, and the slope is 1.1.
As shown in Figure 7A ▶, the rate constants for rearrangement observed for the four circularly permuted forms of BPTI, as well as the circular protein, all follow the correlation observed for the mutants with amino acid replacements. This observation suggests that the rearrangement process for these proteins also involves extensive unfolding of II′. This conclusion is somewhat at odds with the evidence indicating that the II′ form of cp46 BPTI is already at least partially unfolded, which might have been expected to result in a larger rearrangement rate constant. It is possible, however, that this species is still structured in the vicinity of one or both of the remaining disulfides, contributing to a significant barrier to rearrangement.
In contrast to the rates for rearrangement, those for direct reduction of the cp16, cp27, and cp46 variants were markedly higher than predicted by the correlation with the stability of II′ (Fig. 7B ▶). The case of the cp46 variant is particularly striking, with the observed rate approximately 800-fold faster than expected. For this variant and cp27 BPTI, the rates of reduction are at least as large as those for reduction of a fully exposed disulfide. It thus appears that any structure present in the II′ forms of these proteins does not impede the intermolecular reaction of at least one disulfide with the thiol reagent.
From the principle of microscopic reversibility, the transition states for reduction and rearrangement of II′ must be equivalent to those for the formation of this species. We have suggested previously that the barrier to forming II′ directly may be due to the presence of structure in its immediate precursor, most likely the [30–51] intermediate that is incompatible with the transition state for the thiol–disulfide exchange reaction (Mendoza et al. 1994; Zhang and Goldenberg 1997b). For the cp27 and cp46 variants, the rate of directly forming II′ was dramatically greater than for the wild-type protein or any replacement mutant we have examined. Like the Asn43 replacements, these permutations may allow an alternative path for directly forming II′ with less extensive disruption or distortion of structure present in its precursor. Alternatively, or in addition, the modifications may destabilize this structure.
Together with the results obtained with replacement mutants, the behavior of the circularly permuted proteins suggests that the striking preference for the rearrangement pathway in the folding of wild-type BPTI is due to constraints imposed by at least three factors: native-like structure in the major intermediates, the connectivity of the polypeptide chain, and the structure of the chemical transition state for thiol–disulfide exchange. These same factors lead to a protein that is unusually stable, both thermodynamically and kinetically. Although some destabilizing substitutions, especially replacements of Asn43, can increase the rate of the direct folding pathway, this mechanism predominates for all of the replacement mutants we have examined. Only when the connectivity of the polypeptide chain within the β-sheet was broken, did the rate of directly forming II′ approach that for forming other two-disulfide intermediates.
Comparison with circularly permuted proteins without disulfide bonds
Comparisons of the folding kinetics of small proteins with different three-dimensional structures have recently revealed striking correlations between folding rates and parameters such as the relative contact order, which represents the average sequence distance between pairs of interacting atoms in the native structure, normalized by the total number of residues (Plaxco et al. 1998; Ivankov et al. 2003). Because circular permutation has the potential to change a short-range interaction into a long-range one, it might be expected that this type of modification would generally cause substantial changes in RCO and folding rate. Miller et al. (2002) have found, however, that the RCO for many proteins is relatively insensitive to circular permutation and, further, that the folding rate for one protein for which RCO can be substantially changed, ribosomal protein S6, is not greatly affected by at least three permutations. On the other hand, permutation of this protein has been shown to cause a substantial change in folding mechanism, as revealed by mutational analysis of the transition state (Lindberg et al. 2002). Circular permutation of the α-spectrin SH3 domain also appears to alter the transition state for folding (Viguera et al. 1996), as does circularization of src SH3 domain by introducing a disulfide between the N- and C-terminal residues (Grantcharova and Baker 2001).
To explore the possible relationships between backbone topology and folding mechanism in the context of the disulfide-coupled folding of BPTI, RCO was calculated for each of the 58 possible permutations, assuming that each retained the native structure of the wild-type protein. The calculated values are plotted in Figure 8A ▶, with the filled circles representing the sequences of the wild-type protein and the four variants examined here. The wild-type protein has a relatively large RCO, 0.18 when compared to the range of 0.05 to 0.22 calculated for the natural proteins recently analyzed by Ivankov et al. (2003). The large RCO reflects the prevalence of β-sheet structure in native BPTI, as well as the prominent role of interactions between residues in the N- and C-terminal segments. Of the circular permutations examined in this study, one (cp46) left the RCO unchanged, two (cp16 and cp41) decreased RCO slightly, to 0.17, and one (cp27) increased the value to 0.25. Remarkably, the two modifications that most promoted the direct disulfide formation pathway, cp46 and cp27, were associated with quite different values of RCO, 0.18 and 0.25. In calculating the values of RCO plotted in Figure 8A ▶, the sequence separation between interacting atoms were based on the linear sequence, without accounting for the tendency of disulfides present in the folding intermediates or the native protein to shorten the effective separation. Relative contact order for the wild-type and circularly permuted proteins were also calculated with the sequence distance determined by the shortest path through one, two, or three of the native disulfides. As expected, when these pathways were accounted for, generally lower RCO values were obtained, but there was still no obvious correlation with the experimentally observed effects on the folding mechanism. Analysis of values of “long-range order,” as defined by Gromiha and Selvarage (2001) led to qualitatively similar results. It thus appears the effects on folding kinetics and mechanism are uncorrelated with backbone topology as measured by these parameters.
Figure 8.

Relative contact order (RCO) calculated for wild-type BPTI and for each of the 57 possible circularly permuted variants. In A, the curve represents the RCO calculated for all types of interactions, as described by Plaxco et al. (1998), with the filled circles identifying the wild-type sequence and the four variants examined in this study. In B, the values of RCO were calculated from only the disulfide bonds.
As a specific measure of the topological constraints imposed by the native disulfide bonds on the wild-type and circularly permuted proteins, we also calculated RCO considering only the disulfides. These values are plotted in Figure 8B ▶, and show a pattern quite distinct from that observed for RCO calculated using all of the interactions in the native protein. Although the natural sequence gives rise to a relatively low RCO, the same sequence is associated with the highest possible value of RCO calculated for the disulfides, reflecting the relatively large spacings between the disulfide-bonded Cys residues in the natural protein, especially for the 5–55 disulfide. All of the permutations lead to closer average spacings among the disulfide-bonded Cys residues, and very similar values were calculated for the four modified proteins examined here. Thus, the disulfide topology of the native protein also appears to be a poor predictor of the folding mechanism.
From the limited data available at present for proteins with and without disulfides, it appears that the effects of circular permutation on folding mechanism reflect the structural features of specific proteins, rather than general topological parameters such as RCO. In the case of ribosomal protein S6, the effects can be accounted for by a model in which joining the natural termini gives rise to a particularly strong set of local interactions that direct formation of a transition state containing a concentration of interactions in this region, compared to the transition state for the natural protein, which involves weaker interactions throughout a more globally folded structure. For the disulfide-coupled folding of BPTI, the most dramatic changes in folding kinetics arose from circular permutations that separate the strands of the β-sheet, cp27 and cp46. As well as greatly destabilizing the native protein, opening the chain at these sites was found to facilitate the direct disulfide–formation pathway, presumably by removing steric constraints that normally inhibit direct formation and reduction of the II′ intermediate. More generally, changes in polypeptide connectivity may commonly lead to redistributions among parallel folding pathways, with the effects on observed folding rates determined by the relative rates of the affected processes. This view is supported by recent theoretical analyses of circularly permuted proteins (Clementi et al. 2001; Li and Shakhnovich 2001; Weikl and Dill 2003), and further studies with permuted proteins may be an especially valuable approach to dissecting the roles of alternative folding pathways that contribute to the folding of natural proteins, but are otherwise difficult to distinguish experimentally.
Materials and methods
Preparation of circularly permuted BPTI variants
Genes encoding the circularly permuted proteins were constructed from the natural BPTI gene using the polymerase chain reaction method described by Mullins et al. (1994). Each of these sequences was introduced into an expression plasmid, pAED4, derived from the pET plasmids of Studier and colleagues (Studier et al. 1990; Doering 1992; Staley and Kim 1994). In these plasmids, the DNA sequences encoding the N-termini of the permuted sequences were fused to the TrpLE Δ143 fragment, a fusion of portions of the E. coli TrpL and TrpE genes (Miozzari and Yanofsky 1978), a construct that has been found to help stabilize some proteins as inclusion bodies in the E. coli cytoplasm (Kleid et al. 1981). To allow chemical cleavage of the fusion proteins by cyanogen bromide (CNBr), the gene was modified to introduce a Met codon immediately preceding the BPTI-encoding sequence, and the codon for Met52 of the natural sequence was modified to encode Arg. All of the constructs were confirmed by DNA sequencing.
Cultures of E. coli BL21 transformed with the plasmids were grown to mid-log phase at 37°C and expression of the genes was induced by the addition of 0.4 mM isopropyl β-Dthiogalactoside. The bacteria were harvested by centrifugation 2–3 h after induction and the cell paste was stored frozen at −70°C. The frozen bacteria from a 500-mL culture were resuspended in 40 mL of lysis buffer containing 50 mM Tris-HCl (pH 8.0), 0.1 M NaCl, 10% (w/v) sucrose, 1 mM EDTA, and 0.5 mM phenylmethane-sulfonyl fluoride. After the cell were resuspended, hen lysozyme was added to a concentration of 0.6 mg/mL, and the suspension was incubated on ice for 20 min and then subjected to ultrasonic disruption. The resulting extract was centrifuged for 20 min at 13,000 RPM in a Beckman JA-14 rotor. The insoluble fraction was washed twice in approximately 20 mL of 1% (w/v) Triton X-100, 1 mM EDTA (pH 8.0), followed by centrifugation as above, and then dissolved in 20 mL 6 M GuHCl, 25 mM dithiothreitol, 2 mM EDTA, 0.1 M Tris-HCl (pH 8.0). After incubating for 2–3 h at room temperature, any insoluble material remaining was removed by centrifugation, and the solution was dialyzed against 0.1% (w/v) acetic acid. To protect the protein thiols during the subsequent cleavage reaction, the proteins were reacted with oxidized glutathione (GSSG) to form mixed disulfides. This reaction was carried out at room temperature for 2 h in the presence of 0.5 mM GSSG, 6 M GuHCl, 0.1 M Tris-HCl (pH 8.0), and 2 mM EDTA. Insoluble material was removed by centrifugation, and the sample was again dialyzed against 0.1% (w/v) acetic acid, followed by lyophilization.
For cleavage with CNBr, the lyophilized protein was dissolved in 3 mL of 6 M GuHCl, 0.1 mM HCl, and CNBr was added to a final concentration of 3.3 mg/mL, corresponding to a molar excess of approximately 35-fold. The solution was incubated overnight at room temperature and then dialyzed against 0.1 M acetic acid. The BPTI polypeptide was separated from the TrpLE fragment and uncleaved molecules by gel permeation chromatography using a Sephadex G-50 column equilibrated and eluted with 0.1 M acetic acid. The fractions containing the BPTI were identified by reversed-phase HPLC, pooled and dialyzed against 0.1 M acetic acid, and lyophilized. The glutathione mixed disulfides were then reduced by dissolving the lyophilized protein in 1 mL of a solution containing 50 mM dithiothreitol, 6 M GuHCl, 0.1 M Tris-HCl (pH 8.7), and 1 mM EDTA. After incubating at room temperature for approximately 1 h, the solution was acidified by the addition of 0.1 mL concentrated formic acid. The fully reduced protein was purified by reversed-phase HPLC on a Vydac C18 column eluted with a gradient of acetonitrile in 0.1% trifluoroacetic acid. The identities of the proteins were confirmed by electrospray ionization mass spectrometry and by Edman sequencing of the N-terminal four residues. From a 500-mL bacterial culture, the yield of purified reduced BPTI was typically 1–2 mg.
Samples to be used for in vitro folding experiments were lyophilized in the fully reduced state and stored frozen at −70°C. To prepare samples of native protein, lyophilized and reduced samples were dissolved in 10 mM HCl to a concentration of 1 μM and then diluted fivefold into a refolding solution yielding final concentrations of 0.1 M Tris-HCl (pH 8.7), 0.2 M KCl, and 1 mM EDTA. For the cp16 and cp41 variants, which folded very efficiently, the solutions also contained 0.1 mM GSSG to promote disulfide formation. The cp27 and cp46 variants folded less efficiently, and a mixture of 0.5 mM GSSG and 2 mM GSH was used to minimize the accumulation of forms with nonnative disulfides. The reactions were incubated for 25 min at room temperature, and the native proteins were purified by reversed-phase HPLC.
Folding and unfolding kinetics
Folding or unfolding reactions contained 30 μM protein, 0.1 M Tris-HCl (pH 8.7), 0.2 M KCl, 1 mM EDTA, and various concentrations of DTTSS and DTTSHSH. These reactions were carried out under a nitrogen atmosphere at 25°C. At various times after initiating the reaction, samples were withdrawn and reacted with iodoacetic acid (0.1 M at pH 8.0) and analyzed by nondenaturing gel electrophoresis. Gels were stained with Coomassie blue and the band intensities were quantified by video densitometry.
The observed rates at which the various species appeared and disappeared during folding or unfolding were compared with those predicted by numerical integration of the rate expressions making up the mechanism illustrated in Scheme 1 ▶. For each protein variant, the individual rate constants in the model were adjusted until all of the folding and unfolding experiments, using a range of reagent concentrations, could be simulated with a single set of rate constants. These parameters are estimated to be reliable within a factor of 2, based on attempts to adjust their values while maintaining consistency with the experimental data. Because the rate constants are derived from a global fitting procedure, the uncertainties in the derived equilibrium constants are significantly less than would be expected by simply multiplying the uncertainties in the rate constants. The rate constants are linked to one another so that increasing or decreasing the value of one by a factor of two typically requires a compensating change in the rate constant for the reverse reaction, or an adjacent forward reaction in the scheme, to account for the experimental data. For the overall equilibrium constant for folding, the uncertainty is estimated to be about fourfold, corresponding to a free energy difference of about 1 kcal/mole.
Further details of the kinetic experiments and their analysis can be found in Zhang and Goldenberg (1997b).
Model of circular BPTI and calculation of relative contact order
A model of the circularized wild-type BPTI was constructed using the crystallographically determined atomic coordinates of a circular form of BPTI (1K6U in the Protein Data Bank), which also differs from the wild-type sequence by the replacement of four residues in the trypsin binding region (Botos et al. 2001). These replacements lead to only localized structural changes, which are also observed in a linear protein with the same sequence and natural termini (Addlagatta et al. 2001). It thus appears that the replacements in the 1K6U structure are unlikely to affect the conformation of the cyclization site, which is located at the opposite end of the folded structure. To create a model of circular wild-type BPTI, the 16KU structure was superimposed on that of the natural protein (4PTI), and residues 1–4 and 56–58 of of 16KU were used to replace the equivalent segments of 4PTI.
Relative contact order (RCO) was calculated as described by Plaxco et al. (1998) using a program generously provided by Dr. Plaxco (University of California, Santa Barbara), in which inter-residue contacts are defined by the presence of nonhydrogen atoms within 6 Å of one another. To calculate RCO for the permuted sequences, the model of circular BPTI described above was modified by simply rearranging and renumbering the residues in the atomic coordinate file.
Scheme 2.

Acknowledgments
We thank Emily M. Kimmel for assistance with some of the kinetic experiments, Dr. Scott A. Beeser for collecting the NMR spectra, and Dr. Robert Schackmann for the synthesis of oligo-nucleotides and protein sequencing. Oligonucleotides, protein sequencing, and mass spectrometry analyses were provided by the Protein/DNA core facility of the Utah Regional Cancer Center (Supported by U.S. NCI Grant No. 5 P30 CA 42014). NMR spectra were collected at the Biomolecular NMR Facility of the University of Utah Health Science center, supported by NIH Grants RR13030, RR06262, and RR14768, and NSF Grant DBI-0002806. This work was supported by grant number GM42594 from the NIH.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Abbreviations
BPTI, bovine pancreatic trypsin inhibitor
cBPTI, a circular form of BPTI generated by forming a peptide bond between the natural termini
cpBPTI, circularly permuted BPTI.
The specific variants are designated cp16, cp27, cp41, and cp46, where the numbers indicate the new N-terminal residues; amino acid replacements are identified by the wild-type residue, using the standard one-letter abbreviations, the residue number, and the residue type in the mutant protein; disulfide bonds are identified by the numbers of the Cys residues, and disulfide-bonded folding intermediates are indicated by square brackets enclosing the designations of the disulfides they contain.
GSSG and GSH, the oxidized and reduced forms of glutathione, respectively; DTTSS and DTTSHSH, the oxidized and reduced forms of dithiothreitol, respectively; GuHCl, guanidinium chloride; Tris-HCl, tris(hydroxymethyl)-aminomethane hydrochloride; EDTA, ethylenediaminetetraacetic acid; HPLC, high-performance liquid chromatography.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.03563704.
References
- Addlagatta, A., Krzywda, S., Czapinska, H., Otlewski, J., and Jaskölski, M. 2001. Ultrahigh-resolution structure of a BPTI mutant. Acta Crystallogr. D. 57649–663. [DOI] [PubMed] [Google Scholar]
- Beeser, S.A., Goldenberg, D.P., and Oas, T.G. 1997. Enhanced protein flexibility caused by a destabilizing amino acid replacement in BPTI. J. Mol. Biol. 269 154–164. [DOI] [PubMed] [Google Scholar]
- Beeser, S.A., Oas, T.G., and Goldenberg, D.P. 1998. Determinants of backbone dynamics in native BPTI: Cooperative influence of the 14–38 disulfide and the Tyr 35 side chain. J. Mol. Biol. 284 1581–1596. [DOI] [PubMed] [Google Scholar]
- Botos, I., Wu, Z., Lu, W., and Wlodawer, A. 2001. Crystal structure of a cyclic form of bovine pancreatic trypsin inhibitor. FEBS Lett. 509 90–94. [DOI] [PubMed] [Google Scholar]
- Buchwalder, A., Szadkowski, H., and Kirschner, K. 1992. A fully active variant of dihydrofolate reductase with a circularly permuted sequence. Biochemistry 31 1621–1630. [DOI] [PubMed] [Google Scholar]
- Buczek, O., Krowarsch, D., and Otlewski, J. 2002. Thermodynamics of single peptide bond cleavage in bovine pancreatic trypsin inhibitor (BPTI). Protein Sci. 11 924–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulaj, G. and Goldenberg, D.P. 1999. Early events in the disulfide-coupled folding of BPTI. Protein Sci. 8 1825–1842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ———. 2001a. Mutational analysis of hydrogen bonding residues in the BPTI folding pathway. J. Mol. Biol. 313 639–656. [DOI] [PubMed] [Google Scholar]
- ———. 2001b. φ-Values for BPTI folding intermediates and implications for transition states. Nat. Struct. Biol. 8 326–330. [DOI] [PubMed] [Google Scholar]
- Chazin, W.J., Goldenberg, D.P., Creighton, T.E., and Wüthrich, K. 1985. Comparative studies of conformation and internal mobility in native and circular basic pancreatic trypsin inhibitor by 1H nuclear magnetic resonance in solution. Eur. J. Biochem. 152 429–437. [DOI] [PubMed] [Google Scholar]
- Clementi, C., Jennings, P.A., and Onuchic, J.N. 2001. Prediction of folding mechanism for circular-permuted proteins. J. Mol. Biol. 311 879–890. [DOI] [PubMed] [Google Scholar]
- Creighton, T.E. 1974. The single-disulphide intermediates in the refolding of reduced pancreatic trypsin inhibitor. J. Mol. Biol. 87 603–624. [DOI] [PubMed] [Google Scholar]
- ———. 1975. The two-disulfide intermediates and the folding pathway of reduced pancreatic trypsin inhibitor. J. Mol. Biol. 95 167–199. [DOI] [PubMed] [Google Scholar]
- ———. 1977. Effects of urea and guanidine-HCl on the folding and unfolding of pancreatic trypsin inhibitor. J. Mol. Biol. 113 313–328. [DOI] [PubMed] [Google Scholar]
- ———. 1983. An emperical approach to protein conformation, stability and flexibility. Biopolymers 22 49–58. [DOI] [PubMed] [Google Scholar]
- ———. 1992a. Folding pathways determined using disulfide bonds. In Protein folding (ed. T. Creighton), pp. 301–351. W.H. Freeman, New York.
- ———. 1992b. Protein folding pathways determined using disulfide bonds. BioEssays 14 195–199. [DOI] [PubMed] [Google Scholar]
- Creighton, T.E. and Goldenberg, D.P. 1984. Kinetic role of a meta-stable native-like two-disulphide species in the folding transition of bovine pancreatic trypsin inhibitor. J. Mol. Biol. 179 497–526. [DOI] [PubMed] [Google Scholar]
- Dadlez, M. and Kim, P.S. 1996. Rapid formation of the native 14–38 disulfide bond in the early stages of BPTI folding. Biochemistry 35 16153–16164. [DOI] [PubMed] [Google Scholar]
- Darby, N.J. and Creighton, T.E. 1993. Dissecting the disulfide-coupled folding pathway of bovine pancreatic trypsin inhibitor. Forming the first disulfide bonds in analogs of the reduced protein. J. Mol. Biol. 232 873–896. [DOI] [PubMed] [Google Scholar]
- Deschrevel, B., Vincent, J.-P., Ripoll, C., and Thellier, M. 2003. Thermodynamic parameters monitoring the equilibrium shift of enzyme-catalyzed hydrolysis/synthesis reactions in favor of synthesis in mixtures of water and organic solvent. Biotechnol. Bioeng. 81 167–177. [DOI] [PubMed] [Google Scholar]
- Doering, D. 1992. “Functional and structural studies of a small F-actin binding domain.” Ph.D. thesis, Massachusetts Institute of Technology, Cambridge, MA.
- Estell, D.A., Wilson, K.A., and Laskowski Jr., M. 1980. Thermodynamics and kinetics of the hydrolysis of the reactive-site peptide bond in pancreatic trypsin inhibitor (Kunitz) by Dermasterias imbricata Trypsin 1. Biochemistry 19 131–137. [DOI] [PubMed] [Google Scholar]
- Fersht, A.R. 2000. Transition-state structure as a unifying basis in protein-folding mechanisms: Contact order, chain topology, stability, and the extended nucleus mechanism. Proc. Natl. Acad. Sci. 97 1525–1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldenberg, D.P. 1985. Dissecting the roles of individual interactions in protein stability: Lessons from a circularized protein. J. Cell. Biochem. 29 321–335. [DOI] [PubMed] [Google Scholar]
- ———. 1992. Native and non-native intermediates in the BPTI folding pathway. Trends Biochem. Sci. 17 257–261. [DOI] [PubMed] [Google Scholar]
- ———. 1999. Finding the right fold. Nat. Struct. Biol. 6 987–990. [DOI] [PubMed] [Google Scholar]
- Goldenberg, D.P. and Creighton, T.E. 1983. Circular and circularly permuted forms of bovine pancreatic trypsin inhibitor. J. Mol. Biol. 165 407–413. [DOI] [PubMed] [Google Scholar]
- ———. 1984. Folding pathway of a circular form of bovine pancreatic trypsin inhibitor. J. Mol. Biol. 179 527–545. [DOI] [PubMed] [Google Scholar]
- Goldenberg, D.P., Frieden, R.W., Haack, J.A., and Morrison, T.B. 1989. Mutational analysis of a protein folding pathway. Nature 338 127–132. [DOI] [PubMed] [Google Scholar]
- Grantcharova, V.P. and Baker, D. 2001. Circularization changes the folding transition state of the src SH3 domain. J. Mol. Biol. 306 555–563. [DOI] [PubMed] [Google Scholar]
- Gromiha, M.M. and Selvaraj, S. 2001. Comparison between long-range interactions and contact order in determining the folding rate of two-state proteins: Application of long-range order to folding rate prediction. J. Mol. Biol. 310 27–32. [DOI] [PubMed] [Google Scholar]
- Hanson, W.M., Beeser, S.A., Oas, T.G., and Goldenberg, D.P. 2003. Identification of a residue critical for maintaining the functional conformation of BPTI. J. Mol. Biol. 333 425–441. [DOI] [PubMed] [Google Scholar]
- Henneke, J., Sebbel, P., and Glockshuber, R. 1999. Random circular permutation of DsbA reveals segments that are essential for protein folding and stability. J. Mol. Biol. 286 1197–1215. [DOI] [PubMed] [Google Scholar]
- Homandberg, G.A., Mattis, J.A., and Laskowski Jr., M. 1978. Synthesis of peptide bonds by proteinases. Addition of organic cosolvents shifts peptide bond equilibria toward synthesis. Biochemistry 17 5220–5227. [DOI] [PubMed] [Google Scholar]
- Itzhaki, L.S., Otzen, D.E., and Fersht, A.R. 1995. The structure of the transition state for folding of chymotrypsin inhibitor 2 analyzed by protein engineering methods: Evidence of a nucleation-condensation mechanism for protein folding. J. Mol. Biol. 254 260–288. [DOI] [PubMed] [Google Scholar]
- Ivankov, D.N., Garbuzynskiy, S.O., Alm, E., Plaxco, K.W., Baker, D., and Finkelstein, A.V. 2003. Contact order revisited: Influence of protein size on the folding rate. Protein Sci. 12 2057–2062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwakura, M., Nakamura, T., Yamane, C., and Maki, K. 2000. Systematic circular permutation of an entire protein reveals essential folding elements. Nat. Struct. Biol. 7 580–585. [DOI] [PubMed] [Google Scholar]
- Johnson, R.E., Adams, P., and Rupley, J.A. 1978. Thermodynamics of protein cross-links. Biochemistry 17 1479–1484. [DOI] [PubMed] [Google Scholar]
- Jung, J. and Lee, B. 2001. Circularly permuted proteins in the protein structure database. Protein Sci. 10 1881–1886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleid, D.G., Yansura, D., Small, B., Dowbenko, D., Moore, D.M., Grubman, M.J., McKercher, P.D., Morgan, D.O., Robertson, B.H., and Bachrach, H.L. 1981. Cloned viral protein vaccine for foot-and-mouth disease: Responses in cattle and swine. Science 214 1125–1129. [DOI] [PubMed] [Google Scholar]
- Li, L. and Shakhnovich, E.I. 2001. Different circular permutations produced different folding nuclei in proteins: A computational study. J. Mol. Biol. 306 121–132. [DOI] [PubMed] [Google Scholar]
- Lin, T.Y. and Kim, P.S. 1989. Urea dependence of thiol-disulfide equilibria in thioredoxin: Confirmation of the linkage relationship and a sensitive assay for structure. Biochemistry 28 5282–5287. [DOI] [PubMed] [Google Scholar]
- Lindberg, M.O., Tångrot, J., Otzen, D.E., Dolgikh, D.A., Finkelstein, A.V., and Oliveberg, M. 2001. Folding of circular permutants with decreased contact order: General trend balanced by protein stability. J. Mol. Biol. 314 891–900. [DOI] [PubMed] [Google Scholar]
- Lindberg, M., Tångrot, J., and Oliveberg, M. 2002. Complete change of the protein folding transition state upon circular permutation. Nat. Struct. Biol. 9818–822. [DOI] [PubMed] [Google Scholar]
- Lindqvist, Y. and Schneider, G. 1997. Circular permutations of natural protein sequences: Structural evidence. Curr. Opin. Struct. Biol. 7 422–427. [DOI] [PubMed] [Google Scholar]
- Luger, K., Hommel, U., Herold, M., Hofsteenge, J., and Kirschner, K. 1989. Correct folding of circularly permuted variants of a βα barrel enzyme in vivo. Science 243 206–210. [DOI] [PubMed] [Google Scholar]
- Makarov, D.E. and Plaxco, K.W. 2003. The topomer search model: A simple, quantitative theory of two-state protein folding kinetics. Protein Sci. 12 17–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumura, M., Becktel, W.J., Levitt, M., and Matthews, B.W. 1989. Stabilization of phage T4 lysozyme by engineered disulfide bonds. Proc. Natl. Acad Sci. 86 6562–6566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendoza, J.A., Jarstfer, M.B., and Goldenberg, D.P. 1994. Effects of amino acid replacements on the reductive unfolding kinetics of pancreatic trypsin inhibitor. Biochemistry 33 1143–1148. [DOI] [PubMed] [Google Scholar]
- Miller, E.J., Fischer, K.F., and Marqusee, S. 2002. Experimental evaluation of topological parameters determining protein-folding rates. Proc. Natl. Acad. Sci. 99 10359–10363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millet, O., Loria, J.P., Kroenke, C.D., Pons, M., and Palmer III, A.G. 2000. The static magnetic field dependence of chemical exchange linebroadening defines the NMR chemical shift time scale. J. Am. Chem. Soc. 122 2867–2877. [Google Scholar]
- Miozzari, G.F. and Yanofsky, C. 1978. Translation of the leader region of the E. coli tryptophan operon. J. Bacteriol. 133 1457–1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirny, L. and Shakhnovich, E. 2001. Protein folding theory: From lattice to all-atom models. Annu. Rev. Biophys. Biomol. Struct. 30 361–396. [DOI] [PubMed] [Google Scholar]
- Mullins, L.S., Wesseling, K., Kuo, J.M., Garrett, J.B., and Raushel, F.M. 1994. Transposition of protein sequences: Circular permuation of ribonuclease T1. J. Am. Chem. Soc. 116 5529–5533. [Google Scholar]
- Naderi, H.M., Thomason, J.F., Borgias, B.A., Anderson, S., James, T.L., and Kuntz, I.D. 1991. 1H NMR assignments and three-dimensional structure of Ala14/Ala38 bovine pancreatic trypsin inhibitor based on two-dimensional NMR and distance geometry. In Conformations and forces in protein folding (eds. B. Nall and K. Dill), pp. 86–114. American Association for the Advancement of Science, Washington, DC.
- Oas, T.G. and Kim, P.S. 1988. A peptide model of a protein folding intermediate. Nature 336 42–48. [DOI] [PubMed] [Google Scholar]
- Paci, E., Vendruscolo, M., Dobson, C.M., and Karplus, M. 2002. Determination of a transition state at atomic resolution from protein engineering data. J. Mol. Biol. 324 151–163. [DOI] [PubMed] [Google Scholar]
- Plaxco, K.W., Simons, K.T., and Baker, D. 1998. Contact order, transition state placement and the refolding rates of single domain proteins. J. Mol. Biol. 277 985–994. [DOI] [PubMed] [Google Scholar]
- Pritchard, L. and Dufton, M.J. 1999. Evolutionary trace analysis of the Kunitz/BPTI family of proteins: Functional divergence may have been based on conformational adjustment. J. Mol. Biol. 285 1589–1607. [DOI] [PubMed] [Google Scholar]
- Schellman, J.A. 1955. Stability of hydrogen bonded peptide structures in aqueous solution. C. R. Trav. Lab. Carlsberg [Chim.] 29 230–259. [PubMed] [Google Scholar]
- Siekmann, J., Wenzel, H.R., Matuszak, E., von Goldammer, E., and Tschesche, H. 1988. The pH dependence of the equilibrium constant KHyd for the hydrolysis of the Lys15-Ala16 reactive-site peptide bond in bovine pancreatic trypsin inhibitor (Aprotinin). J. Protein Chem. 7 633–644. [DOI] [PubMed] [Google Scholar]
- Sosnick, T.R., Mayne, L., and Englander, S.W. 1996. Molecular collapse: The rate-limiting step in two-state cytochrome c folding. Proteins 24 413–426. [DOI] [PubMed] [Google Scholar]
- Staley, J.P. and Kim, P.S. 1994. Formation of a native-like subdomain in a partially folded intermediate of bovine pancreatic trypsin inhibitor. Protein Sci. 3 1822–1832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Studier, F.W., Rosenberg, A.H., Dunn, J.J., and Dubendorff, J.W. 1990. Use of T7 RNA polymerase to direct expression of cloned genes. Methods Enzymol. 185 60–89. [DOI] [PubMed] [Google Scholar]
- Szyperski, T., Luginbühl, P., Otting, G., Güntert, P., and Wüthrich, K. 1993. Protein dynamics studied by rotating frame 15N spin relaxation times. J. Biomol. NMR 3 141–164. [DOI] [PubMed] [Google Scholar]
- Uliel, S., Fliess, A., and Unger, R. 2001. Naturally occurring circular permutations in proteins. Protein Eng. 14 533–542. [DOI] [PubMed] [Google Scholar]
- van den Berg, B., Chung, E.W., Robinson, C.R., Mateo, P.L., and Dobson, C.M. 1999. The oxidative refolding of hen lysozyme and its catalysis by protein disulfide isomerase. EMBO J. 18 4794–4803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viguera, A.R., Serrano, L., and Wilmanns, M. 1996. Different folding transition states may result in the same native structure. Nat. Struct. Biol. 3 874–880. [DOI] [PubMed] [Google Scholar]
- Wagner, G., Braun, W., Havel, T.F., Schaumann, T., Go, N., and Wüthrich, K. 1987a. Protein structures in solution by nuclear magnetic resonance and distance geometry: The polypeptide fold of the basic pancreatic trypsin inhibitor determined using two different algorithms, DISGEO and DISMAN. J. Mol. Biol. 196 611–639. [DOI] [PubMed] [Google Scholar]
- Wagner, G., Brühwiler, D., and Wüthrich, K. 1987b. Reinvestigation of the aromatic side-chains in the basic pancreatic trypsin inhibitor by hetero-nuclear two-dimensional nuclear magnetic resonance. J. Mol. Biol. 196 227–231. [DOI] [PubMed] [Google Scholar]
- Wedemeyer, W.J., Welker, E., Narayan, M., and Scheraga, H.A. 2000. Disulfide bonds and protein folding. Biochemistry 39 4207–4216. [DOI] [PubMed] [Google Scholar]
- Weikl, T.R. and Dill, K.A. 2003. Folding kinetics of two-state proteins: Effect of circularization, permutation and crosslinks. J. Mol. Biol. 332 953–963. [DOI] [PubMed] [Google Scholar]
- Weissman, J.S. and Kim, P.S. 1991. Reexamination of the folding of BPTI: Predominance of native intermediates. Science 253 1386–1393. [DOI] [PubMed] [Google Scholar]
- Wlodawer, A., Deisenhofer, J., and Huber, R. 1987. Comparison of two highly refined structures of bovine pancreatic trypsin inhibitor. J. Mol. Biol. 193 145–156. [DOI] [PubMed] [Google Scholar]
- Yang, Y.R. and Schachman, H.K. 1993. Aspartate transcarbamoylase containing circularly permuted catalytic polypeptide chains. Proc. Natl. Acad Sci. 90 11980–11984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zdanowski, K. and Dadlez, M. 1999. Stability of the residual structure in unfolded BPTI in different conditions of temperature and solvent composition measured by disulfide kinetics and double mutant cycle analysis. J. Mol. Biol. 287 923–942. [DOI] [PubMed] [Google Scholar]
- Zhang, J.-X. and Goldenberg, D.P. 1997a. Mutational analysis of the BPTI folding pathway: I. Effects of aromatic →Leu substitutions on the distribution of folding intermediates. Protein Sci. 6 1549–1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ———. 1997b. Mutational analysis of the BPTI folding pathway: II. Effects of aromatic →Leu substitutions on folding kinetics and thermodynamics. Protein Sci. 6 1563–1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, T., Bertelsen, E., Benvegnu, D., and Alber, T. 1993. Circular permutation of T4 lysozyme. Biochemistry 32 12311–12318. [DOI] [PubMed] [Google Scholar]
- Zhou, H.-X. 2003. Effect of backbone cyclization on protein folding stability: Chain entropies of both the unfolded and the folded state are restricted. J. Mol. Biol. 332 257–264. [DOI] [PubMed] [Google Scholar]