

Abstract
We identified in Salmonella enterica serovar Typhi a cluster of four genes encoding a deoxyribokinase (DeoK), a putative permease (DeoP), a repressor (DeoQ), and an open reading frame encoding a 337 amino acid residues protein of unknown function. We show that the latter protein, called DeoM, is a hexamer whose synthesis is increased by a factor over 5 after induction with deoxyribose. The CD spectrum of the purified recombinant protein indicated a dominant contribution of βtype secondary structure and a small content of α-helix. Temperature and guanidinium hydrochloride induced denaturation of DeoM indicated that the hexamer dissociation and monomer unfolding are coupled processes. DeoM exhibits 12.5% and 15% sequence identity with galactose mutarotase from Lactococcus lactis and respectively Escherichia coli, which suggested that these three proteins share similar functions. Polarimetric experiments demonstrated that DeoM is a mutarotase with high specificity for deoxyribose. Site-directed mutagenesis of His183 in DeoM, corresponding to a catalytically active residue in GalM, yielded an almost inactive deoxyribose mutarotase. DeoM was crystallized and diffraction data collected for two crystal systems, confirmed its hexameric state. The possible role of the protein and of the entire gene cluster is discussed in connection with the energy metabolism of S. enterica under particular growth conditions.
Keywords: Salmonella enterica, DeoM protein, deoxyribose mutarotase, site-directed mutagenesis, structural characterization
The genome sequencing projects revealed that over one-third of the putative proteins encoded by the newly found genes have no known function. Despite the significant progress in automation and large-scale manipulations made in the proteomics disciplines, finding a precise biological function for a protein of hypothetic role remains a tedious task, requiring intuition, a comprehensive biological background, and a variety of experimental tests (Fields and Song 1989; Burley et al. 1999; Orengo et al. 1999; Rigaut et al. 1999; Saveanu et al. 2002). On the other hand, a still significant number of well-characterized proteins isolated in the pregenomic era are awaiting assignment to their corresponding genes (Riley and Serres 2000). This was also the case of deoxyribokinase (dRK) from Salmonella enterica, encoded by the deoK gene, and whose expression is inducible by 2-D-deoxyribose (dR). dRK catalyses the ATP-dependent phosphorylation of dR to deoxyribose 5 phosphate (dR-5P), and allows S. enterica, in contrast to Escherichia coli K12, to use this sugar as the sole carbon source (Schimmel et al. 1974). Partial purification of dRK and N-terminal sequencing of the protein allowed us to identify the corresponding gene in S. enterica. Along with the deoK gene three open reading frames were identified, and putative functions have been assigned (Tourneux et al. 2000). The deoP gene product shares 34.5% sequence identity with the fucP gene product of E. coli, and corresponds most probably to the dR transporter described by Hoffee (1968). The product of another gene, deoQ, exhibits 40% identity with the deoR gene product of E. coli suggesting that DeoQ is a repressor specific for deoK expression. A third gene, encoding a 337 amino acids protein of unknown function was initially called deoX. As deoK, deoP, and deoX are transcribed in the same direction, we hypothesized that they belong to the same operon whose expression is controlled by deoQ. To assign a function to deoX, we first wanted to know if the corresponding protein was coexpressed with either deoK or deoP, or both. As shown here deoX encodes a soluble, oligomeric protein that is indeed induced by dR. Its biochemical function is presumably that of a mutarotase, and hence, we propose to rename the corresponding structural gene deoM.
Results
Deoxyribose induces both synthesis of DeoM and dRK
Strain LT2 of serovar Typhimurium was grown in LB medium at 37°C. When the optical density at 600 nm reached 0.8 the medium was supplemented with dR (1g/liter) and the growth was continued for another 2 h. Bacteria collected by centrifugation were assayed for dRK, thymidine phosphorylase (TP), and phosphopentose aldolase (PPA) activities. In parallel, a control culture was grown in the absence of dR. The specific activity of the three enzymes after induction was 0.45 U/mg prot (TP), 0.64 U/mg prot (PPA), and 0.12 U/mg prot (dRK), which represented a 6.5- (TP), 30-(PPA), and 10 (dRK)-fold increase over the control. dRK and DeoM were identified on the 2D-gel electrophoresis maps of soluble extract of S. enterica by comigration of crude extract with purified recombinant proteins. The spots corresponding to the two proteins were increased by a factor over 5 after induction with dR (data not shown).
Characterization of recombinant DeoM
The His-tagged protein overproduced in strain BL21 (DE3) was purified by Nickel-nitriloacetic acid affinity chromatography and by Sephacryl S-300HR chromatography. The molecular mass determined by ESI-MS was 40,076.75 ± 2.74 Daltons. The 132.02-Daltons mass difference from that calculated from the sequence (40,207.77) accounted for the missing N-terminal Met residue. The molecular mass of the native protein determined by gel permeation chromatography (242 kD) indicated that DeoM is a hexamer. The CD spectrum of DeoM in the far-UV region (200–250 nm) shows a large negative band with a minimum at 214 nm, indicating a dominant contribution from β-type secondary structure and a small content of α-helix (Fig. 1A ▶). The thermal denaturation curve of DeoM at 214 nm showed a highly cooperative transition with a midpoint at 66.8°C (Fig. 1B ▶). Fitting the data to a two-state denaturation process gave a van’t Hoff enthalpy (ΔHvH) of 196 ± 16 kcal • mole−1. The CD signal in the near-UV range also shows a transition step in the same temperature range, indicating that the tertiary and secondary structure unfolding are parallel. The thermal stability of DeoM was further characterized using differential scanning calorimetry (DSC). At pH 7.4 and protein concentrations above 0.1 mg/mL large exothermic aggregation occurred near Tm rendering the unfolding transition irreversible. At concentrations below 0.1 mg/ mL (routinely 0.04 mg/mL, i.e., 0.16 μM in terms of the hexamer), the DSC endotherms were complete and reversible (Fig. 2 ▶). The denaturation enthalpy obtained by integration of the Cp curve at pH 7.4 (ΔHcal) was of 1169 ± 9 kcal • mole−1. The Cp profile could be precisely predicted using a non-two-state denaturation model with one single transition giving ΔHvH of 195 ± 2 kcal • mole−1, which agreed with the CD value. The relationship between the measured ΔHcal and calculated ΔHvH values provides further information on the thermal denaturation process of DeoM (Freire 1994). The ratio, ΔHcal/ΔHvH of 5.99, suggested that DeoM is hexameric and that it unfolds as six cooperative units per mole of hexamer. DSC endotherms of DeoM recorded at different pH values showed that the protein was more stable at pH 7.4 (Tm = 65.7°C) than at pH 6.0 (Tm = 63°C) or at pH 8.3 (Tm = 63.4°C; data not shown).
Figure 1.

Physico-chemical characterization of DeoM. (A) Far-UV CD spectrum of DeoM from S. enterica before (continuous line) and after (dotted line) the thermal denaturation; (B) thermal denaturation curve of DeoM, monitored by the CD band intensity at 214 nm; (C) chemical denaturation curve of DeoM by GdmCl. The continuous line represents the best fit to a simple two-state unfolding model.
Figure 2.

Excess heat capacity of DeoM determined by differential scanning calorimetry. The protein (40 μg/mL, i.e., 0.16 μM in terms of hexamer) in 50 mM phosphate buffer (pH 7.4) was heated at a constant rate (1°C/min) up to 80°C. The continuous line represents the fitting of the experimental data to a nontwo-state model. Thermodynamic parameters derived from the fit are given in the text.
Protein denaturation by GdmCl, monitored by the far-UV CD signal, was also highly cooperative, and took place around 1.7 M denaturant concentration (Fig. 1C ▶). Assuming that the chemical denaturation is a two-state process, the experimental points were fitted to a simple transition model giving a structural stability free energy of −11.9 ± 1.2 kcal • mole−1. Existence of a unique unfolding transition step in both temperature and chemical denaturations suggests again that hexamer dissociation and unfolding of the monomers occur simultaneously.
Assignment of DeoM as a deoxyribomutarotase
PSI-BLAST search did not indicate any protein in the databases sharing sequence similarity with DeoM. Among known proteins involved in sugar metabolism and with similar size to DeoM we identified galactose mutarotase (GalM) from E. coli (Bouffard et al. 1994) and Lactococcus lactis (Thoden and Holden 2002), xylose mutarotase (XylM) from L. lactis (Erlandson et al. 2001), whose biochemical function is the interconversion of the α and β anomers of several carbohydrates. Pairwise amino acid sequence alignment of DeoM to GalM from two sources indicated that these proteins exhibit sequence identities ranging from 12.5% to 15.1% (Fig. 3 ▶). The mutarotases themselves showed no amino acid sequence similarities to other proteins in the databanks. The crystal structure of the enzyme from L. lactis complexed with galactose showed that residues responsible for anchoring the sugar to the protein were R71, H96, H170, D243, and E304 (Thoden and Holden 2002; Thoden et al. 2002). H96 and H170 are conserved as H104 and H175 in GalM from E. coli and as H111 and H183 in DeoM from S. enterica. Site-directed mutagenesis experiments targeting these two His residues in E. coli and L. lactis GalM decreased the kcat or kcat/Km values by several orders of magnitude compared to the wild-type enzymes (Beebe and Frey 1998; Thoden et al. 2003).
Figure 3.

Sequence alignment of DeoM with GalM from L. lactis and E. coli. The identical residues over all sequences are shown on a black background, and the conservative substitutions on gray. Asterisks indicate the two conserved His residues submitted to site-directed mutagenesis.
With these considerations in mind we tested DeoM for mutarotase activity on various sugars including D-deoxyribose, D-galactose, D-glucose, D-xylose, or L-arabinose. In parallel, recombinant GalM from E. coli was assayed for mutarotase activity on the same sugars. At 24°C the first-order rate constant of uncatalyzed mutarotation of β to α isomers of dR was 0.32 ± 0.02 min−1, in agreement with previous data (Lemieux et al. 1971). This value is approximately 10 times higher than that observed with nonenzymatic mutarotation of α-D-glucose to β-D-glucose. DeoM was found active only with dR as substrate, the increase of the “spontaneous” isomerization rate of β to α isomers being related to the protein concentration. Because the initial rates of both uncatalyzed and enzyme-driven reactions were missed, accurate measurements of Vm and Km were not possible. Thus, the specific activities were calculated from the first-order rate constants and the initial concentration of sugar (Table 1). Measurements at two concentrations of dR (75 and 150 mM) suggested that the Km of DeoM for deoxyribose is significantly higher that the Km for glucose observed with GalM from L. lactis or E. coli. On the other hand, the Vm/Km ratio of DeoM for deoxyribose calculated from the first-order rate constants is closely similar to that of GalM from L. lactis with α-D-glucose as the substrate. Site-directed mutagenesis of H183 yielded a DeoM variant inactive for dR anomerization (Table 1). In contrast, E82A, H111N, and E245A substitutions did not affect or affected moderately the reaction rate of DeoM. Recombinant GalM from E. coli was active as expected with D-glucose, D-galactose, D-xylose, or L-arabinose but not with dR (data not shown).
Table 1.
Specific activity of wild-type DeoM from S. enterica and of several variants obtained by site-directed mutagenesisa
| Enzyme | Activity (3mole/min/mg protein)a |
| Wild-type | 5521 ± 492 |
| E82A | 5614 ± 258 |
| E245A | 2193 ± 188 |
| H111N | 4438 ± 350 |
| H183N | 53 ± 35 |
2-deoxy-D-ribose was freshly dissolved at room temperature in 10 mM HEPES buffer (pH 6.8) at 20 mg/mL (which corresponds to 150-mM final concentration). The optical rotation readings were taken at every 10–15 sec until equilibrium within 45–60 sec after dissolution. Under these conditions the rate constant of uncatalyzed mutarotation was 0.32 ± 0.02 min−1 (half-time 130 sec). Enzyme solution (between 8 and 50 μg protein/mL final concentration) was added to the freshly dissolved sugar. The maximal reaction rate constant still accurately measurable corresponds to 2.5 times the background.
a To compare the activities of wild-type DeoM and of variants obtained by site-directed mutagenesis the first-order rate constant calculated after sub-straction of uncatalyzed reaction was multiplied with the initial concentration of the sugar (150 mM) and reported to 1 mg of protein.
Crystallization, X-ray diffraction studies, and self-rotation calculations for DeoM
Crystals obtained in the presence of PEG 4000 belong to the monoclinic space group P21. On the other hand, crystals obtained with ammonium sulfate as a precipitant belong to a different system: they are trigonal, space group P3. Assuming that the unit cell contains six molecules per asymmetric unit, the calculated Matthews coefficient (VM) is 2.51 Å3.Da−1 (corresponding to a solvent content of 50.5%) for P21 crystals, and 2.39 Å3.Da−1 (solvent content 48.1%) for P3 crystals. These VM values, typical of protein crystals (Matthews 1968), show that the crystal packing is compatible with the hexameric structure inferred from gel permeation chromatography and DSC experiments. Table 2 summarizes the useful data collection statistics. To get stronger evidence that DeoM is hexameric in the crystals, we performed rotation function calculations. For the P21 data set (left part of Fig. 4 ▶), in addition to the crystallographic (lower part, black arrows) and the two additional noncrystallographic twofold symmetry (κ value 180°) axes (clear arrows), there is a threefold noncrystallographic symmetry axis (upper part, κ value 120°) that is orthogonal to the twofold one. Accordingly, in the case of the P3 data set (right part of Fig. 4 ▶), in addition to the crystallographic threefold symmetry axis (upper part), there are three non-crystallographic axes perpendicular to the threefold axis (lower part). Therefore, in both crystals studied a threefold axis is found, perpendicular to three twofold axes. This generates a hexamer in the crystals, regardless of the crystal packing associated to each space group. Whatever the crystal system was, no peak appeared for a kappa value of 60° (not shown), indicating the absence of a sixfold axis. The X-ray fluorescence scan obtained at the European Synchrotron Radiation Facility (ESRF) beamline for crystals of the selenomethionine protein confirmed the presence of Se. However, current programs used (CNS, Brünger et al. 1998; SHARP, De La Fortelle and Bricogne 1997; SnB, Miller et al. 1994; and SOLVE, Terwilliger and Berendzen 1999) could not locate the large number of selenium sites (15 per monomer, leading to 90 sites per asymmetric unit to be found for the hexameric protein).
Table 2.
Data-collection statistics
| Space group | P21 | P3 |
| Unit-cell parameters (Å) | a = 85.3, b = 143.3, c = 101.7 | a = b = 140.1, c = 101.3 |
| α = γ = 90, β = 104.3 | α = β = 90, γ = 120 | |
| Resolution (Å) | 2.4 | 2.2 |
| Observed reflexions | 1,350,182 | 729,675 |
| Unique reflexions | 94,206 | 113,239 |
| Completeness (%) | 94.7 (78.2) | 90.4 (83.5) |
| Rsym (%)a | 7.8 (27) | 8.8 (22.1) |
| I/σ (I) | 13.9 (3.1) | 9.5 (3.1) |
Values for the highest resolution shell are given in parentheses (10 shells).
a Rsym = ∑hkl [(∑i|Ii − 〈I〉|)/∑iIi] for equivalent observations.
Figure 4.

Self-rotation function plots for observed structure factors. (Left) In the case of the monoclinic P21 data set. (Right) For the trigonal system P3. In the set of polar angles (ψ, ϕ, κ), ψ measures the declination from the b (for P21) or b* (for P3) axis and is plotted as lines of latitude; ϕ measures the angle between the a and c* axes (longitudes). a and b axes refer to the unit cell, whereas b* is the axis perpendicular to (a, c) plane, and c* the axis perpendicular to (a, b) plane. Black arrows indicate the crystallographic axes, whereas clear arrows indicate the noncrystallographic axes. Drawn with GLRF (Otwinowski and Minor 1993).
Discussion
Carbohydrates, the most abundant group of compounds in the living world, perform a wide range of functions as major cellular constituents or as sources of energy. The large number of the simple building blocks, the monosaccharides, and their derivatives, explain the variety as well as the importance of carbohydrates as foods, textiles, cosmetics, or pharmaceuticals. In nature, dR appears exclusively as a component of DNA or its degradation products. The only reaction in which a ribosyl group is transformed into a deoxyribosyl group is catalyzed by ribonucleotide reductases with the overall stoichiometry:
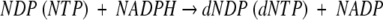 |
where NDP and NTP correspond to ribonucleoside di- and triphosphates (Jordan and Reichard 1998). Free dR may result from phosphorolysis of deoxynucleosides catalyzed by nucleoside phosphorylases, followed by hydrolysis of the resulting phosphate ester catalyzed by either specific or nonspecific phosphatases. The physiological relevance of such reaction has never been established.
Identification of dRK and an entire gene cluster controlled by DeoQ (Christensen et al. 2003), reopened the question of the role of dR as an energy source, at least under particular circumstances. Thus, Finkel and Kolter (2001) demonstrated that isolated homo- or hetero-specific DNA fragments can support E. coli growth as the sole source of carbon and energy. Within the environments inhabited by bacteria like the mammalian gastrointestinal tract or lung mucosa, extracellular DNA concentrations may rise to several mg/mL (Potter et al. 1963; Lorenz and Wackernagel 1994). It is therefore conceivable that under certain conditions the concentration of free dR originating from DNA degradation can reach the millimolar range, making the sugar a transient but essential nutrient for bacterial survival. In this scenario, deoM, along with the whole operon, has its role to play. E. coli bearing the serovar Typhimurium deoK gene alone can grow in mineral medium containing dR as carbon source. However, when deoP and deoM genes were inserted together with deoK, growth was significantly improved (Christensen et al. 2003).
Assuming that these hypotheses are valid in vivo, the next question is how the enzymatic mutarotation offers an advantage for dR utilization in view of the significant rate of uncatalyzed anomerization. At equilibrium, dR exists predominantly in the pyranose form (42% α, 43% β), but the five-membered furanose form (5% α, 10% β) exists in significant amounts, as demonstrated by NMR spectroscopy (Lemieux et al. 1971). The availability of the furanose anomer(s) will most probably dictate the overall phosphorylation rate of dR to dR-5P, which in turn, depends on the pyranose–furanose interconversion rate. Are all these “spontaneous” equilibria also enzymatically driven? We are tempted to suggest that DeoM plays a role in speeding up one or more of the spontaneous anomerization reactions that are important for optimal catabolism of dR.
The low sequence identity between DeoM and GalM or XylM renders difficult the 3D structure prediction of DeoM. On the other hand, GalM is a dimer, whereas DeoM is a hexamer, while XylM lacks any structural characterization. In addition, the crystal structure of a monomeric mutarotase has been recently deposited in the Protein Data Bank (PDB code 1LUR). Up to now, we failed to solve the structure of DeoM, either by molecular replacement using the recently published crystal structures of GalM from L. lactis (Erlandson et al. 2001; Thoden et al. 2003), and from Caenorhabditis elegans (PDB entry 1LUR) or by MAD phasing. However, the high content of β-type secondary structure of DeoM as inferred from CD spectra is in good agreement with the structure of GalM, which contains 28 β-strands, forming a β-sandwich connected by only three α-helices and nine reverse turns. With regard to the key residues involved in catalysis, some were identified from the crystal structure of GalM from L. lactis, and some by site-directed mutagenesis of GalM from L. lactis. They include R71, H96, H170, D243, and E304 in GalM. In the structure of GalM from this organism H96 and H170 were found to be responsible for anchoring the sugar. The imidazole nitrogen of these residues was located within hydrogen bonding distance to the C-5 oxygen of galactose. An interesting conclusion arising from combined X-ray and site-directed mutagenesis studies of GalM from L. lactis complexed with several sugar substrates is that there are distinct binding modes of individual monosaccharides, depending on the spatial orientation of their C4 hydroxyl group. For this reason only some residues (E304 or H170) are important for catalysis with all sugars, whereas other residues are “specific” for a particular group. These characteristics might explain why site-directed mutagenesis of “conserved” residues in DeoM yielded in some cases active enzymes. We expect to answer these questions using better crystals of DeoM to solve the structure by MAD and by following the kinetics of mutarotation at temperatures close to 0°C, where the uncatalyzed “background” represents less than 10% of the overall reaction.
Materials and methods
Chemicals
Nucleotides, restriction enzymes, T4 DNA ligase, and coupling enzymes were from Roche Applied Sciences. Vent DNA polymerase was from New England Biolabs, Inc. Tfu DNA polymerase was from Q-Biogene. Oligonucleotides were synthesized according to the phosphoramidate method using a commercial DNA synthesizer (Cyclone TM Bioresearch). HiPrep 26/60 Sephacryl S-300 HR column was from Amersham Pharmacia Biotech. D-glucose, D-galactose, D-xylose, L-arabinose, and 2-D-deoxyribose of highest purity were from Sigma.
Bacterial strains, plasmids, growth conditions, and DNA manipulations
The deoM gene and galM gene were PCR-amplified from S. enterica serovar Typhimurium genomic DNA and E. coli genomic DNA, respectively, using the following primers:
5′-deoM: 5′-GGAATTCCATATGACAACACGTATTACATT ATGGC-3′;
3′-deoM: 5′-CGGCGCTCGAGTCAGACAATGCCTGTCGTG ACG-3′ and
5′-galM: 5′-GGAATTCCATATGCTGAACGAAACTCCCGC ACTG-3′;
3′-galM: 5′-CCGCTCGAGTTACTCAGCAATAAACTGATA TTCCGT-3′. The PCR products were cloned at the NdeI and XhoI sites (underlined) of the vector pET28a (Novagen) containing a T7 promoter and a His-tag. The H111N, H183N, and E245A DeoM mutants were constructed by the one-tube PCR-based mutagenesis method (Picard and Bock 1997) using Tfu DNA polymerase and the following mutagenic oligonucleotides:
3′-deoM H111N: 5′-GCAGGGCATCTCGCCATTCAGCGGA TGTGTATC-3′; 3′-deoM H183N: 5′-ATAGGCATAGTTCATGTTGCACATATACTGTAGCGGCATGGC-3; 3′-deoM E245A: 5′-GGCGAAGAACACGATCGCCGGGTCGTAAAAGTC-3′ (modified codons are underlined). The resulting plasmids pLA542 and pLA2202 (harboring the deoM and galM genes, respectively) and the plasmids pLA543, pLA544, and pLA549 (harboring the respective mutated deoM genes) were used for transforming the E. coli strain BL21(DE3)pDIAI7 (Munier et al. 1991) for expression. The recombinant strain was grown in a 2YT medium supplemented with kanamycin (70 μg/mL) and chloramphenicol (30 μg/ mL) at 37°C to an optical density of 1.5 at 600 nm; then the overproduction of the recombinant proteins was induced by the addition of isopropyl-1-β-D-thiogalactoside (1 mM final concentration). After 3 h, bacteria was harvested by centrifugation for 20 min at 10,000 × g and 4°C.
Purification of DeoM and activity assay
Cells were disrupted by sonication in 50 mM sodium phosphate buffer, pH 8.0 containing 300 mM NaCl (buffer A) and the homogenate was clarified by centrifugation at 10,000 × g for 45 min. The supernatant was loaded onto a Nickel-nitriloacetic acid resin column (Qiagen) previously equilibrated with buffer A. The column was then washed with 8 volumes of buffer A containing 5 mM imidazole. The protein was eluted with buffer A supplemented with 250 mM imidazole. Fractions containing the His-tagged protein were pooled and loaded onto a Sephacryl S-300 HR gel filtration column (1.5 × 110 cm) equilibrated with 50 mM Tris-HCl (pH 7.4). The molecular mass of DeoM was determined by gel permeation chromatography onto a HiPrep 26/60 Sephacryl S-300 column equilibrated with 50 mM Tris-HCl pH 7.4 and previously calibrated with blue dextran 2000 (2000 kD), thyroglobulin (669 kD), ferritin (440 kD), pyruvate kinase (240 kD), and lactate dehydrogenase (140 kD). Polarimetric determination of the mutarotation rate of various sugars in the presence or absence of DeoM was made on a Perkin-Elmer model 241MC apparatus at room temperature (24°C). Sugars were freshly dissolved in 10 mM HEPES (pH 6.8). The background reaction was subtracted from the enzyme-catalyzed reaction. Other enzyme activities such as thymidine phosphorylase (TP), phosphopentose aldolase (PPA) or dRK were determined using published procedures (Hoffee 1968; Schimmel et al. 1974; Schwartz 1978). One unit of enzyme activity corresponds to 1 μmole substrate transformed in 1 min at 30°C.
Analytical procedures
Protein concentration was measured according to Bradford (1976) using a Bio-Rad kit or by amino acid analysis on a Beckman system 6300 high-performance analyzer after 6 N HCl hydrolysis for 22 h at 115°C. SDS-PAGE was performed as described by Laemmli (1970). 2D gel electrophoresis was run on 100-μg soluble proteins loaded onto Bio-Rad IPG strips (pH gradient 4–7); the second dimension gels were performed onto 12.5% slab gels. Proteins were visualized by silver nitrate staining. Ion mass spectra were recorded on an API365 triple-quadrupole mass spectrometer (Applied Biosystems-MDS-Sciex) equipped with a nano-electro-spray source (Protana). The circular dichroism (CD) spectra were acquired on a Jasco715 spectropolarimeter equipped with a device for automatic temperature control. The denaturation curves were recorded on a 2.5-μM protein solution (in terms of hexamer) in 50 mM MOPS buffer (50 mM), at pH 7.4 by monitoring the ellipticity at 214 nm as a function of temperature (1°C/min), or guanidinium hydrochloride (GdmCl) concentration. Supposing that the chemical unfolding is an equilibrium process, one can fit the experimental data to a two-state model with the free energy of unfolding (ΔG°) given by the expression ΔG° = −RT lnK, where K is the equilibrium constant of unfolding at a given denaturant concentration. The free energy of unfolding is considered to be linearly dependent on the GdmCl concentration: ΔG° = ΔG° H2O − m [GdmCl] (Pace et al. 1989). Differential scanning calorimetry (DSC) endotherms were obtained using the VP-DSC from Micro-Cal Inc. DeoM stock solution dialyzed overnight at 4°C against 50 mM sodium phosphate buffer pH 7.4 was diluted with the same buffer (between 0.04 and 1.3 mg/mL). Samples were degassed under vacuum for 10 min with gentle stirring prior to being loaded into the calorimetric cell (0.5 mL). Samples held in situ under a constant external pressure of 29 psi to avoid bubble formation, and evaporation at high temperatures were equilibrated 30 min at 25°C, then heated at a constant scan rate of 1°C/min. Experimental data were collected with a 16-sec filter and the instrument baseline removed by subtraction of scans of the same buffer, prior to data analysis. After normalizing to concentration, a chemical baseline calculated from the progress of the unfolding transition was subtracted. The excess heat capacity functions were analyzed using the software package Origin 7 provided by the manufacturer (Plotnikov et al. 1997). Data at various pH values were collected in the same manner. Thermodynamic parameter values are the mean of two independent experiments; errors estimations are deviations from the means.
Crystallization, data collection, processing, and self-rotation calculations
Two types of crystals (monoclinic and trigonal) of the His-tagged DeoM were grown at 20°C by vaporphase diffusion, using the hanging-drop method. First, 3 μL of protein solution at 10 mg/mL were mixed with 3 μL of a reservoir solution containing 0.4 M lithium sulfate, 19% (w/v) polyethylene glycol 4000 and 10% glycerol in a 50 mM Tris-HCl buffer pH 8.5. Monoclinic P21 crystals were obtained. Second, the selenomethionine protein was used, in an attempt to use MAD (multiple anomalous diffusion) phasing. Three microliters of a 20 mg/mL protein solution were mixed with 3 μL of a reservoir solution containing 1.1 M ammonium sulfate and 10 mM dithiothreitol in a 50 mM Tris-HCl buffer pH 8.5. This resulted in trigonal P3 crystals. All crystals were flash-frozen in nitrogen gas at 100 K (Oxford Cryosystems Cryostream Cooler), after 2 min soaking in a cryoprotectant solution containing 30% to 35% glycerol. Data were collected at ESRF in Grenoble (France) on the BM14 line for the monoclinic P21 crystal, and on the ID14–4 beamline for the trigonal P3 crystal. The raw data were indexed (using hexagonal axes for P3 crystal), processed and scaled using the HKL package (Otwinowski and Minor 1993). For the monoclinic crystal used in data collection, systematic absences were in accordance with a P21 space group. For the trigonal crystal, no systematic absences related to a 31 or a 32 screw axis were seen. Therefore, the remaining possible trigonal groups were P3, P312, and P321. Among them, P3 gave the best Rsym value (these values were even worse with space groups P6 or P622). Self-rotation analysis used the GLRF program (Tong and Rossmann 1990). Reflections between 10.0 and 3.5 Å resolution were used, and the radius of integration was 30 Å.
Acknowledgments
We thank Michel Yvan Popoff for the culture of S. enterica serovar typhimurium and many inspiring discussions, Nathalie Joly for growing crystals, Chantal Le Bouguenec for fruitful discussion, Akuavi Amoussou for enthusiastic participation in the preliminary experiments, and Régine Lambrecht for excellent secretarial help. Marcel and Liliane Pollack are acknowledged for the generous donation of funds for the acquisition of the DSC station. This work was supported by grants from the Centre National de la Recherche Scientifique (URA2185) and Institut Pasteur (Programme Transversal de Recherche [PTR] 53).
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Article published ahead of print. Article and publication date are at http://www.proteinscience.org/cgi/doi/10.1110/ps.03566004.
References
- Beebe, J.A. and Frey, P.A. 1998. Galactose mutarotase: Purification, characterization, and investigations of two important histidine residues. Biochemistry 37 14989–14997. [DOI] [PubMed] [Google Scholar]
- Bouffard, G.G., Rudd, K.E., and Adhya, S.L. 1994. Dependence of lactose metabolism upon mutarotase encoded in the gal operon in Escherichia coli. J. Mol. Biol. 244 269–278. [DOI] [PubMed] [Google Scholar]
- Bradford, M.M. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye-binding. Anal. Biochem. 72 248–254. [DOI] [PubMed] [Google Scholar]
- Brünger, A.T., Adams, P.D., Clore, G.M., DeLano, W.L., Gros, P., Grosse-Kunstleve, R.W., Jiang, J.S., Kusziewski, J., Nilges, M., Pannu, N.S., et al. 1998. Crystallography & NMR system: A new software suite for macro-molecular structure determination. Acta Crystallogr. D 54 905–921. [DOI] [PubMed] [Google Scholar]
- Burley, S.K., Almo, S.C., Bonanno, J.B., Capel, M., Chance, M.R., Gaasterland, T., Lin, D., Sali, A., Studier, F.W., and Swaminathan, S. 1999. Structural genomics: Beyond the human genome project. Nat. Genet. 23 151–157. [DOI] [PubMed] [Google Scholar]
- Christensen, M., Borza, T., Dandanell, G., Gilles, A.-M., Bârzu, O., Keller, R., and Neuhard, J. 2003. Regulation of expression of the 2-deoxy-D-ribose utilization regulon, deoQRPX, from Salmonella enterica serovar Typhimurium. J. Bacteriol. 185 6042–6050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De La Fortelle, E. and Bricogne, G. 1997. Maximum-likelihood heavy-atom parameter refinement for multiple isomorphous replacement and multi-wavelength anomalous diffraction methods. Methods Enzymol. 276 472–494. [DOI] [PubMed] [Google Scholar]
- Erlandson, K.A., Delamarre, S.C., and Batt, C.A. 2001. Genetic evidence for a defective xylan degradation pathway in Lactococcus lactis. Appl. Environ. Microbiol. 67 1445–1452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fields, S. and Song, O. 1989. A novel genetic system to detect protein–protein interactions. Nature 340 245–246. [DOI] [PubMed] [Google Scholar]
- Finkel, S.E. and Kolter, R. 2001. DNA as a nutrient: Novel role for bacterial competence gene homologs. J. Bacteriol. 183 6288–6293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freire, E. 1994. Statistical thermodynamic analysis of differential scanning calorimetry data: Structural deconvolution of heat capacity function of proteins. Methods Enzymol. 240 502–530. [DOI] [PubMed] [Google Scholar]
- Hoffee, P.A. 1968. 2-Deoxyribose gene–enzyme complex in Salmonella typhimurium. I. Isolation and enzymatic characterization of 2-deoxyribose-negative mutants. J. Bacteriol. 95 449–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan, A. and Reichard, P. 1998. Ribonucleotide reductases. Annu. Rev. Biochem. 67 71–98. [DOI] [PubMed] [Google Scholar]
- Laemmli, U.K. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227 680–685. [DOI] [PubMed] [Google Scholar]
- Lemieux, R.U., Anderson, L., and Conner, A.H. 1971. The mutarotation of 2-deoxy-D-erythro-pentose (“2-deoxy-D-ribose”). Conformations, kinetics, and equilibria. Carbohydr. Res. 20 59–72. [DOI] [PubMed] [Google Scholar]
- Lorenz, M.G. and Wackernagel, W. 1994. Bacterial gene transfer by natural genetic transformation in the environment. Microbiol. Rev. 58 563–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews, B.W. 1968. Solvent content of protein crystals. J. Mol. Biol. 33 491–497. [DOI] [PubMed] [Google Scholar]
- Miller, R., Gallo, S.M., Khalak, H.G., and Weeks, C.M. 1994. SnB: Crystal structure determination via Shake-and-Bake. J. Appl. Crystallogr. 27 613–621. [Google Scholar]
- Munier, H., Gilles, A.-M., Glaser, P., Krin, E., Danchin, A., Sarfati, R.S., and Bârzu, O. 1991. Isolation and characterization of catalytic and calmodulin-binding domains of Bordetella pertussis adenylate cyclase. Eur. J. Biochem. 196 469–474. [DOI] [PubMed] [Google Scholar]
- Orengo, C.A., Todd, A.E., and Thornton, J.M. 1999. From protein structure to function. Curr. Opin. Struct. Biol. 9 374–382. [DOI] [PubMed] [Google Scholar]
- Otwinowski, Z. and Minor, W. 1993. DENZO, a film processing program for macromolecular crystallography. Yale University Press, New Haven, CT.
- Pace, C.N., Shirley, B.A., and Thomson, J.A. 1989. Measuring the conformational stability of a protein. In Protein structure: A practical approach (ed. E.T. Creighton), pp. 311–329. IRL Press, Oxford, UK.
- Picard, V. and Bock, S. 1997. Rapid and efficient one-tube PCR-based mutagenesis method. Methods Mol. Biol. 67 183–188. [DOI] [PubMed] [Google Scholar]
- Plotnikov, V.V., Brandts, J.M., Lin, L.-N., and Brandts, J.F. 1997. A new ultrasensitive scanning calorimeter. Anal. Biochem. 250 237–244. [DOI] [PubMed] [Google Scholar]
- Potter, J., Spector, S., Matthews, L., and Lemm, J. 1963. Studies on pulmonary secretions from patients with cystic fibrosis, bronchiectasis, and laryngectomy. Am. Rev. Respir. Dis. 99 909–916. [DOI] [PubMed] [Google Scholar]
- Rigaut, G., Shevchenko, A., Rutz, B., Wilm, M., Mann, M., and Seraphin, B. 1999. A generic protein purification method for protein complex characterization and proteome exploration. Nat. Biotechnol. 17 1030–1032. [DOI] [PubMed] [Google Scholar]
- Riley, M. and Serres, M.H. 2000. Interim report on genomics of Escherichia coli. Annu. Rev. Microbiol. 54 341–411. [DOI] [PubMed] [Google Scholar]
- Saveanu, C., Miron, S., Borza, T., Craescu, C.T., Labesse, G., Gagyi, C., Popescu, A., Schaeffer, F., Namane, A., Laurent-Winter, C., et al. 2002. Structural and nucleotide-binding properties of YajQ and YnaF, two Escherichia coli proteins of unknown function. Protein Sci. 11 2551–2560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schimmel, S.D., Hoffee, P., and Horecker, B.L. 1974. Deoxyribokinase from Salmonella typhimurium. Arch. Biochem. Biophys. 164 560–570. [DOI] [PubMed] [Google Scholar]
- Schwartz, M. 1978. Thymidine phosphorylase from Escherichia coli. Methods Enzymol. 51 442–445. [DOI] [PubMed] [Google Scholar]
- Terwilliger, T.C. and Berendzen, J. 1999. Automated MAD and MIR structure solution. Acta Crystallogr. D 55 849–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thoden, J.B. and Holden, H.M. 2002. High resolution X-ray structure of ga-lactose mutarotase from Lactococcus lactis. J. Biol. Chem. 277 20854–20861. [DOI] [PubMed] [Google Scholar]
- Thoden, J.B., Kim, J., Raushel, F.M., and Holden, H.M. 2002. Structural and kinetic studies of sugar binding to galactose mutarotase from Lactococcus lactis. J. Biol. Chem. 277 45458–45465. [DOI] [PubMed] [Google Scholar]
- ———.2003. The catalytic mechanism of galactose mutarotase. Protein Sci. 12 1051–1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong, L.A. and Rossmann, M.G. 1990. The locked rotation function. Acta Crystallogr. A 46 783–792. [DOI] [PubMed] [Google Scholar]
- Tourneux, L., Bucurenci, N., Saveanu, C., Kaminski, P.A., Bouzon, M., Pistotnik, E., Namane, A., Marlière, P., Bârzu, O., Li de la Sierra, I., et al. 2000. Genetic and biochemical characterization of Salmonella typhi deoxyribokinase. J. Bacteriol. 182 869–873. [DOI] [PMC free article] [PubMed] [Google Scholar]