

Abstract
A systematic study of helix–helix packing in a comprehensive database of protein structures revealed that the side chains inside helix–helix interfaces on average are shorter than those in the noninterface parts of the helices. The study follows our earlier study of this effect in transmembrane helices. The results obtained on the entire database of protein structures are consistent with those obtained on the transmembrane helices. The difference in the length of interface and noninterface side chains is small but statistically significant. It indicates that helices, if viewed along their main axis, statistically are not circular, but have a flattened interface. This effect brings the helices closer to each other and creates a tighter structural packing. The results provide an interesting insight into the aspects of protein structure and folding.
Keywords: protein modeling, structure prediction, protein folding, secondary structure packing, docking
Packing of protein structures in general and packing of secondary structure elements in particular have been extensively studied in structural biology. Surface complementarity between elements of protein structure is an important factor in protein stability. Our earlier systematic study (Jiang et al. 2003) confirmed the major role of geometric match between secondary structure elements (α-helices, β-strands, and loops). Low-resolution docking studies (Vakser 1996b; Vakser et al. 1999) of helix–helix interactions (Vakser 1996a; Vakser and Jiang 2002) indicated the existence of geometrically preferred sides/faces in helices for the interaction with other helices. Our earlier study of helix bundles in integral membrane proteins (Jiang and Vakser 2000) revealed that side chains inside helix–helix interfaces, on average, are shorter than those in the noninterface parts of the helices. Thus, transmembrane helices, if viewed along the main axis, on average, are not circles but rather ellipses. This effect brings main axes of helices closer to each other and maximizes the surface contact between the helices (thus enhancing the packing/stability of the protein). The membrane-spanning helices were chosen because they are well packed, aligned, and almost parallel to each other in the transmembrane bundles, thus being an ideal object for such a study. These results obtained on a few available crystal structures of transmembrane proteins raised a natural question: whether the same phenomenon applies to helix–helix pairs in all protein structures.
In the present study, a comprehensive nonredundant set of interacting helices from the entire PDB was used for the analysis of side-chain length distribution. The results show that similar to the earlier analyzed transmembrane subset, on average, the side chains in helices are shorter inside the helix–helix interfaces than those outside the interface areas. This small but statistically significant difference places helices closer to each other and enhances the packing of the structure. This phenomenon provides a new insight into the packing and stability of protein structures.
Data set of helices
Selection of proteins
A comprehensive nonredundant set of protein chains was obtained from PDB_SELECT (Hobohm and Sander 1994). In the data set all protein chains had less than 25% sequence identity to each other. Structures determined by NMR and low-resolution (>2.5 Å) structures were excluded. To compare the results with our earlier study of transmembrane proteins (Jiang and Vakser 2000), the transmembrane proteins were also excluded. The final data set contained 886 protein chains.
Selection of helices
Helical fragments in the protein structures were identified by DSSP (Kabsch and Sander 1983). To guarantee at least two turns in all faces of a helix, only those with ≥8 residues were selected for the next step. Each selected helix was checked against the rest of the helices in the protein for interaction. Two helices were considered interacting if each had ≥4 residues in contact with the other helix. Following our earlier study (Jiang and Vakser 2000), two residues were considered to be in contact if they had atoms within the sum of their van der Waals radii plus 0.2 Å.
Unlike most transmembrane helices that are roughly parallel to each other, helices in globular proteins are packed at the variety of angles (Reddy and Blundell 1993; Walther et al. 1996; Hespenheide and Kuhn 2003). The helix packing angle was used to determine the interacting segments in the helices. The outstanding parts of the helices were deleted. The definition of the interacting segments is shown in Figure 1A ▶. All pairs of helical segments satisfying the above criteria were considered, including more than one segment–segment interaction per helix (where the helix interacted with more than one helix).
Figure 1.

Parameters of helices used in the analysis. (A) Definition of the interacting segments. A pair of packed helices is represented by cylinders A and B, with the main axes defined by aa′ and bb′, and ab orthogonal to the main axes. The helix packing angle α is the dihedral angle a ′abb′. Point a is the center of mass of seven Cα atoms in helix A with the center Cα being the closest to a Cα atom in helix B. Point a′ is the center of mass of Cα atoms in a fragment from point a to the C or N terminus of the helix, whichever is longer. Points b and b′ in helix B are defined similarly. The length L of packed helical fragments (shown as solid rectangles) is calculated as L = 2Rcot(α/2), where R = 5Å is the helix radius. Outstanding parts (dotted lines) are deleted. (B) Definition of the helix–helix interface. Viewed along the axis a′ a of helix A, vector ab is in the center of a 90° interface sector (shaded).
Interface definition
All residues in the packed pair of helical fragments were separated into interface and noninterface ones. Because the side-chain length difference is the object of this study the interface definition had to be independent of the side-chain length. Thus, the residue–residue contact criteria, which implicitly take into account the residue size, were deemed inappropriate. The criterion used was based on pure geometric considerations (Fig. 1B ▶). A residue was considered belonging to the interface if its Cα atom was in the interface. The criterion was applied regardless of the number of helix–helix interfaces per helix. If a segment of a helix was involved in more than one helix–helix interaction, a residue in that segment could be in the interface in one interaction and outside the interface in the other interaction. In such cases the residue was considered to be the interface one.
Results and Discussion
The sizes of the 20 residue types (Table 1) taken from Levitt (1976) were defined as average distances from the Cα atom to the side-chain center of mass in a database of protein structures. Although the number of available structures in that database was limited, the number of occurrences of each amino acid type was sufficient for a simple length quantification scheme. Sizes of interface side chains and noninterface side chains were averaged over all helices in the data set. The average size of 6749 interface residues was 1.92 Å, and the average size of 12,806 noninterface residues was 2.07 Å (Fig. 2 ▶). The 0.15-Å difference is statistically significant, with a P-value = 10−29 in the Student t-test, assuming equal variance.
Table 1.
Side-chain length
| Residue | Length |
| Ala | 0.77 |
| Arg | 3.72 |
| Asn | 1.98 |
| Asp | 1.99 |
| Cys | 1.38 |
| Gln | 2.58 |
| Glu | 2.63 |
| Gly | 0.00 |
| His | 2.76 |
| Ile | 1.83 |
| Leu | 2.08 |
| Lys | 2.94 |
| Met | 2.34 |
| Phe | 2.97 |
| Pro | 1.42 |
| Ser | 1.28 |
| Thr | 1.43 |
| Trp | 3.58 |
| Tyr | 3.36 |
| Val | 1.49 |
Figure 2.

Average length of helix–helix interface and noninterface side chains. The difference is small but statistically significant (P value = 10−29; see text).
A comparison with the results of the previous study of transmembrane helices (Jiang and Vakser 2000) shows that the difference between noninterface and interface side chains in helices of soluble proteins is, to some extent, smaller than in transmembrane helices (0.15 Å and 0.18 Å respectively). However, both the noninterface and interface side-chain sizes (2.07 Å and 1.92 Å respectively) are larger than those in transmembrane helices (1.92 Å and 1.74 Å respectively). This data indicates that in addition to an obvious difference in residue content determined by the environment (hydrophobic for integral membrane proteins and hydrophilic for soluble proteins), on average, the transmembrane helices are packed more tightly than the helices in soluble proteins.
The existence of a small but statistically significant difference in length between helix–helix interface and noninterface side chains in soluble proteins determined in this study is consistent with a similar effect discovered in the transmembrane helices (Jiang and Vakser 2000). The example of this effect is shown in Figure 3 ▶. It is important to emphasize though that this difference is determined statistically, on a large number of helix–helix pairs; thus, the existence and the extent of such a difference in individual helix–helix pairs vary. There might be even entire classes of protein structures and structural motifs where this general rule does not necessarily apply (e.g., in leucine zippers the helix–helix interfaces are known to be formed mostly by residues with long side chains–Leu, Lys, Arg, Glu, Gln). However, on average, for the entire data set of protein structures the rule still holds.
Figure 3.
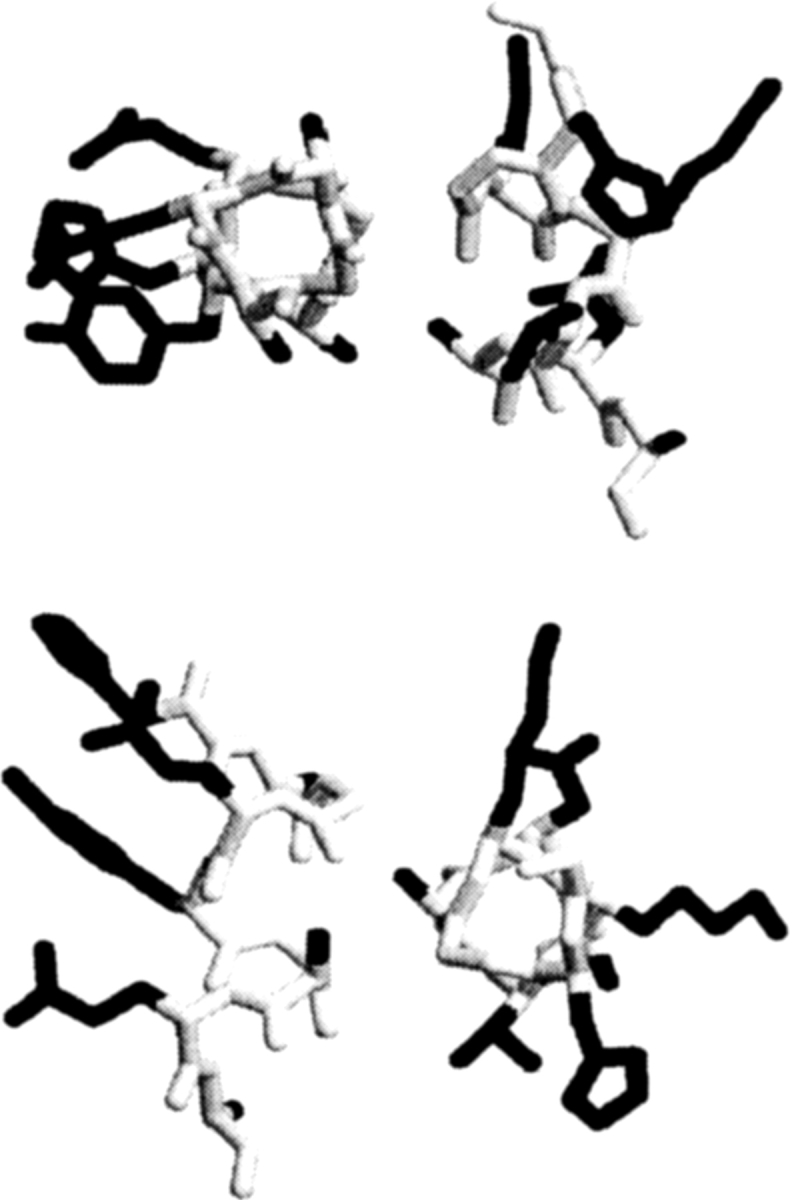
Example of a helix–helix interface. The backbone is in gray, and the side chains are in black. The same helix–helix pair is shown in two different views, along the main axis of the first helix and the second helix. The interface side chains are shorter than those outside the interface.
Shorter side chains at helix–helix interfaces mean that helices have preferred sides/faces for interaction with other helices, which is interesting scientifically, and can be potentially utilized in structure prediction algorithms (although the effect is small and thus its utility for prediction procedures is unclear). From the physicochemical point of view, the helix–helix interface residues tend to be more hydrophobic than the noninterface ones (Li and Woodward 1999). Because the hydrophobic residues, to a certain extent, on average are shorter than the hydrophilic ones, the hydrophobicity factor appears to be correlated with the difference in the interface/noninterface side-chain length.
Such interface/noninterface difference enhances the packing of the helices by flattening the interface and bringing the helices closer to each other (Fig. 4 ▶), and thus, may have resulted from evolutionary pressure to create tighter packing of protein structures. The effect provides an interesting insight into aspects of protein structure and folding.
Figure 4.

Schematic illustration of helices. The side chains are in gray areas. The helices viewed along the main axes look not like circles (A) but have a flatter interface (B) created by shorter side chains (and by correspondingly longer side chains outside the interface). The effect brings them closer to each other and increases the helix–helix packing.
Acknowledgments
This study was supported by NIH R01 GM61889 and NSF DBI-9808093.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Article published ahead of print. Article and publication date are at http://www.proteinscience.org/cgi/doi/10.1110/ps.03505804.
References
- Hespenheide, B.M. and Kuhn, L.A. 2003. Discovery of a significant, nontopological preference for antiparallel alignment of helices with parallel regions in sheets. Protein Sci. 12 1119–1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hobohm, U. and Sander, C. 1994. Enlarged representative set of protein structures. Protein Sci. 3 522–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang, S. and Vakser, I.A. 2000. Side chains in transmembrane helices are shorter at helix–helix interfaces. Proteins 40 429–435. [DOI] [PubMed] [Google Scholar]
- Jiang, S., Tovchigrechko, A., and Vakser, I.A. 2003. The role of geometric complementarity in secondary structure packing: A systematic docking study. Protein Sci. 12 1646–1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch, W. and Sander, C. 1983. Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 22 2577–2637. [DOI] [PubMed] [Google Scholar]
- Levitt, M. 1976. A simplified representation of protein conformations for rapid simulation of protein folding. J. Mol. Biol. 104 59–107. [DOI] [PubMed] [Google Scholar]
- Li, R. and Woodward, C. 1999. The hydrogen exchange core and protein folding. Protein Sci. 8 1571–1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy, B.V.B. and Blundell, T.L. 1993. Packing of secondary structure elements in proteins. Analysis and prediction of inter-helix distances. J. Mol. Biol. 233 464–479. [DOI] [PubMed] [Google Scholar]
- Vakser, I.A. 1996a. Long-distance potentials: An approach to the multiple-minima problem in ligand–receptor interaction. Protein Eng. 9 37–41. [DOI] [PubMed] [Google Scholar]
- ———. 1996b. Low-resolution docking: Prediction of complexes for underdetermined structures. Biopolymers 39 455–464. [DOI] [PubMed] [Google Scholar]
- Vakser, I.A. and Jiang, S. 2002. Strategies for modeling the interactions of the transmembrane helices of G-protein coupled receptors by geometric complementarity using the GRAMM computer algorithm. Methods Enzymol. 343 313–328. [DOI] [PubMed] [Google Scholar]
- Vakser, I.A., Matar, O.G., and Lam, C.F. 1999. A systematic study of low-resolution recognition in protein–protein complexes. Proc. Natl. Acad. Sci. 96 8477–8482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walther, D., Eisenhaber, F., and Argos, P. 1996. Principles of helix–helix packing in proteins: The helical lattice superposition model. J. Mol. Biol. 255 536–553. [DOI] [PubMed] [Google Scholar]