

Abstract
Gram-negative bacteria have five major subcellular localization sites: the cytoplasm, the periplasm, the inner membrane, the outer membrane, and the extracellular space. The subcellular location of a protein can provide valuable information about its function. With the rapid increase of sequenced genomic data, the need for an automated and accurate tool to predict subcellular localization becomes increasingly important. We present an approach to predict subcellular localization for Gram-negative bacteria. This method uses the support vector machines trained by multiple feature vectors based on n-peptide compositions. For a standard data set comprising 1443 proteins, the overall prediction accuracy reaches 89%, which, to the best of our knowledge, is the highest prediction rate ever reported. Our prediction is 14% higher than that of the recently developed multimodular PSORT-B. Because of its simplicity, this approach can be easily extended to other organisms and should be a useful tool for the high-throughput and large-scale analysis of proteomic and genomic data.
Keywords: subcellular localization, support vector machine, Gram-negative bacteria, machine-learning method, proteome, genome, n-peptide compositions
The subcellular location of a protein is closely correlated to its biological function (Jensen et al. 2002). With the rapid increase of sequenced genomic data, the need for an automated and accurate tool to predict protein subcellular localization becomes increasingly important. Many efforts have been made to predict protein subcellular localization. There are methods (Nakai and Kanehisa 1992; Nielsen et al. 1997; Emanuelsson et al. 1999, 2000; Nakai 2000) based on the observation that sequences targeted to specific locations rely on the N-terminal sorting or signal sequences. For example, TargetP (Emanuelsson et al. 2000), a useful tool for analysis of signal peptides, predicts protein subcellular localization for eukaryotic sequences. On the other hand, a number of studies (Cedano et al. 1997; Andrade et al. 1998; Reinhardt and Hubbard 1998; Yuan 1999; Chou 2001; Hua and Sun 2001; Chou and Cai 2002) have shown that amino acid compositions are useful in discriminating protein sub-cellular localization sites. Cedano et al. (1997) developed a predictive system ProtLock based on a correlation analysis of the amino acid compositions and the cellular locations for five protein classes. Reinhardt and Hubbard (1998) developed a neural network approach, NNPSL, based on amino acid compositions for both eukaryotic and prokaryotic sequences. For the same data sets, Hua and Sun (2001) also developed SubLoc based on support vector machine (SVM) techniques. Chou (2001) developed approaches based on the pseudo amino acid compositions that include sequence-order information.
Gram-negative bacteria have five major subcellular localization sites that include the cytoplasm, the inner membrane, the outer membrane, the periplasm, and the extracellular space. PSORT I (Nakai and Kanehisa 1991) has been the most widely used predictive tool for Gram-negative bacteria. However, it does not predict extracellular sequences, and its predictive performance reaches only 61% in overall prediction accuracy for a standard data set (Gardy et al. 2003). Recently Gardy et al. (2003), combining different algorithms and input information, developed a multimodular method PSORT-B. This approach comprises six modules examining the query sequence specifically for different characteristics such as amino acid composition, similarity to proteins of known localization, presence of a signal peptide, transmembrane α-helices, and motifs corresponding to specific localizations. This program then constructs a Bayesian network to generate a final probability value for each localization site. This approach yields an overall prediction accuracy of 75% for all location sites, significantly improving on the previous results of PSORT I by 14%. However, despite the great improvement, PSORT-B gives modest prediction for some subcellular locations. For example, it gives a poor predictive accuracy of 58% for periplasmic sequences and of 69% for cytoplasmic sequences. In this work, we present an approach using a single module, the SVM classifier, based on the multiple feature vectors (Yu et al. 2003), to predict the subcellular localization for Gram-negative bacteria.
Materials and methods
Support vector machines
The SVM (Vapnik 1995) tries to find the separating hyper-plane with the largest distance between two classes, measured along a line perpendicular to this hyperplane. However, in practice, these data to be classified may not be linearly separable. To overcome this difficulty, SVM nonlinearly transforms the original input space into a higher dimensional feature space by the so-called kernel functions. When the training data are mapped into a vector in a higher dimensional space, it is possible that data can be linearly separated. In the training process, only part of the training data are used to construct the hyperplane, hence avoiding the overfitting problem usually plaguing other machine learning methods. These data constructing the classifier are called support vectors. Preliminary tests show that the radial basis function (RBF) kernel gives results better than other kernels. Therefore, in this work we use the RBF kernel for all the experiments.
An important issue of optimizing SVMs is the selection of parameters. For SVM training, a few parameters such as the penalty parameter and the kernel parameter of the RBF function must be determined in advance. Choosing optimal parameters for SVM is an important step in SVM design. We use the cross-validation on different parameters for the model selection (Duan et al. 2003). In this work, all SVM calculations are performed by using LIBSVM (Chang and Lin 2001), a general library for support vector classification and regression.
Sequence coding schemes
We have shown in the previous work (Yu et al. 2003) that protein descriptors based on the generalized n-peptide compositions are effective in predicting protein three-dimensional folds. If n = 1, then the n-peptide composition reduces to the amino acid composition, and if n = 2, the n-peptide composition gives dipeptide composition. When n gets larger, the n-peptide compositions will cover more global sequence information, but at the same time, such a coding scheme becomes not only impractical from a computational viewpoint but also undoable from a learning viewpoint. However, the size problem can be overcome if we regroup the amino acids into smaller groups of classes according to their physicochemical properties or structural properties. In this work, we use the notation An to denote the n-peptide composition of amino acids, Fn to denote the reduced amino acid composition in which 20 amino acids are classified into four groups (charged, polar, aromatic and nonpolar), and Xk to denote the partitioned amino acid composition in which the sequence is partitioned into k regions of equal length. Similar sequence coding schemes such as the n-gram hashing function has also been successfully applied to the protein classification (Wu et al. 1992, 1996).
SVM raining and testing
For multiclass SVM classification, we use the one-against-one method (Yu et al. 2003). For five classes of subcellular locations, we can construct 5(5 - 1)/2 = 10 SVM classifiers for a given type of input vector. Each classifier is trained with proteins from two different subcellular locations. For each penalty parameter and kernel parameter, cross-validation combining with the one-against-one method is used for estimating the performance of the model. Therefore, for each model, 10 decision functions share the same parameter. Each protein in the test set will always get a vote from each binary classifier. In this work we use four sequence coding schemes (A1, A2, X4, and F3X5); therefore, we have constructed 10 × 4 = 40 SVM classifiers. We combine votes from these classifiers and use the jury votes to determine the final assignment. In the case of identical votes, we will give more weight to the votes from A1. The general architecture of our predictive system is shown in Figure 1 ▶. Note that the program SubLoc, which is based on amino acid compositions, can be seen as a special case of our predictive system. For convenience, we will refer to our Subcellular Localization Predictive System as CELLO.
Figure 1.

The query sequence is encoded by different coding schemes to obtain (a1a2•), (b1b2•), and (c1c2•), which are used to train the SVM classifiers. We combine votes from these classifiers and use the jury votes to determine the final assignment. We use four coding schemes in this work, which are A1, A2, X4, and F3X5. Because we use the one-against-one methods, we construct 40 SVM classifiers for the prediction of five subcellular localization sites.
Evaluation of the predictive performances
We assess the performance of the classifiers by the leave-one-out tests, which measure the prediction accuracy systematically by singling out one sequence as a test case from the data set during the training process and then testing the classifiers against this single protein. Performances are measured as percentage accuracy and the overall prediction accuracy given by
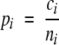 |
(1) |
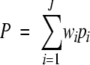 |
(2) |
where ci is the number correctly predicted in the ith sub-cellular location, ni its number of sequences, J the number of locations, w = ni/N and N the total number of sequences. We also use Matthews’ correlation coefficient (MCC; Matthews 1975) as a measure of the predictive performance for each location:
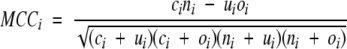 |
(3) |
where ni is the number of correctly predicted sequences not of location i, ui is the number of underpredicted sequences, and oi is the number of overpredicted sequences. The value of MCCi is one for a perfect prediction and zero for a completely random assignment. Following the method of Gardy et al. (2003), for the sequences with dual locations, if one of their locations is predicted, we will consider them as correctly predicted. Such consideration will lead to a slight overestimation of the prediction accuracy (~1% of protein sequences of the data set are multiple localization). At present, CELLO does not predict multiple subcellular sites for protein sequences.
Data sets
We use the same data set of Gardy et al. (2003), extracted from SWISS-PROT release 40.29 (Bairoch and Apweiler 2000). This data set consists of 1443 protein sequences: 1302 proteins localized in a single subcellular site, which are 248 cytoplasmic, 268 inner membrane, 244 periplasmic, 352 outer membrane, and 190 extracellular. This data set also includes a further 141 proteins resident at multiple localization sites: 14 cytoplasmic/inner membrane, 50 inner membrane/periplasmic, and 77 outer membrane/extracellular.
Results and Discussion
In Table 1, we compares the predictive performances of CELLO, PSORT I, PSORT-B, and SubLoc for five subcellular localization sites. Because the original SubLoc for prokaryotes predicts only three subcellular localization sites (cytoplasmic, periplasmic, and extracellular), we will use the A1 SVM classifier for the current data set. The results are obtained with fivefold cross-validation. The overall prediction accuracy of CELLO reaches 89%, which is 14% higher than that of PSORT-B, 28% higher than that of PSORT I, and 10% higher than that of SubLoc. In general, CELLO achieves better prediction accuracy for all subcellular localization sites than do the other approaches. Noticeably, our prediction accuracy for cytoplasmic location (p = 91%) is 22% higher than that of PSORT-B, and for periplasmic location (p = 87%) is 30% higher. These are very significant improvements on the previous results. In CELLO, the only prediction <80% is for extracellular location (p = 79%), but it is still 9% higher than that of PSORT-B. Although the prediction accuracy p offers a convenient measure for predictive performances, one should be careful in drawing hasty conclusion from p, because it overlooks overpredictions (equation 1). MCC (equation 2), taking into account of both under- and overpredictions, offers a complementary measure for the predictive performances. For example, PSORT I gives a remarkable prediction accuracy, p = 95%, for inner membrane, but, due to overpredictions, it gives a less impressive MCC = 0.64, which is much lower than CELLO (MCC = 0.92) and other approaches. CELLO also performs better than other approaches in terms of MCCs. The MCCs of CELLO ranges consistently between 0.80 and 0.92, but the MCCs of PSORT-B deviate greatly among location sites (the difference between MCCs could reach 0.24). PSORT-B gives a particularly poor prediction for periplasmic location (MCC = 0.69), compared with that of CELLO (MCC = 0.80). The inconsistent prediction accuracies of PSORT-B for different localization sites may reflect the uneven predictive performances of different modules in PSORT-B. It is also worth noting that even though PSORT-B uses different modules and input information tuned up for specific localization sites, CELLO, a single module approach, achieves better predictive performances. For example, PSORT-B uses HMMTOP (Tusnady and Simon 1998,Tusnady and Simon 2001) to predict inner membrane sequences, HMMTOP being a well-known hidden Markov model approach specifically designed to identify transmembrane proteins, but CELLO still gives better results, p = 88% and MCC = 0.92, compared with p = 79% and MCC = 0.85 obtained by PSORT-B. It is interesting to note that SubLoc shows a better overall performance than the more complicated multimodular PSORT-B. SubLoc can be seen as a special case of CELLO, because SubLoc uses amino acid compositions as the only input vectors. This surprisingly good predictive performances support previous observations that amino acid composition is indeed a good discriminator for subcellular localization.
Table 1.
The comparison of predictive performances of different approaches in the prediction of subcellular localization for Gram-negative bacteria
| CELLO | PSORT-B | PSORT I | SubLoca | |||||
| Localization | Accuracy | MCC | Accuracy | MCCb | Accuracy | MCCb | Accuracy | MCC |
| Cytoplasmic | 90.7 | 0.85 | 69.4 | 0.79 | 75.4 | 0.58 | 75.0 | 0.74 |
| Inner membrane | 88.4 | 0.92 | 78.7 | 0.85 | 95.1 | 0.64 | 82.8 | 0.89 |
| Periplasmic | 86.9 | 0.80 | 57.6 | 0.69 | 66.4 | 0.55 | 68.9 | 0.71 |
| Outer membrane | 94.6 | 0.90 | 90.3 | 0.93 | 54.5 | 0.47 | 89.1 | 0.86 |
| Extracellular | 78.9 | 0.82 | 70.0 | 0.79 | — | — | 69.5 | 0.78 |
| Overall | 88.9 | — | 74.8 | — | 60.9 | — | 78.5 | — |
a The original SubLoc for prokaryotes predicts only three subcellular localization sites, therefore, we retrained the A1 SVM for this data set using the one-against-one method, which is different from the original one-against-all method.
bMCCs are calculated using the precision and recall values reported in Gardy et al. (2003).
Accuracy is in %.
Conclusion
CELLO is a simple, straightforward implementation of a single module (SVM) based on multiple n-peptide composition to predict subcellular localization. It does not need specialized algorithms or particular input vectors for each subcellular localization site. Compared with CELLO, PSORT-B comprises six modules, with different modules examining specific localization sites, the results of which are then used to construct a Bayesian network to generate a final probability for localization sites. However, it is remarkable that CELLO gives significantly better predictive performances. Because CELLO is a simple straightforward implementation of SVM classifiers, one can easily extend CELLO to other organisms. For example, we have applied our method to a data set comprising 2280 eukaryotic sequences of 12 subcellular localization sites (Chou and Elrod 1999). Our method yields an overall predictive performance of 83% compared with the previous results of 75% (Cai et al. 2002). An interesting question is whether CELLO, trained specifically for Gram-negative bacteria, can also predict heterologous expression of proteins in prokaryotic hosts. The availability of such predictive system would surely be helpful to researchers working on recombinant protein expression. Unfortunately, such study is presently hindered by the relatively scant amount of relevant testing data. However, it is expected that with more data accumulated in the future, such study will become more feasible. We have implemented a CELLO Web server, which is available at http://cello.life.nctu.edu.tw.
Acknowledgments
This work is supported by grants of National Science Council, Taiwan for J.K.H. and C.J.L.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.03479604.
References
- Andrade, M.A., O’Donoghue, S.I., and Rost, B. 1998. Adaptation of protein surfaces to subcellular location. J. Mol. Biol. 276 517–525. [DOI] [PubMed] [Google Scholar]
- Bairoch, A. and Apweiler, R. 2000. The SWISS-PROT protein sequence database and its supplement TrEMBL in 2000. Nucleic Acids Res. 28 45–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai, Y.D. Liu, X.J., Xu, X.B., and Chou, K.C. 2002. Support vector machines for prediction of protein subcellular location by incorporating quasi-sequence-order effect. J. Cell. Biochem. 84 343–348. [DOI] [PubMed] [Google Scholar]
- Cedano, J., Aloy, P., Perez-Pons, J.A., and Querol, E. 1997. Relation between amino acid composition and cellular location of proteins. J. Mol. Biol. 266 594–600. [DOI] [PubMed] [Google Scholar]
- Chang, C.-C. and Lin, C.-J. 2001. LIBSVM: A library for support vector machines. Software. http://www.csie.ntu.edu.tw/~cjlin/libsvm.
- Chou, K.C. 2001. Prediction of protein cellular attributes using pseudo-amino acid composition. Proteins 43 246–255. [DOI] [PubMed] [Google Scholar]
- Chou, K.C. and Cai, Y.D. 2002. Using functional domain composition and support vector machines for prediction of protein subcellular location. J. Biol. Chem. 277 45765–45769. [DOI] [PubMed] [Google Scholar]
- Chou, K.C. and Elrod, D.W. 1999. Protein subcellular location prediction. Protein Eng. 12 107–118. [DOI] [PubMed] [Google Scholar]
- Duan, K., Keerthi, S.S., and Poo, A.N. 2003. Evaluation of simple performance measures for tuning SVM hyperparameters. Neurocomputing 51 41–59. [Google Scholar]
- Emanuelsson, O., Nielsen, H., and von Heijne, G. 1999. ChloroP: A neural network-based method for predicting chloroplast transit peptides and their cleavage sites. Protein Sci. 8 978–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emanuelsson, O., Nielsen, H., Brunak, S., and von Heijne, G. 2000. Predicting subcellular localization of proteins based on their N-terminal amino acid sequence. J. Mol. Biol. 300 1005–1016. [DOI] [PubMed] [Google Scholar]
- Gardy, J.L., Spencer, C., Wang, K., Ester, M., Tusnady, G.E., Simon, I., Hua, S., deFays, K., Lambert, C., Nakai, K., et al. 2003. PSORT-B: Improving protein subcellular localization prediction for Gram-negative bacteria. Nucleic Acids Res. 31 3613–3617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua, S. and Sun, Z. 2001. Support vector machine approach for protein sub-cellular localization prediction. Bioinformatics 17 721–728. [DOI] [PubMed] [Google Scholar]
- Jensen, L.J., Gupta, R., Blom, N., Devos, D., Tamames, J., Kesmir, C., Nielsen, H., Staerfeldt, H.H., Rapacki, K., Workman, C., et al. 2002. Prediction of human protein function from post-translational modifications and localization features. J. Mol. Biol. 319 1257–1265. [DOI] [PubMed] [Google Scholar]
- Matthews, B.W. 1975. Comparison of the predicted and observed secondary structure of T4 phage lysozyme. Biochim. Biophys. Acta. 405 442–451. [DOI] [PubMed] [Google Scholar]
- Nakai, K. 2000. Protein sorting signals and prediction of subcellular localization. Adv. Protein Chem. 2000 277–344. [DOI] [PubMed] [Google Scholar]
- Nakai, K. and Kanehisa, M. 1991. Expert system for predicting protein localization sites in Gram-negative bacteria. Proteins 11 95–110. [DOI] [PubMed] [Google Scholar]
- ———. 1992. A knowledge base for predicting protein localization sites in eukaryotic cells. Genomics 14 897–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen, H., Engelbrecht, J., Brunak, S., and von Heijne, G. 1997. Identification of prokaryotic and eukaryotic signal peptides and prediction of their cleavage sites. Protein Eng. 10 1–6. [DOI] [PubMed] [Google Scholar]
- Reinhardt, A. and Hubbard, T. 1998. Using neural networks for prediction of the subcellular location of proteins. Nucleic Acids Res. 26 2230–2236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tusnady, G.E. and Simon, I. 1998. Principles governing amino acid composition of integral membrane proteins: Application to topology prediction. J. Mol. Biol. 283 489–506. [DOI] [PubMed] [Google Scholar]
- ———. 2001. The HMMTOP transmembrane topology prediction server. Bioinformatics 17 849–850. [DOI] [PubMed] [Google Scholar]
- Vapnik, V. 1995. The nature of statistical learning theory. Springer, New York.
- Wu, C., Whitson, G., McLarty, J., Ermongkonchai, A., and Chang, T.C. 1992. Protein classification artificial neural system. Protein Sci. 1 667–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, C.H., Zhao, S., Chen, H.L., Lo, C.J., and McLarty, J. 1996. Motif identification neural design for rapid and sensitive protein family search. Comput. Appl. Biosci. 12 109–118. [DOI] [PubMed] [Google Scholar]
- Yu, C.-S., Wang, J.-Y., Yang, J.-M., Lyu, P.C., Lin, C.-J., and Hwang, J.-K. 2003. Fine-grained protein fold assignment by support vector machines using generalized npeptide coding schemes and jury voting from multiple-parameter sets. Proteins 50 531–536. [DOI] [PubMed] [Google Scholar]
- Yuan, Z. 1999. Prediction of protein subcellular locations using Markov chain models. FEBS Lett. 451 23–26. [DOI] [PubMed] [Google Scholar]