

Abstract
Cu/Zn superoxide dismutase (SOD) mutations are involved in about 20% of all cases of familial amyotrophic lateral sclerosis (FALS). Recently, it has been proposed that aberrant copper activity may be occurring within SOD at an alternative binding, and cysteine 111 has been identified as a potential copper ligand. Using a commercial source of human SOD isolated from erythrocytes, an anomalous absorbance at 325 nm was identified. This unusual property, which does not compromise SOD activity, had previously been shown to be consistent with a sulfhydryl modification at a cysteine residue. Here, we utilized limited trypsin proteolysis and mass spectrometry to show that the modification has a mass of 32 daltons and is located at cysteine 111. The reaction of SOD with sodium sulfide, which can react with cysteine to form a persulfide group, and with potassium cyanide, which can selectively remove persulfide bonds, confirmed the addition of a persulfide group at cysteine 111. Gel electrophoresis and glutaraldehyde cross-linking revealed that this modification makes the acid-induced denaturation of SOD fully irreversible. Furthermore, the modified protein exhibits a slower acid-induced unfolding, and is more resistant to oxidation-induced aggregation caused by copper and hydrogen peroxide. Thus, these results suggest that cysteine 111 can have a biochemical and biophysical impact on SOD, and suggest that it can interact with copper, potentially mediating the copper-induced oxidative damage of SOD. It will be of interest to study the role of cysteine 111 in the oxidative damage and aggregation of toxic SOD mutants.
Keywords: Cu/Zn superoxide dismutase, SOD, cysteine modification, persulfide, copper, oxidative damage, ALS
Copper/zinc (Cu/Zn) human superoxide dismutase (SOD) is a dimeric protein with two identical subunits arranged in a Greek key configuration with eight β-stands connected by seven loops. Each subunit contains a zinc atom (Zn) whose main role is to stabilize the protein (Rotilio et al. 1972; Mach et al. 1991) and a copper (Cu) atom responsible for its dismutase activity of converting two superoxide molecules into hydrogen peroxide and oxygen. SOD also contains two cysteines (Cys 57 and Cys 148) involved in an intramolecular disulfide bond, two free cysteines (Cys 6 and Cys 111), and a single tryptophan at position 32 (Fig. 1 ▶).
Figure 1.

Ribbon diagram of human SOD showing the antiparallel β-barrel configuration of each monomer (PDB file #1SPD). Labeled are the Cu and Zn ion, the single Trp residue at position 32, the disulfide bond (C57–C146), and the two free cysteines (C6 and C111).
SOD was discovered several decades ago (McCord and Fridovich 1969) prior to recombinant DNA technology, and therefore, for years the human protein was often isolated from red blood cells (McCord and Fridovich 1969). It was noticed that after treatment with chloroform and ethanol to isolate hemoglobin from other blood proteins, the final purified SOD had a unique absorbance band at 325 nm (Carrico and Deutsch 1969). However, the modification did not interfere with SOD’s ability to bind copper at the native site (Calabrese et al. 1975). Furthermore, the resulting electron spin resonance spectrum was very similar to the native protein (Calabrese et al. 1975) and the activity of the protein was not compromised (Briggs and Fee 1978). The modification could be removed by treatment with reducing agents, suggesting that the addition to SOD is an oxidative product occurring at one of the free cysteines (McCord and Fridovich 1969). Attempts to recreate the 325 nm band were mixed. Treatment of SOD with pyrophosphate (Calabrese et al. 1975) and cysteine trisulfide (Briggs and Fee 1978) produced an absorbance band at 325 nm, while treatment with sodium sulfite (Calabrese et al. 1975) and glutathione (Briggs and Fee 1978) caused no change in the absorbance spectra of SOD. Because Cys 6 is not highly solvent exposed, it has been suggested (Hallewell et al. 1991; Liu et al. 2000) that cysteine modification of native SOD occurs exclusively at the solvent-exposed Cys 111.
Over the past 10 years SOD has been linked to some familial cases of amyotrophic lateral sclerosis (FALS), a fatal neurodegenerative disease that kills motor neurons (Cleveland and Rothstein 2001). Over 100 FALS-related SOD missense mutations located throughout the protein have been discovered to date (Andersen et al. 2003), and it has been established that these SOD mutants cause FALS by acquiring a toxic property (Gurney et al. 1994; Ripps et al. 1995; Wong et al. 1995; Reaume et al. 1996). Two general hypotheses have been suggested to explain how SOD mutations may cause FALS (reviewed in Cleveland and Rothstein 2001). The “copper hypothesis” proposes that aberrant chemistry by copper either bound to or released by mutant SOD generates free radicals that cause oxidative damage to motor neurons. Alternatively, the “aggregation hypothesis” suggests that SOD mutations cause the protein to misfold and self-assemble into toxic species. The evidence supporting both, oxidative damage and SOD aggregation in FALS, has led to the suggestion that these aberrant SOD properties may not be exclusive of each other (Valentine and Hart 2003).
It has been recently suggested that Cu may play an adverse role in SOD mutants by binding to an alternative binding site (Bush 2002). This idea is supported by recent evidence of Cu binding to an alternative binding site involving Cys 111 in the FALS-related H46R SOD mutant (Liu et al. 2000). Motivated by our findings of the abnormal 325 nm band in commercially purchased SOD (SOD-C), and the potential link of Cys 111 to FALS, we sought to confirm the location and identity of the modifying group responsible for this spectral property, and determine the biophysical consequences of such a modification. Through mass spectrometry and limited trypsin proteolysis, the site of modification in SOD and the size of the group were determined. Potassium cyanide, which selectively reduces persulfide groups, removed the SOD-C absorbance peak at 325 nm, and sodium sulfide was then used to restore the band. Together, these results revealed a persulfide modification in SOD-C at Cys 111. To further probe the biochemical and biophysical consequences of the Cys 111 modification, we determined its unfolding kinetic, ability to refold after acid-induced denaturation, and its ability to aggregate after treatment with Cu and hydrogen peroxide. We found that these properties were altered in SOD-C compared to unmodified SOD, with potential implications for the role of Cys 111 in FALS.
Results and Discussion
Altered spectral properties of SOD-C is due to the oxidative modification of cysteine
Purified SOD over-expressed in Escherichia coli (SOD-E) exhibits a maximum absorbance at 270 nm that drops off steeply resulting in little to no absorbance above 310 nm. In contrast, SOD-C has a much higher initial absorbance at 250 nm, a steep slope from 250 nm to 300 nm and a band with a maximum absorbance at approximately 325 nm (Fig. 2 ▶). The treatment of SOD-C with 2-mercaptoethanol (BMe) causes the UV/VIS spectrum to become almost identical to that of SOD-E (Fig. 2 ▶). The presence of tryptophan at position 32 makes it possible to probe the fluorescence properties of SOD. SOD-E and SOD-C have a fluorescence maximum at about 350 nm (Fig. 3 ▶), consistent with the largely exposed tryptophan residue. However, the fluorescence intensity of SOD-C was about half of that for SOD-E. This is likely caused by energy transfer due to the Cys-linked adduct, whose absorbance profile overlaps with the fluorescence spectrum of tryptophan. As expected, when SOD-C is treated with BMe, the fluorescence spectrum became exactly the same as the one observed for SOD-E. The sensitivity of the SOD-C modification to a sulfhydryl reductant such as BMe, is consistent with a cysteine modification, as previously suggested for SOD purified from erythrocytes (Calabrese et al. 1975; Briggs and Fee 1978).
Figure 2.

UV Spectra of SOD-E, SOD-C, and SOD-C samples (0.5 mg/ mL in 20 mM PB, pH 7.4) BMe-treated. Wavelength scans were measured from 250 nm to 350 nm.
Figure 3.

Fluorescence spectra of SOD-E, SOD-C, and SOD-C treated with BMe. Emission of SOD samples (0.05 mg/mL, 20 mM PB, pH 7.4) was monitored from 300 to 450 nm after excitation at 278 nm.
We used electrospray mass spectrometry (ES-MS) to determine the mass of the modifying group in SOD-C. ES-MS analysis confirmed the monomer mass of SOD-E to be 15,804 daltons, which corresponds to the predicted mass based on the SOD amino acid sequence (Table 1). The mass determined (15,877) for SOD-C was 73 daltons greater than that of SOD-E, but when treated with BMe it was reduced to 15,845, indicating that the oxidative modification is due to a group with a mass of 32 daltons (Table 1). The remaining 41 daltons difference between SOD-E and BMe-treated SOD-C is accounted for by the N-terminal acetylation of the protein, which only occurs in eukaryotic cells. Having determined the mass of the modifying group, we turned our attention toward identifying the exact location of the modification and the chemical nature of the group.
Table 1.
Mass of SOD proteins with and without BMe
| Protein | Experimental MW (daltons) | Theoretical MW (daltons) |
| SOD-E | 15,804 | 15,804 |
| SOD-C | 15,877 | 15,845a |
| SOD-C + BMe | 15,845 | N/A |
a The additional mass of 41 daltons in SOD-C is due to N-terminal acetylation.
Oxidative modification of SOD-C is localized to cysteine 111
From the crystal structure of SOD it is apparent that Cys 6 is buried while Cys 111 is highly solvent-exposed and accessible to possible modification (Parge et al. 1992). To confirm that Cys 111 is modified in SOD-C, we combined limited trypsin digestion and matrix-assisted laser desorption ionization (MALDI) mass spectrometry (Table 2). The digestion pattern of SOD-E (Fig. 4A ▶) reveals a 3663–3664-dalton proteolytic fragment consistent with residues 80–115 containing an unmodified Cys 111. The digestion pattern of SOD-C not treated with BMe (Fig. 4B ▶) revealed a 3663-dalton peak corresponding to the unmodified 80–115 fragment, and a new 3695-dalton fragment that is 32 daltons larger in mass. When the modified SOD-C was treated with BMe (Fig. 4C ▶), the 3695-dalton fragment disappears. These results directly establish that the oxidative modification in SOD-C is occurring at Cys 111 (Table 2). The presence of both the 3695- and 3663-dalton fragments at almost identical intensity, suggests that only one of the two Cys 111 residues per dimer has been modified. This is consistent with previous work with H46R SOD, where only one Cys 111 per dimer can be modified by 5,5-Dithiobis (2-nitrobenzoic acid) and 4-vinylpyridine (Liu et al. 2000), presumably due to steric effects.
Table 2.
Trypsin digestion fragments of SOD
| Protein | Fragment | Calculated MW (daltons) | Experimental MW (daltons) |
| SOD-E | 4–36 | 3496 | 3495 |
| 37–69 | 3462 | 3462 | |
| 80–115 | 3664 | 3664 | |
| 92–128 | 3850 | 3849 | |
| SOD-C | 4–36 | 3496 | 3495 |
| 37–69 | 3462 | 3462 | |
| 80–115 | 3664 | 3663 | |
| 80–115a | 3664 | 3695 | |
| 92–128 | 3850 | 3849 | |
| SOD-C + BMe | 4–36 | 3496 | 3495 |
| 37–69 | 3462 | 3462 | |
| 80–115 | 3664 | 3664 | |
| 92–128 | 3850 | 3849 |
a Fragment containing the modification in SOD-C.
Figure 4.
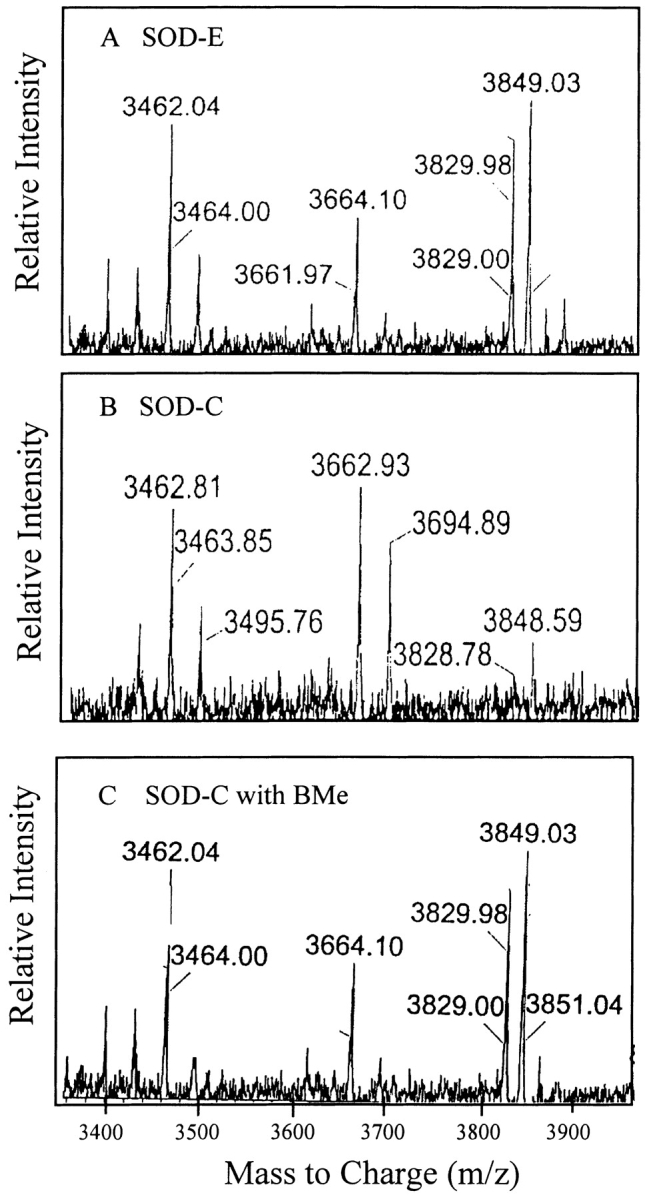
The location of the SOD-C modification was determined by using MALDI MS. Postdigestion fragment patterns were observed for SOD-E (A) modified SOD-C (B) and reduced SOD-C (C). All samples were desalted with Zip Tip and analyzed at 1.0 mg/mL SOD in 90% acetonitrile 0.1% trifluoroacetic acid.
Chemical analyses suggest a persulifide modification at Cys 111
Previous attempts to recreate the unique absorbance were only successful when the samples were treated with cysteine trisulfide (Briggs and Fee 1978) and pyrophosphate (Calabrese et al. 1975). Therefore, it was speculated that the modification was a persulfide or polysulfide group (Briggs and Fee 1978). Based on our ES-MS data, which indicates a decrease of about 32 daltons in the mass of SOD-C upon reduction, possible cysteine modifications that could account for this change in mass include, nitroso thiol (S-NO, 30 daltons), sulfinic acid (S-O-O, 32 daltons), and persulfide (S-S, 32 daltons). Nitrosilation of proteins is BMe-reversible, but has an absorbance maximum at 335 nm (Gergel and Cederbaum 1996; Simon et al. 1996) instead of 325 nm. A sulfinic acid modification has an absorbance maximum at 325 nm, but it is not BMe reversible (Barrett et al. 1999). The persulfide modification, which has been previously identified in rhodanese (Panda and Horowitz 2000), is BMe-reversible and its UV/VIS absorbance spectrum has a maxima at 323 nm. Thus, the properties of the SOD modification suggest that it is likely due to a persulfide group. To confirm this we treated the unmodified SOD-E with sodium sulfide (Na2S), which can react with cysteine residues to form persulfide groups (Berni et al. 1991), and were able to reproduce, although to a lesser extent, the distinctive bump at 325 nm and the increase in slope from 290 to 250 nm (Fig. 5B ▶). When an SOD-C sample was treated with potassium cyanide (KCN), which reacts selectively with persulfide modifications at cysteine residues (Calabrese et al. 1975), both the absorbance band at 325 nm and the increase in slope at from 290 nm to 250 nm disappeared (Fig. 5A ▶). Thus, the mass, spectroscopic features, and chemical reactivity of the modifying group in SOD-C, are consistent with a persulfide group modification at Cys 111.
Figure 5.

SOD-C modification identified through addition and selective removal of the Cys 111 modifying group. (A) SOD-C treated with KCN to selectively remove persulfide group. (B) SOD-E and SOD-E treated with Na2S to add a persulfide group. The SOD samples were at 1.0 mg/mL in 20 mM PB (PH 7.4).
Persulfide-modified SOD-C exhibits altered biophysical properties
Irreversible acid-induced unfolding of modified SOD-C
We observe that in the absence of reductant, dimeric SOD is resistant to denaturation by sodium dodecyl sulfate (SDS). When the SOD sample is not heated, the protein migrates with an apparent molecular weight (MW) of 60 kD instead of 32 kD, consistent with the reduced binding of SDS to SOD that results in a diminished overall net negative charge (Fig. 6A ▶, lane 2). When SOD-E is incubated overnight at pH 2 and then returned back to pH 7 (pH7 → 2 → 7), approximately 25% of the SOD-E appears as a dimer and the other 75 % of the protein migrates as the monomer (Fig. 6A ▶, lane 3). The presence of BMe makes no difference on the migration of SOD-E on the gel or the percent of folding reversibility (Fig. 6A ▶, lanes 4,5). In contrast, when SOD-C is incubated at pH 2 overnight and then returned to pH 7, no native dimer band is observed (Fig. 6A ▶, lane 7), indicating that the pH-induced denaturation of SOD-C is 100% irreversible. Interestingly, about 80% of SOD-C migrates as the monomer while the other 20% migrates as a 32-kD specie due to the formation of an intermolecular disulfide cross-linked SOD-C dimer, as implied by the disappearance of this dimeric band when the sample is treated with BMe (data not shown). Remarkably, when SOD-C is first treated with BMe before unfolding, about 80% of the sample appeared as a native-like SDS-resistant dimer, while 20% ran as unfolded monomer (Fig. 6A ▶, lane 9). Surprisingly, for reasons that are not yet known, in the presence of BMe the yield of SOD-C refolding is much higher than that of SOD-E.
Figure 6.

Reversibility of acid-induced SOD unfolding analyzed by SDS-PAGE. (A) SOD samples were unfolded at pH 2 and returned to pH 7 as described in Materials and Methods. Unheated samples were mixed with SDS sample buffer lacking reductant and analyzed on a 14% acrylamide gel. Labels represent the native SOD dimer (D) (which migrates as a higher MW species), and monomeric SOD bands (M). (B) Following identical unfolding/refolding treatments as in (A), the samples were cross-linked with glutaraldehyde, mixed with sample buffer containing BMe, boiled, and analyzed on a 14% acrylamide gel.
Because metal-deficient SOD is not SDS resistant and migrates as a monomer on SDS-PAGE, it is not possible to determine from the experiments shown in Figure 6A ▶ whether any dimeric apo SOD may be present after rising the pH back to 7. It is plausible that acid-unfolded SOD may refold and dimerize, but not be able to properly rebind Cu and Zn. To help address this issue, we used glutaraldehyde cross-linking to trap the species present after the pH was raised from 2 back to 7 (Fig. 6B ▶). While overall the cross-linking data is quite consistent with Figure 6A ▶, several features are worth noting. First, it is clear that a significant amount of SOD-E is not able to refold back into a dimer, although the sharp and darker dimer bands suggests that the refolding yield is higher than suggested in Figure 6A ▶. Second, the presence of a broad dimer band in lane 7 indicates that while metal binding is irreversible for SOD-C, some apo SOD dimer may be reformed that is not being detected on regular SDS-PAGE. Finally, the similar cross-linking pattern in lanes 8 and 9 (Fig. 6B ▶) and the sharp bands confirm the results shown in Figure 6A ▶ that when SOD-C is treated with BMe to remove the modification at Cys 111, the protein is largely able to refold and rebind Cu and Zn, thereby becoming SDS-resistant. It is puzzling how an extra sulfur atom at the surface of a 16,000-dalton protein can have such a dramatic effect on the reversibility of acid-unfolded SOD-C. We speculate that the irreversibility of SOD-C may be caused by the formation of nonnative intramolecular disulfide bonds and disulfide cross-linked dimer. Also, it is unclear at this time why SOD-E does not achieve a similar level of reversibility (Fig. 6 ▶, lane 5), but it is possible that it may be related to the lack of N-terminal acetylation. Because the N-terminus of SOD is near Cys111 and the dimer interface, it is possible that the acetyl group in SOD-C may interact intra- or intermolecularly with Cys111 or across the dimer interface to favor the correct folding of SOD-C.
Increased kinetic stability of SOD-C
The resistance of SOD to SDS-induced denaturation suggests that SOD may be very rigid and kinetically stable. Kinetically stable proteins are characterized by their very slow unfolding rate under native conditions, virtually being trapped in their native conformation due to a high-energy barrier towards unfolding (Jaswal et al. 2002). Because the SOD-C modification is not pH labile, the kinetic stability of SOD was probed by measuring the kinetics of acid-induced (pH 2.0–2.1) unfolding. After manually mixing the SOD samples into acidic solution, the tryptophan fluorescence was monitored over time, and the rate of unfolding was determined. SOD-E and SOD-C treated with BMe unfold in a single step with similar rates of 1.1 × 10−2 sec−1 and 2.2 × 10−2 sec−1, respectively (Fig. 7 ▶). In contrast, the unfolding of SOD-C consisted of a faster phase with a rate similar to SOD-E, and a slower phase with a rate of 3.0 × 10−3 sec−1, with relative amplitudes of 46% and 54%, respectively. Thus, unfolding of SOD-C is about four times slower due to the persulfide modification. The mechanism by which the persulfide modification in SOD-C results in the appearance of a slow second unfolding phase is not clear. It is possible that the enhanced kinetic stability of persulfide-modified SOD, as indicated by the appearance of the slower unfolding phase, might be due to inter- or intra-molecular hydrogen bonding. Although the Cys 111 residues in the SOD dimer are about 9 Å from each other and near the dimer interface (Parge et al. 1992), it seems unlikely that Cys 111 can interact with the other subunit without some local conformational change. Alternatively, the extra sulfur atom in the persulfide group may be involved in intramolecular hydrogen bonding, leading to an increase in the thermodynamic, and consequently, kinetic stability of SOD-C.
Figure 7.

Unfolding kinetics of SOD measured by fluorescence spectroscopy. Unfolding of SOD-E, modified SOD-C and reduced SOD-C was monitored following manual mixing in 0.01 N HCl (pH 2). SOD samples were at 0.05 mg/mL. Fluorescence was monitored at 355 nm after excitation at 278 nm.
Persulfide modification blocks the copper-induced aggregation of SOD
There is evidence of a link between oxidative damage and aggregation of SOD mutants in FALS (Bowling et al. 1995; Andrus et al. 1998; Rakhit et al. 2002; Chung et al. 2003). Because oxidative damage of SOD can occur via hydroxyl radicals upon incubation with Cu and H2O2 (Stadtman and Berlett 1997), and because Cys 111 has been proposed to be part of an alternative copper binding-site in FALS mutants, we decided to investigate the effect of the Cys 111 modification on the ability of SOD-C to aggregate in the presence of Cu and H2O2. We monitored the aggregation of SOD by measuring the light scattered at 90° using a fluorescence spectrometer (Fig. 8 ▶). The SOD-E sample lacking any modification exhibited modest aggregation with the scattering increasing at a rate 4.6 × 10−4 sec−1. Surprisingly, SOD-C containing the persulfide is resistant to aggregation under the same experimental conditions. As expected, when the SOD-C sample was treated with BMe, the aggregation profile converts to the same rate as the SOD-E sample. Oxidative-induced aggregation has previously been shown to occur in various proteins, including human relaxin (Khossravi et al. 2000) and hamster prion protein (Turnbull et al. 2003). Rakhit et al. utilizing ascorbate and copper to generate hydroxyl radicals were able to show that zinc-deficient SOD mutants suffer oxidative damage, leading to SOD aggregation (Rakhit et al. 2002). The inhibition of SOD-C aggregation by the persulfide modification appears to support the hypothesis that Cys 111 may function as an alternative copper binding site.
Figure 8.

Oxidative aggregation of SOD-E, modified SOD-C and reduced SOD-C measured by light scattering. Samples were at 0.3 mg/mL in 20 mM Tris (pH 7.4) and contained 1.0 mM Cu2SO4. Oxidative aggregation was induced by rapidly mixing in 1.0 mM H2O2. Light scattering was measured with excitation and emission wavelengths set at 350 nm.
Potential implications for FALS
Postmortem brain deposits of FALS patients have been shown to contain the presence of oxidized SOD (Bowling et al. 1995), suggesting that perhaps oxidative damage is playing a direct role in disease progression. Recently, we showed (Chung et al. 2003) that the oxidation of apo wild-type SOD and several FALS-related mutants results in the formation of potentially toxic pore-like structures reminiscent of those seen β-amyloid and α-synuclein, which are implicated in Alzheimer’s and Parkinson’s diseases, respectively (Lashuel et al. 2002). In this study, we have shown that in holo WT SOD, persulfide modification to Cys 111 inhibits its oxidative-induced aggregation. It will be of much interest to determine what effect a persulfide modification would have on the oxidative-induced aggregation of FALS mutants, as this could lead to a target region within SOD for the design of molecule to block this potentially toxic effect.
Materials and methods
Reagents
Human SOD (SOD-C), purchased from Sigma Chemical Co., was used in this study. The lyophilized powder was dissolved with 20 mM potassium phosphate buffer (PB) at pH 7.0 and used without any further processing. Activities of the purified and purchased SOD samples were measured using the Calbiochem SOD assay kit based on the spectroscopic method developed by Nebot et al. (Nebot et al. 1993). Unless noted, all other chemicals were from Fisher Scientific.
Expression and purification of SOD
Human SOD was cloned into PET21 vector between BamHI and NcoI sites and transformed into E. coli strain BL21 pLysS-competent cells as previously described (Crow et al. 1997). E. coli cells were grown at 37°C in Luria broth (LB) media until the O.D.600 was 0.8, at which time cells were induced at 30°C through the addition of 0.3 mM isopropyl-D-thiogalactoside and supplemented with 1.0 mM CuSO4 and 0.05 mM ZnSO4. After 90 min of induction, cells were pelleted by centrifugation for 15 min (3200 g) and resuspended in 100 mL 20 mM PB (pH 7.4), and frozen at −80°C. Cells were lysed by two freeze/thaw cycles, digested with DNAase and RNAase to cleave nucleotide, and centrifuged (20,000 × g) for 30 min to pellet debris. The protein extract was loaded onto a DE52 ion exchange resin (Whatman) and eluted with a linear gradient of 5–200 mM PB. Fractions containing SOD were pooled, concentrated, and dialyzed against 10 mM PB with 100 mM NaCl prior to loading on an ACA54 size exclusion resin (Sigma). Fractions containing purified SOD were pooled, concentrated and dialyzed against 20 mM PB prior to use.
Sample preparation
The metal content of SOD expressed in E. coli (SOD-E) and SOD-C was determined using the metal chelater 4-pyridylazoresorcinol (PAR) as outlined by Crow et al. (1997) and confirmed using atomic absorption (Hitachi Instruments). The SOD proteins used in these studies were in their holo active form and contained above 90% of Cu and Zn. UV/Vis wavelength scans of SOD revealed the absorption spectra characteristic of SOD-C purified from erythrocytes using chloroform and ethanol precipitation (McCord and Fridovich 1969). The extinction coefficient for SOD purified by this method has been previously determined to be 29,000 M−1 cm−1 at 265 nm (based on the MW of the dimer) (Briggs and Fee 1978). The concentration of SOD-E purified without oxidative conditions was determined using an extinction coefficient of 10,300 M−1 cm−1 at 280 nm (Battistoni et al. 1998). Concentrations were confirmed using the BCA protein assay (Pierce) with bovine serum albumin as a standard.
BMe-treated SOD samples were incubated in 20 mM PB with 2.0 mM BMe for 30 min following dialysis against PB overnight. For unfolding and refolding of SOD, samples at pH 7.0 were measured initially in 20 mM PB. To unfold, the samples were brought to pH 2.0 with 6 N HCl, and left to equilibrate for 1.5 to 2.0 h. At this time the spectra of the unfolded protein were measured. The unfolded sample was returned to pH 7.0 with 5 N NaOH and left to equilibrate for 1.5 to 2.0 h before a final measurement was taken.
SOD absorbance and fluorescence spectroscopy
UV-Vis absorbance measurements were recorded from 250 nm to 350 nm with a Hitachi 3010 dual-beam spectrophotometer using a cell with a 1.0-cm path length. The final concentration of SOD samples was 0.4 mg/mL in 20 mM PB. Tryptophan fluorescence wavelength scans of SOD samples were recorded with a Hitachi F4500 spectrophotometer at 20°C in a 1.0-cm path length quartz cuvette, using an excitation and emission bandwidth of 5 nm and 10 nm, respectively. Excitation and emission wavelengths were 280 nm and 355 nm, respectively. The final concentration of SOD samples was 0.05 mg/mL in 20 mM in PB.
Mass spectrometry
To determine the exact mass of the SOD-E, SOD-C and the BMe-treated SOD-C, samples were analyzed using electrospray mass spectrometry. Samples prepared at 0.5 mg/mL in 20 mM PB were first desalted with a C4 Zip Tip (Millipore). Samples eluted from the Zip Tip with 90% acetonitrile and 0.1% TFA were analyzed at the Mass Spectrometry Center of the University of Massachusetts at Amherst, using a Brucker daltonics Esquire-LC Mass Spectrometer set up with an ESI source and positive ion polarity. Scanning was carried out between 500 m/z and 2200 m/z, and the final spectra obtained were an average of 25 individual spectra.
Oxidative modification of SOD-C was located utilizing MALDI MS following trypsin proteolysis. SOD samples (0.3 mg/mL SOD, 50 mM Tris hydroxymethyl aminomethane hydrochloride (Tris), pH 8.1) were boiled for 4 min and rapidly cooled on ice. Trypsin was added at trypsin:protein ratio of 1:30 and incubated for 16 h at 37°C, at which time a second dose of (1:30) trypsin was added and left to incubate further for 6 h. Digestion was stopped by making the sample up to 0.2% TFA. Samples were mixed with equal volume of sinapinic acid and allowed to dry before analyzing samples.
Persulfide formation with Na2S and its removal with KCN
Persulfide formation of holo WT SOD-E was attempted on SOD samples at 0.5 mg/mL in 20 mM PB. Degassed samples were treated with 1.0 mM Na2S for 16 h at room temperature. Afterwards, the samples were analyzed by UV/Vis spectroscopy. To confirm that the adduct formed in SOD-C and the SOD-E modified by Na2S was a persulfide group, the samples were incubated with 10 mM KCN, 50 mM carbonate buffer, pH 9.5 for 18 h at room temperature. Following the specific removal of persulfides with KCN, the UV/Vis scans were repeated.
SOD quaternary structural changes monitored by SDS-PAGE and glutaraldehyde cross-linking
To examine variations in SOD quaternary structure, we used an SDS-PAGE method that takes advantage of holo SOD’s resistance to dissociation by SDS. A dimeric SOD sample at 0.2 mg/mL in 20 mM PB was incubated with SDS sample buffer (2X nonreducing) at room temperature and loaded unboiled, onto a 14% SDS-PAGE gel. Because SOD is resistant to dissociation by SDS, it migrated as a dimer. In contrast, SOD dimer was denatured after incubation at low pH and migrated as a monomer. Because SOD lacking the metals is not resistant to SDS and runs as a monomer on SDS-PAGE, glutaraldehyde cross-linking was employed to examine the possible reversibility of SOD dimerization that may have been missed by SDS-PAGE. SOD samples were made at 0.4 mg/mL in 20 mM PB, pH 7.4. Cross-linking was carried out at 0.5% (w/v) glutaraldehyde for 1.0 min, at which time the reaction was stopped with 0.7% sodium borohydride and then precipitated with 0.1% deoxycholic acid and 1.0% trichloroacetic acid. The precipitate was resuspended in 20 mM PB, mixed with equal volume 2× SDS-PAGE sample buffer containing dithiothreitol, boiled for 5 min, and analyzed using 16% SDS-PAGE.
Unfolding kinetics of SOD
Unfolding kinetics of SOD samples was monitored following manual mixing with 0.01 N HCl (pH 2). The final pH was about 2.1. Fluorescence measurements were recorded for 30 min with a Hitachi F4500 spectrophotometer at 20°C in a 1.0-cm path length quartz cuvette, using an excitation wavelength of 280 nm, and an emission wavelength 355 nm. The final concentration of SOD samples was 0.05 mg/mL in 20 mM PB (pH 7.4). Unfolding kinetics was monitored for SOD-E and SOD-C in the absence and in the presence of 2.0 mM BMe.
Oxidation-induced aggregation of SOD monitored by light scattering
To address the effect of Cys 111 modification on the oxidative-induced aggregation, SOD samples were oxidized in the presence of Cu and H202. SOD samples were prepared at 0.3 mg/mL in 20 mM Tris (pH 7.4), and contained 1.0 mM Cu2SO4. Control light scattering was measured in the absence of H2O2 and no change in signal was observed. Oxidative aggregation was induced by rapidly adding H2O2 to a final concentration of 1.0 mM. Light scattering was measured at 37°C for 60 min with excitation and emission wavelengths set at 350 nm, and using a 2-nm bandwidth.
Acknowledgments
Our thanks to Dr. Dmitri Zagorevski for help with MALDI experiments. The MALDI-TOF mass spectrometer was obtained through NSF Grant CHE-0078056. This work was supported by grants from NIH (R01NS42915) and The ALS Association, and a Research Corporation Innovation Award to W.C.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Abbreviations
ALS, amyotrophic lateral sclerosis
ANS, 1-anilinonaphthalene-8-sulfonate
BMe, 2-mercaptoethanol
EDTA, ethylenediamine tetraacetic acid
ES-MS, electrospray mass spectrometry
FALS, familial amyotrophic lateral sclerosis
GuHCl, guanidine hydrochloride
H2O2, hydrogen peroxide
KCN, potassium cyanate
MALDI, matrix-assisted laser desorption ionization mass spectrometry
MW, molecular weight
NTA, nitrilotriacetic acid
PAR, 4-pyridylazaresorcinol
PB, potassium phosphate buffer
SDS-PAGE, sodium dodecyl sulfate-polyacrylamide gel electrophoresis
SOD, Cu/Zn superoxide dismutase
WT, wild type
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.03576904.
References
- Andersen, P.M., Sims, K.B., Xin, W.W., Kiely, R., O’Neill, G., Ravits, J., Pioro, E., Harati, Y., Brower, R.D., Levine, J.S., et al. 2003. Sixteen novel mutations in the Cu/Zn superoxide dismutase gene in amyotrophic lateral sclerosis: A decade of discoveries, defects and disputes. Amyotroph. Lateral Scler. Other Motor Neuron Disord. 4 62–67. [DOI] [PubMed] [Google Scholar]
- Andrus, P.K., Fleck, T.J., Gurney, M.E., and Hall, E.D. 1998. Protein oxidative damage in a transgenic mouse model of familial amyotrophic lateral sclerosis. J. Neurochem. 71 2041–2048. [DOI] [PubMed] [Google Scholar]
- Barrett, W.C., DeGnore, J.P., Konig, S., Fales, H.M., Keng, Y.F., Zhang, Z.Y., Yim, M.B. and Chock, P.B. 1999. Regulation of PTP1B via glutathionylation of the active site cysteine 125. Biochemistry 38 6699–6705. [DOI] [PubMed] [Google Scholar]
- Battistoni, A., Folcarelli, S., Cervoni, L., Polizio, F., and Desideri, A. 1998. Role of the dimeric structure in the Cu, Zn superoxide dismutase. J. Biol. Chem. 273 5655–5661. [DOI] [PubMed] [Google Scholar]
- Berni, R., Musci, G., Pallini, R., and Cannella, C. 1991. Chemical modification of rhodanese with sulphite. Free Radic. Res. Commun. 15 203–209. [DOI] [PubMed] [Google Scholar]
- Bowling, A.C., Barkowski, E.E., McKenna-Yasek, D., Sapp, P., Horvitz, H.R., Beal, M.F. and Brown, R.H. 1995. Superoxide dismutase concentration and activity in familial amyotrophic lateral sclerosis. J. Neurochem. 64 2366–2369. [DOI] [PubMed] [Google Scholar]
- Briggs, R.G. and Fee, J.A. 1978. Sulfhydryl reactivity of human erythrocyte superoxide dismutase. On the origin of the unusual spectral properties of the protein when prepared by a procedure utilizing chloroform and ethanol for the precipitation of hemoglobin. Biochim. Biophys. Acta 537 100–109. [DOI] [PubMed] [Google Scholar]
- Bush, A.I. 2002. Is ALS caused by an altered oxidative activity of mutant superoxide dismutase? Nat. Neurosci. 5 919. [DOI] [PubMed] [Google Scholar]
- Calabrese, L., Federici, G., Bannister, W.H., Bannister, J.V., Rotilio, G., and Finazzi-Agro, A. 1975. Labile sulfur in human superoxide dismutase. Eur. J. Biochem. 56 305–309. [DOI] [PubMed] [Google Scholar]
- Carrico, R.J. and Deutsch, H.F. 1969. Isolation of human hepatocuprein and cerebrocuprein. Their identity with erythrocuprein. J. Biol. Chem. 244 6087–6093. [PubMed] [Google Scholar]
- Chung, J., Yang, H., de Beus, M.D., Ryu, C.Y., Cho, K., and Colón, W. 2003. Cu/Zn superoxide dismutase can form pore-like structures. Biochem. Biophys. Commun. 312 873–876. [DOI] [PubMed] [Google Scholar]
- Cleveland, D.W. and Rothstein, J.D. 2001. From Charcot to Lou Gehrig: Deciphering selective motor neuron death in ALS. Nat. Rev. Neurosci. 2 806–819. [DOI] [PubMed] [Google Scholar]
- Crow, J.P., Sampson, J.B., Zhuang, Y., Thompson, J.A., and Beckman, J.S. 1997. Decreased zinc affinity of amyotrophic lateral sclerosis-associated superoxide dismutase mutants leads to enhanced catalysis of tyrosine nitration by peroxynitrite. J. Neurochem. 69 1936–1944. [DOI] [PubMed] [Google Scholar]
- Gergel, D. and Cederbaum, A.I. 1996. Inhibition of the catalytic activity of alcohol dehydrogenase by nitric oxide is associated with S nitrosylation and the release of zinc. Biochemistry 35 16186–16194. [DOI] [PubMed] [Google Scholar]
- Gurney, M.E., Pu, H., Chiu, A.Y., Dal Canto, M.C., Polchow, C.Y., Alexander, D.D., Caliendo, C., Hentati, A., Kwon, Y.W., Deng, H., et al. 1994. Motor neuron degeneration in mice that express a human Cu, Zn superoxide dismutase mutation. Science 264 1772–1775. [DOI] [PubMed] [Google Scholar]
- Hallewell, R.A., Imlay, K.C., Lee, P., Fong, N.M., Gallegos, C., Getzoff, E.D., Tainer, J.A., Cabelli, D.E., Tekamp-Olson, P., Mullenbach, G.T., et al. 1991. Thermostabilization of recombinant human and bovine CuZn super-oxide dismutases by replacement of free cysteines. Biochem. Biophys. Commun. 181 474–480. [DOI] [PubMed] [Google Scholar]
- Jaswal, S.S., Sohl, J.L., Davis, J.H., and Agard, D.A. 2002. Energetic landscape of α-lytic protease optimizes longevitiy through kinetic stability. Nature 415 343–346. [DOI] [PubMed] [Google Scholar]
- Khossravi, M., Shire, S.J., and Borchardt, R.T. 2000. Evidence of the involvement of histidine A(12) in the aggregation and precipitation of human relaxin induced by metal-catalyzed oxidation. Biochemistry 39 5876–5885. [DOI] [PubMed] [Google Scholar]
- Lashuel, H.A., Hartley, D., Petre, B.M., Walz, T., and Lansbury, Jr., P.T. 2002. Neurodegenerative disease: Amyloid pores from pathogenic mutations. Nature 418 291. [DOI] [PubMed] [Google Scholar]
- Liu, H., Zhu, H., Eggers, D., Nersissian, A., Faull, K., Goto, J., Ai, J., Sanders-Loehr, J., Gralla, E., and Valentine, J. 2000. Copper(2+) binding to the surface residue cysteine 111 of His46Arg human copper–zinc superoxide dismutase, a familial amyotrophic lateral sclerosis mutant. Biochemistry 39 8125–8132. [DOI] [PubMed] [Google Scholar]
- Mach, H., Dong, Z., Middaugh, C.R., and Lewis, R.V. 1991. Conformational stability of Cu,Zn-superoxide dismutase, the apoprotein, and its zinc-substituted derivatives: Second-derivative spectroscopy of phenylalanine and tyrosine residues. Arch. Biochem. Biophys. 287 41–47. [DOI] [PubMed] [Google Scholar]
- McCord, J.M. and Fridovich, I. 1969. Superoxide dismutase. An enzymic function for erythrocuprein (hemocuprein). J. Biol. Chem. 244 6049–6055. [PubMed] [Google Scholar]
- Nebot, C., Moutet, M., Huet, P., Xu, J.-Z., Yadan, J.-C., and Chaudiere, J. 1993. Spectrophotometric assay of superoxide dismutase activity based on the activated autooxidation of a tetracyclic cathecol. Anal. Biochem. 214 442–451. [DOI] [PubMed] [Google Scholar]
- Panda, M. and Horowitz, P.M. 2000. Active-site sulfhydryl chemistry plays a major role in the misfolding of urea-denatured rhodanese. J. Protein Chem. 19 399–409. [DOI] [PubMed] [Google Scholar]
- Parge, H.E., Hallewell, R.A., and Tainer, J.A. 1992. Atomic structures of wild-type and thermostable mutant recombinant human Cu,Zn superoxide dismutase. Proc. Natl. Acad. Sci. 89 6109–6113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakhit, R., Cunningham, P., Furtos-Matei, A., Dahan, S., Qi, X.F., Crow, J.P., Cashman, N.R., Kondejewski, L.H., and Chakrabartty, A. 2002. Oxidation-induced misfolding and aggregation of superoxide dismutase and its implications for amyotrophic lateral sclerosis. J. Biol. Chem. 277 47551–47556. [DOI] [PubMed] [Google Scholar]
- Reaume, A.G., Elliot, J.L., Hoffman, E.K., Kowall, N.W., Ferrante, R.J., Siwek, D.F., Wilcox, H.M., Flood, D.G., Beal, M.F., Brown, Jr., R.H., et al. 1996. Motor neurons in Cu/Zn superoxide dismutase-deficient mice develop normally but exhibit enhanced cell death after axotomy. Nat. Genet. 13 43–47. [DOI] [PubMed] [Google Scholar]
- Ripps, M.E., Huntley, G.W., Hof, P.R., Morrison, J.H., and Gordon, J.W. 1995. Transgenic mice expressing an altered murine superoxide-dismutase gene provide an animal-model of amyotrophic-lateral-sclerosis. Proc. Natl. Acad. Sci. 92 689–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rotilio, G., Calabrese, L., Bossa, F., Barra, D., Agro, A.F., and Mondovi, B. 1972. Properties of the apoprotein and role of copper and zinc in protein conformation and enzyme activity of bovine superoxide dismutase. Biochemistry 11 2182–2187. [DOI] [PubMed] [Google Scholar]
- Simon, D.I., Mullins, M.E., Jia, L., Gaston, B., Singel, D.J., and Stamler, J.S. 1996. Polynitrosylated proteins: Characterization, bioactivity, and functional consequences. Proc. Natl. Acad. Sci. 93 4736–4741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadtman, E.R. and Berlett, B.S. 1997. Reactive oxygen-mediated protein oxidation in aging and disease. Chem. Res. Toxicol. 10 485–494. [DOI] [PubMed] [Google Scholar]
- Turnbull, S., Tabner, B.J., Brown, D.R., and Allsop, D. 2003. Copper-dependent generation of hydrogen peroxide from the toxic prion protein fragment PrP106–126. Neurosci. Lett. 336 159–162. [DOI] [PubMed] [Google Scholar]
- Valentine, J.S. and Hart, P.J. 2003. Misfolded CuZnSOD and amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. 100 3617–3622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong, P.C., Pardo, C.A., Borchelt, D.R., Lee, M.K., Copeland, N.G., Jenkins, N.A., Sisodia, S.S., Cleveland, D.W., and Price, D.L. 1995. An adverse property of a familial ALS-linked SOD1 mutation causes motor neuron disease characterized by vacuolar degeneration of mitochondria. Neuron 14 1105–1116. [DOI] [PubMed] [Google Scholar]