

Abstract
To further understand oligomeric protein assembly, the folding and unfolding kinetics of the H3–H4 histone tetramer have been examined. The tetramer is the central protein component of the core nucleosome, which is the basic unit of DNA compaction into chromatin in the eukaryotic nucleus. This report provides the first kinetic folding studies of a protein containing the histone fold dimerization motif, a motif observed in several protein–DNA complexes. Previous equilibrium unfolding studies have demonstrated that, under physiological conditions, there is a dynamic equilibrium between the H3–H4 dimer and tetramer species. This equilibrium is shifted predominantly toward the tetramer in the presence of the organic osmolyte trimethylamine-N-oxide (TMAO). Stopped-flow methods, monitoring intrinsic tyrosine fluorescence and far-UV circular dichroism, have been used to measure folding and unfolding kinetics as a function of guanidinium hydrochloride (GdnHCl) and monomer concentrations, in 0 and 1 M TMAO. The assignment of the kinetic phases was aided by the study of an obligate H3–H4 dimer, using the H3 mutant, C110E, which destabilizes the H3–H3′ hydrophobic four-helix bundle tetramer interface. The proposed kinetic folding mechanism of the H3–H4 system is a sequential process. Unfolded H3 and H4 monomers associate in a burst phase reaction to form a dimeric intermediate that undergoes a further, first-order folding process to form the native dimer in the rate-limiting step of the folding pathway. H3–H4 dimers then rapidly associate with a rate constant of ≥107 M−1sec−1 to establish a dynamic equilibrium between the fully assembled tetramer and folded H3–H4 dimers.
Keywords: kinetics, circular dichroism, fluorescence, chemical denaturation
Studies of the folding mechanisms of monomeric proteins have been a major focus in efforts to describe the protein folding code: how a protein’s primary structure encodes its unique, three-dimensional structure as well as the pathway to this native state from an unfolded, random coil ensemble of states. The folding kinetics of small, single-domain model systems can often be described by a rapid, two-state mechanism (for reviews, see Jackson 1998; Plaxco et al. 2000; Daggett and Fersht 2003). In contrast, the folding kinetics of larger, multidomain monomeric proteins are often more complex, requiring the coordination of long-range interdomain and short-range intradomain interactions (for reviews, see Kim and Baldwin 1990; Matthews 1993; Wallace and Matthews 2002).
Significant insights have been gained from monomeric folding systems regarding the intramolecular forces responsible for the productive formation and stabilization of secondary and tertiary structures as the folding energy landscape is successfully and efficiently traversed. However, it is not yet clear how these insights apply to the intermolecular association reactions that stabilize the quaternary structure of oligomeric proteins. Oligomeric proteins are prevalent in biology and offer evolutionary advantages such as the potential of increased thermostability relative to mono mers and sensitive regulation through allostery and cooper ativity in ligand binding. The folding of oligomeric proteins requires that the protein folding code direct the formation of intramolecular secondary and tertiary structure as well as the productive quaternary interactions necessary for biologi cal function. Folding experiments on large, oligomeric en zymes demonstrated the complexity of this coordination of multiple levels of structure formation (for reviews, see Jaenicke 1987; Jaenicke and Lilie 2000). A lack of reversibil ity, often resulting from aggregation, is a complication in the folding of many proteins, and particularly oligomeric systems. Protein concentration-dependent, nonproductive aggregation reactions often kinetically compete with on pathway, partially folded, kinetic intermediates in large oligomeric proteins (Seckler 2000). In vivo, this competition between productive folding and aggregation is often offset by the assistance of chaperones.
Given these complexities, recent efforts have focused on smaller oligomeric folding model systems. Small (less than 60 residues per monomer), dimeric, two-state folding sys tems have been identified, for example the P22 Arc repres sor (Milla and Sauer 1994; Srivastava and Sauer 2000) and the GCN-4-derived leucine zipper peptides (Zitzewitz et al 1995, 2000). However, just as with monomeric systems, larger dimers, containing subdomains, tend to traverse more complicated folding landscapes, with transient kinetic inter mediates and sometimes parallel pathways. Examples in clude ketosteroid isomerases (Kim et al. 2001a,b), Esche richia coli Trp repressor (Gittelman and Matthews 1990; Mann et al. 1995; Gloss et al. 2001), glutathione transfer ases (Wallace et al. 1998; Wallace and Dirr 1999), and bacterial luciferase (Clark et al. 1997; Noland et al. 1999; Inlow and Baldwin 2002). Both monomeric and dimeric kinetic intermediates have been observed, but as yet, no clear rules are discernable for the prediction of the features of the folding landscape of dimeric proteins.
This article investigates the folding reactions of the his tone fold motif, using the (H3–H4)2 tetramer (Fig. 1 ▶) as model system. The histone fold is a dimerization motif found in several important protein–DNA complexes. The fold was first structurally characterized in the eukaryotic heterodimers of the core nucleosome—the (H3–H4)2 tetramer and the H2A–H2B heterodimer (Arents et al. 1991; Luger et al. 1997a). The motif is also seen in the struc tures of homodimeric archael histones (Starich et al. 1996; Zhu et al. 1998; Decanniere et al. 2000) and some of the TATA-binding protein Associated Factors (TAFs) of the TFIID complex (Xie et al. 1996; Birck et al. 1998). Sequence alignments have suggested that many other DNA binding proteins may also contain the histone fold dimerization motif (Arents and Moudrianakis 1995; Baxevanis et al. 1995).
Figure 1.

Ribbon structure of the histone fold and the dimer–dimer interface of the H3–H4 tetramer. The H3 monomers, from residues 38–135 of 135, are shown in a lighter gray; the darker-colored H4 monomers encompass residues 20–102 of 102 residues. The three helices of the canonical histone fold are shown as cylinders. The H3 Cys-110 Cα atoms are represented by spheres; in this study, Cys 110 is mutated to Glu to disrupt the dimer–dimer interface. The figure was rendered from the coordinates of the tetramer in the core nucleosome (Luger et al. 1997a), using MOLSCRIPT v2.1 (Kraulis 1991).
The histone fold contains a long central α-helix that is flanked on both the N and C termini by short β-loops and shorter α-helices (Arents et al. 1991; Luger et al. 1997a). The histone monomers associate to form dimers in a head-to-tail fashion called the “handshake motif” (Arents and Moudrianakis 1995). Two H3–H4 dimers then dimerize with one another through a four-helix bundle of their H3 C-terminal helical regions. Thus, the folding of the (H3–H4)2 tetramer requires the coordinated folding of four polypeptide chains and the assembly of two dimerization interfaces.
To date, biophysical characterization of the eukaryotic histones has focused on the equilibrium stabilities of the oligomers (Baxevanis et al. 1991; Karantza et al. 1995, 1996 Karantza et al. 2001; Gloss and Placek 2002; Placek and Gloss 2002; Banks and Gloss 2003). The equilibrium stability of recombinant (H3–H4)2 tetramer was determined by GdnHCl-induced denaturation, using fluorescence and circular dichroism spectroscopies (Banks and Gloss 2003). At moderate ionic strength (μ ~ 0.2 M), H3–H4 is unstable, with little native baseline in the unfolding transition. Therefore, tri-methylamine-N-oxide (TMAO) was used to stabilize the tetramer to obtain accurate thermodynamic parameters. The equilibrium unfolding of the (H3–H4)2 tetramer was best described by a three-state mechanism, with well-folded H3–H4 dimers as a populated intermediate. When compared to the structurally similar H2A–H2B dimer, the H3–H3′ tetramer interface and the H3–H4 histone fold are strikingly less stable (Banks and Gloss 2003). This article provides the first report of kinetic folding studies and a proposed folding mechanism for an oligomer containing the histone fold.
Results
The unfolding and folding reactions of the H3–H4 ensemble were collected using stopped-flow methods, monitoring intrinsic Tyr FL and CD at 222 nm. Previous equilibrium studies demonstrated that the H3–H4 species is an equilibrium of tetramer and H3–H4 dimers (Banks and Gloss 2003). TMAO was used to stabilize the ensemble, and shift the equilibrium to predominantly the tetrameric species.
Unfolding reactions of the H3–H4 tetramer
As shown previously, in the absence of TMAO, the unfolding reactions of the H3–H4 tetramer were best described by a fit to a sum of two first-order exponentials, equation 2 (below; Banks and Gloss 2003). The faster kinetic phase was attributed to the unfolding of the H3–H4 dimer, and the slower kinetic phase corresponded to the unfolding of the H3–H4 tetramer. Each kinetic phase exhibited a similar dependence on the final GdnHCl concentration (Fig. 2A ▶; Table 1). Over a range of final GdnHCl concentrations, the relative amplitude of the two unfolding phases is similar and largely independent of denaturant concentration (Fig. 2B ▶). For the CD data, the sum of the two amplitudes from SF-CD agreed reasonably well with the total amplitude expected from equilibrium data, suggesting that no significant “burst-phase” reaction was occurring in the ~5-msec dead time of the stopped flow. Given the differences in the instrumentation detection setup between SF-FL (total FL above ~300 nm) and equilibrium FL measurements (with an emission monochromator), it was not possible to compare the observed kinetic amplitudes to those expected from equilibrium data.
Figure 2.

Folding (open symbols) and unfolding (closed symbols) reactions of the H3–H4 tetramer monitored by SF-FL and SF-CD. In A, C, and D, the solid lines represent the global fits of the CD and FL kinetic traces as a function of GdnHCl to equation 3. Error bars are shown or are less than the size of the data points. (A) GdnHCl concentration dependence of the folding and unfolding reactions in the absence of TMAO. Folding: SF-FL (open triangles), SF-CD (open circles); unfolding: SF-FL (filled triangles and filled down triangles for fast and slow reactions, respectively), SF-CD (filled circles and filled diamonds for fast and slow reactions, respectively). (B) The average relative amplitudes as a function of final [GdnHCl] in 0 M TMAO, from three independent unfolding data sets. Data are represented by the same symbols as used in A. Error bars represent the standard error of the averages. (C) GdnHCl dependence of the folding and unfolding reactions in 1 M TMAO. Folding: SF-FL (open triangle), SF-CD (open circle); unfolding: SF-FL (filled down triangle), SF-CD (filled diamond). (D) GdnHCl dependence of the folding and unfolding global fits overlaid in the presence (open triangle, down filled triangle)and absence (open square, filled square, filled circle) of 1 M TMAO. Symbols are to help identify the global fits and do not represent data points. Conditions: 4μM monomer, 200 mM KCl, 20 mM potassium phosphate at pH 7.2, 1 mM EDTA, 3 mM β-ME, 25°C.
Table 1.
Parameters describing the kinetic folding and unfolding reactions of the H3–H4 ensemble at 4 μM monomer (1 μM tetramer)
| Parameters | 0 M TMAO | 1 M TMAO |
| kunfold fast (s) | 0.18 (0.01) | — |
| m‡unfold fast (kcal mole−1M−1) | −0.71 (0.02) | — |
| kunfold slow (s) | 0.012 (0.005) | 0.081 (0.003) |
| m‡unfold slow (kcal mole−1M−1) | −0.77 (0.01) | −0.38 (0.01) |
| kfold (s) | 3.7 (0.2) | 12.7 (0.2) |
| m‡fold (kcal mole−1M−1) | 0.50 (0.04) | 0.80 (0.04) |
Results of global fitting of the kinetic data to equation 3. Conditions: 200 mM KCl, 20 mM potassium phosphate at pH 7.1, 1 mM EDTA, 3 mM β-ME, at 25°C. The values in parentheses are the standard deviation of global fits.
The relaxation times of both unfolding phases were also determined to be largely independent of final monomer concentration (data not shown). The GdnHCl dependence of unfolding data were globally fitted to equation 3 (below). The results of the global fit of 28 kinetic unfolding traces for [GdnHCl] between 1.4 and 3.5 M is shown as the solid lines in Figure 2, A and D ▶, and the fitted parameters are given in Table 1.
1 M TMAO has a stabilizing effect on the H3–H4 ensemble, shifting the dimer–tetramer equilibrium largely toward the tetramer species, with little isolated dimer present (Banks and Gloss 2003). As reported previously, SF-FL and SF-CD unfolding reactions in 1 M TMAO are adequately fit by a single exponential equation. The observed relaxation times exhibited a dependence on the final [GdnHCl] (Fig. 2C ▶), with relaxation times similar to the slowest kinetic phase observed in the absence of TMAO (Fig. 2D ▶). The GdnHCl dependence was determined from the global fit of 26 kinetic traces to equation 3 (below) for [GdnHCl] between 2 and 3.5 M. The results are shown in Figure 2, C and D ▶, and the fitted parameters given in Table 1.
Folding reactions of the H3–H4 tetramer
Folding reactions, initiated from an equimolar mixture of H3 and H4 monomers unfolded in ≥3 M GdnHCl, were well described by a single, first-order exponential equation, with and without TMAO (Fig. 3 ▶). The observed relaxation times exhibited a significant dependence on the final [GdnHCl] (Fig. 2A,C,D ▶). However, very little dependence on monomer concentration was observed (Fig. 4 ▶), suggesting that the kinetic phase represents a first-order reaction. The small increase in relaxation time at the lowest monomer concentrations reflects the coupling of a first-order reaction to an overall biomolecular process. The final equilibrium conditions and the stability of the burst-phase dimeric intermediate (see below) are protein concentration dependent. Thus, the rate of folding, even for a strictly first-order process, should exhibit a small protein concentration dependence, as seen in the protein concentration dependence of the Fapp curves (e.g., Fig. 6 ▶ [below] or Banks and Gloss 2003).
Figure 3.

Representative stopped-flow refolding kinetic traces of the H3–H4 oligomer to 0.5 M GdnHCl in the absence of TMAO. The thick lines represent the local fits of the data to a single, first order exponential. Shown in the insets are the residuals of the fits. (A) SF-FL monitoring intrinsic Tyr FL. (B) SF-CD at 222 nm. Conditions are described in the Fig. 2 ▶ legend.
Figure 4.

Monomer concentration dependence of the H3–H4 oligomer folding reaction in the absence (open symbols) and presence of 1 M TMAO (filled symbols). Unfolded H3 and H4 monomers were folded to 0.5 M GdnHCl; refolding relaxation times were monitored by SF-FL (open/filled square) and SF-CD (open/filled circle). The monomer concentration dependence expected for the relaxation times of a second-order process are represented by the solid line. The observed relaxation time at 1 μM monomer was used as a reference point. The expected decrease in relaxation time as a function of [monomer] was estimated from the relationship of 1/τ is proportional to kassoc[Umonomer]2; that is, a twofold increase in [monomer] should result in a fourfold decrease in the observed relaxation time. Error bars represent the standard deviation of the relaxation time for the local fits. Buffer conditions are given in the Fig. 2 ▶ legend.
Figure 6.

Equilibrium characterization of the H3–C110E/H4 dimer. Fapp curves for the GdnHCl-induced equilibrium unfolding transitions of the H3–C110E/H4 dimer in 1 M TMAO as a function of dimer concentration. The lines represent the global fits of the data to a two-state dimer unfolding model. 0.5 μM (filled triangle, FL), 1.0 μM (filled square, CD; filled down triangle, FL), 2.0 μM (open circle, CD; open square, FL). Conditions are given in the Fig. 2 ▶ legend. (Inset) HPLC size-exclusion chromatography elution profiles of the wild-type core histone oligomers and H3–C110E/H4 dimer. The proteins were chromatographed on a BioSep-SEC-S 3000 column, equilibrated at room temperature under stabilizing conditions: 1 M KCl, 20 mM potassium phosphate at pH 7.2, 1 mM EDTA, 3 mM β-ME, with detection at 280 nm. H3–H4 tetramer, solid line/triangle; H2A-H2B dimer, dashed line/circle; H3–C110E/wtH4 dimer, solid line/square.
The GdnHCl dependence of the folding reaction was assessed by globally fitting, to equation 3 (below), 33 kinetic traces from 0.5 to 1.3 M GdnHCl and 17 kinetic traces from 0.5 to 1.7 M GdnHCl in 0 and 1 M TMAO respectively. The results of the global fits are shown in Figure 2, A, C, and D ▶, and Table 1. Near the midpoint of the equilibrium transitions (Banks and Gloss 2003), the observed folding relaxation time converges with the faster unfolding relaxation time observed in the absence of TMAO. This convergence suggests that the folding kinetic phase represents the last refolding step that leads to the native H3–H4 dimer.
The observed amplitudes of the SF-CD refolding kinetic traces were less than that expected from the equilibrium data for the conversion of unfolded species to the native oligomers (Fig. 5A ▶). This difference is particularly pronounced in the presence of 1 M TMAO (Fig. 5B ▶). Comparison of the observed initial folding signal with that expected for the unfolded species suggests that there is a rapid formation of secondary structure in the 5-msec mixing dead time of the stopped-flow instrument. This burst-phase signal decreases with increasing [GdnHCl], as expected for a transient kinetic intermediate. The amount of ellipticity developed in 5 msec is also protein concentration dependent, increasing with higher monomer concentrations. The percentages of the initial signal relative to the final signal are 15%, 34%, 44%, and 51% for monomer concentrations of 1, 4, 8, and 10 μM monomer, respectively, when folding to 1 M GdnHCl in 1 M TMAO. A similar but less pronounced dependence is seen in the absence of TMAO. This protein concentration dependence of the burst-phase signal is consistent with a dimerization reaction occurring in the 5-msec SF dead time. The stability of a dimeric intermediate should increase with protein concentration, resulting in greater extent of folding at higher monomer concentrations and the same final [denaturant], as observed in Fapp curves for equilibrium unfolding transitions (e.g., Fig. 6 ▶).
Figure 5.

SF-CD ellipticities of the H3–H4 ensemble in the absence (A) and presence (B) of 1 M TMAO. The data were normalized for buffer/ cuvette contributions. Ellipticity observed after 5 msec refolding (open square); final folding ellipticity (filled square); final unfolding ellipticity (filled circle). Equilibrium unfolding transitions are shown as solid lines and the expected unfolded baseline is shown as a dashed line. Conditions are given in the Fig. 2 ▶ legend.
Characterization of a dimeric H3–H4—the H3–C110E/H4 dimer
To test the role of the dimeric and tetrameric species in the folding and unfolding reactions of the H3–H4 system, protein engineering was used to create an obligatory H3–H4 dimer, incapable of forming a tetramer. A mutation was introduced into the hydrophobic tetramer interface of the H3–H3′ four-helix bundle. The tetramer interface was destabilized by mutation of the largely buried Cys 110 residue to Glu, which introduces two new negative charges into the hydrophobic core of the four-helix bundle. The H3–C110E monomer was reconstituted with wild-type H4 monomer.
The oligomeric state of the H3–C110E/H4 system was examined by HPLC size-exclusion chromatography (Fig. 6 ▶, inset). The elution profiles of H3–C110E/H4 are consistent with a dimeric species, in buffers without TMAO (data not shown) and in buffers with stabilizing concentrations of KCl, 1 to 1.5 M, shown previously to shift the equilibrium of the H3–H4 oligomer to the more fully assembled tetrameric state (Banks and Gloss 2003). No significant difference in far-UV CD mean residue ellipticity spectra was observed between the H3–C110E/H4 dimer and the wild-type dimer–tetramer ensemble (data not shown). This lack of change in secondary structure between dimer and tet-ramer has been reported previously for pH-dependent dissociation of the (H3–H4)2 tetramer (Karantza et al. 1996).
The stability of the H3–C110E/H4 dimer was determined by GdnHCl-induced denaturation titrations (Fig. 6 ▶). Like the wild-type H3–H4 system, the unfolding transitions of this H3–H4 variant are reversible (evidenced by overlapping titrations in the folding and unfolding direction; data not shown). Data were collected at 1 M TMAO, monitoring far-UV CD and intrinsic Tyr FL. The CD and FL equilibrium data were globally fitted to a two-state dimeric model, yielding an excellent agreement between the data and the fit (Fig. 6 ▶). The free energy of unfolding in the absence of denaturant (at a standard state of 1 M oligomer), ΔG° (H2O), was 11.4 ± 0.3 kcal mole−1 with an m-value of 2.6 ± 0.2 kcal mole−1 M−1. These values are similar to those for the unfolding of the wild type H3–H4 dimer in 1 M TMAO: ΔG° (H2O) of 12.4 ± 0.6 kcal mole−1 and an m-value of 2.9 ± 0.3 kcal mole−1 M−1 (Banks and Gloss 2003). The HPLC and stability data demonstrate that the H3–C110E mutation destabilizes the tetramer interface, with little effect on the H3–H4 dimer.
The unfolding reactions of the H3–C110E/H4 dimer were monitored by SF-FL and SF-CD. The kinetic traces were well described by fits to a single exponential in 0 and 1 M TMAO, with [GdnHCl]-dependent relaxation times. In the absence of TMAO, the observed unfolding relaxation times were similar to those of the fast unfolding phase of the wild-type H3–H4 system (Fig. 7 ▶). In 1 M TMAO, the unfolding relaxation times are significantly less, 2.5-fold, than those observed for the wild-type tetramer (data not shown). These data support the attribution of the fast and slow unfolding relaxation times to the unfolding of the H3–H4 dimer and tetramer, respectively.
Figure 7.

[GdnHCl] dependence of the folding and unfolding reactions of the H3–C110E/H4 dimer monitored by SF-FL and SF-CD in 0 M TMAO. Folding: SF-FL (open triangle), SF-CD (open circle); unfolding: SF-FL (filled triangle), SF-CD (filled circle). The individual data points represent the relaxation times determined from local fits of the data; error bars represent the standard deviation of the fitted values. The solid lines represent the global fits of the kinetic traces of the wild-type H3–H4 tetramer as a function of GdnHCl, for comparison. Monomer concentrations and conditions are the same described in the Fig. 2 ▶ legend.
The H3–C110E/H4 species was folded in the absence of TMAO from an equal molar unfolded mixture of H3–C110E and wild-type H4 monomers equilibrated in 3 M GdnHCl. The folding reaction was monitored by both SF-FL and SF-CD and well described by fits to a single exponential equation. The relaxation times for folding of the dimer were similar in magnitude and [GdnHCl] dependence to those of the wild-type H3–H4 ensemble in 0 M TMAO (Fig. 7 ▶). The similarity in the observed folding relaxation time for the wild-type and C110E-containing dimers supports the attribution of this folding phase to the formation of the native H3–H4 dimer.
Association of H3–H4 dimers
No folding kinetic phase, either in SF-FL or SF-CD experiments, was observed that converged in the transition region with the unfolding of the H3–H4 tetramer (i.e., the slower unfolding phase in 0 M TMAO or the single unfolding phase in 1 M TMAO). Fitting the equilibrium unfolding data for the H3–H4 tetramer in 1 M TMAO indicated an ~30% increase in Tyr fluorescence upon tetramer-to-dimer dissociation (Banks and Gloss 2003). Therefore, the dimer-to-tetramer kinetic folding reaction should proceed with an observable FL change. However, if tetramer formation is rapid, compared to dimer folding, the dimer–dimer association reaction would occur after the rate-limiting step of the folding pathway, and thus not exhibit a detectable kinetic phase.
Conditions were chosen to specifically monitor the H3–H4 dimer-to-tetramer association reaction, separated from other folding events. To determine the conditions that yield the largest signal change between tetramer and dimer species, fluorescence wavelength scans of the H3–H4 system were performed at 0 to 0.7 M GdnHCl; 0 and 1 M TMAO were used to alter the equilibrium from dimer–tetramer to largely tetramer. The largest signal change was observed at 0.5 M GdnHCl (Fig. 8A ▶), a 14% decrease at 305 nm upon association to tetramer. The decrease in FL upon dimer association is also supported by the FL spectra of the H3–C110E/H4 dimer, which is very similar to that of the wild type in the absence of TMAO (data not shown). The equilibrium FL at 0 and 1 M TMAO was also compared in the stopped-flow setup (monitoring total FL intensity), confirming a decrease in FL intensity upon dimer association. The kinetics of association, at 0.5 M GdnHCl, were then monitored by SF-FL, jumping from 0 to 1 M TMAO. After the 5-msec mixing dead time, the observed fluorescence signal was coincident with that of the tetramer preequilibrated in the same final solvent conditions (Fig. 8B ▶), and no other kinetic phase was observed after this burst-phase reaction. These data suggest that dimer–dimer association to form the (H3–H4)2 tetramer is very rapid, relative to the rate-determining step of dimer folding.
Figure 8.

Dimer–tetramer conversion monitored by intrinsic Tyr FL at 0.5 M GdnHCl. (A) Fluorescence spectra with excitation at 280 nm of 1.0 μM H3–H4 tetramer in the 0 M (filled triangle) and 1 M (filled down triangle) TMAO. (B) SF-FL kinetic jump from 0 to 1 M TMAO for the H3–H4 ensemble. The upper trace represents the signal observed for 8 μM monomer in 0 M TMAO; the trace was obtained by mixing equal volumes of (1) 16 μM monomer in 0.5 M GdnHCl and buffer, and (2) buffered 0.5 M GdnHCl. The 1 M TMAO data, at 8 μM monomer, are the lower traces: The equilibrium signal is the darker line (filled circle), and the kinetic data is the lighter line (stippled square). The equilibrium trace was obtained by mixing equal volumes of the identical solution—8 μM monomer in 1 M TMAO, 0.5 M GdnHCl, and buffer—from two syringes. The kinetic jump was initiated by mixing equal volumes of (1) 16 μM monomer in 0.5 M GdnHCl and buffer, and (2) buffer with 2 M TMAO and 0.5 M GdnHCl—giving the same final conditions as the equilibrium trace. The SF traces have been buffer corrected. Buffer conditions are described in the Fig. 2 ▶ legend.
Folding–unfolding double jump experiments in 0 M TMAO confirmed this conclusion. In this type of experiment, protein is folded for varying delay times, after which there is a second jump to unfolding conditions, and the unfolding reaction is monitored. The observed unfolding amplitudes reflect the accumulation of the native state(s) as a function of folding delay time. This type of experiment has been described in depth elsewhere (e.g., Gloss and Matthews 1998a b; Gloss et al. 2001; Wallace and Matthews 2002).
Unfolded H3–H4 was refolded to 0.5 M GdnHCl and 6 μM monomer and then unfolded to 2 M GdnHCl and 4 3M monomer. When the refolding delay times were less than the observed, first-order, folding relaxation time (Fig. 2A ▶), there was a diminution of the total, detected unfolding amplitude. With increasingly longer folding delay times, the observed unfolding kinetic amplitude increased, and with folding delay times sufficient for the completion of the observed folding kinetic phase, the total expected unfolding amplitude was observed. These results are further confirmation that the observed first-order folding phase leads to the native oligomers.
Two unfolding kinetic phases were observed after all folding delay times, 50 msec to 3 sec. The relaxation times (1.4 and 10.6 sec) and relative amplitudes (80% and 20% for the fast and slow amplitudes, respectively) are similar to those observed for direct unfolding jumps from these initial folding conditions: relaxation times of 0.43 and 11.6 sec with relative amplitudes of 74% and 26%. This agreement shows that native oligomers are formed by the observed first-order folding reaction and that the equilibrium tetramer:dimer ratio is achieved rapidly, relative to the folding to native H3–H4 dimer.
Discussion
Folding mechanism of the H3–H4 dimer–tetramer
Previously published results have shown that the two unfolding reactions of the H3–H4 ensemble in the absence of TMAO reflect the unfolding of the H3–H4 dimer and (H3–H4)2 tetramer, which are in equilibrium with each other (Banks and Gloss 2003). This conclusion is supported by the kinetic data of the H3–C110E/H4 dimer reported here (Figs. 6 ▶and 7 ▶). The folding reaction of the H3–H4 ensemble appears to have two detectable steps: a burst-phase reaction, completed in ≤5 msec (Fig. 5 ▶) and a slower, first-order folding reaction that leads to the native dimer (Figs. 2 ▶, 3, ▶ and 4 ▶). The further association to native tetramer occurs after this rate-determining step to form native dimer (Fig. 8 ▶). The mechanism in Scheme 1A ▶ is a working model for the kinetic folding of the H3–H4 ensemble. Unfolded H3 and H4 monomers associate in the SF dead time to form a heterodimeric kinetic intermediate; this intermediate is subsequently converted to the native H3–H4 dimer by an observable, first-order folding reaction. The final association reaction of heterodimers to form the native tetramer occurs rapidly, relative to the first-order folding reaction. Discussed below are the details of the reactions postulated in the mechanism of Scheme 1A and how the data support the proposed mechanism.
Scheme 1.

Kinetic folding mechanism of the H3–H4 histone oligomers. (A) Mechanisms of folding and unfolding. The estimated relaxation times are for the reactions in the absence of denaturant (Table 1). (B) Reaction coordinate diagram corresponding to the mechanism in A, at 4 μM monomer (1 μM tetramer). The estimation of the stability of the ground and transition states is described in the text.
SF-CD burst-phase folding intermediate
Within 5 msec of the initiation of folding, a substantial amount of secondary structure has formed. The initial CD signal, at 0.5 M GdnHCl and 4 μM monomer, constitutes ~30% and 50% of the final, native CD signal, in 0 and 1 M TMAO, respectively. It was not possible to make reliable dilutions to lower [GdnHCl] to determine the ellipticity of the burst-phase intermediate under more strongly folding conditions. Therefore, it was not possible to determine a free energy for the folding of the intermediate as has been done for other burst-phase intermediates (e.g., Gloss and Matthews 1998a b; Topping et al. 2003). However, the burst-phase intermediate appears to be significantly less stable than the native H3–H4 dimer, which is itself only marginally stable at micromolar concentrations (Banks and Gloss 2003).
The oligomeric state of the burst-phase intermediate is most likely dimeric. Two lines of evidence support this conclusion. First, the folding reaction observable after the SF dead time shows no significant protein concentration dependence (Fig. 4 ▶), demonstrating that it is a first-order reaction that leads to the native H3–H4 dimer. Therefore, dimerization must have occurred prior to this observed folding step. Second, the magnitude of the burst-phase amplitude increases significantly with protein concentration, particularly under the strongly refolding conditions of 1 M TMAO (see Results). As the association reaction appears to be completed within 5 msec at micromolar monomer concentrations, the unfolded (or partially folded) monomers must associate at a rate ≥107 M−1sec−1. Because the association itself cannot be observed under stopped-flow conditions with wild-type spectral probes, it is not possible to assess the foldedness of the associating monomers, if they are completely unfolded or have partially folded prior to association.
Folding and unfolding of the H3–H4 dimer
The observable folding kinetic phase was well described by a fit to a single exponential, in 0 M and 1 M TMAO (Fig. 3 ▶). A second-order process, such as the association of two monomers, should not be adequately described by a single exponential. Further verification of the first-order nature of this folding kinetic phase is the small protein concentration dependence of the reaction (Fig. 4 ▶). Between 1 and 16 μM monomer, there is a fourfold decrease in the observed relaxation time; a second-order process would predict a thousand-fold decrease.
In the equilibrium transition region, 1.5 to 2 M GdnHCl, the folding relaxation time is similar to that for the unfolding of the H3–H4 dimer, strongly implying that the two processes are the reverse of each other as shown in Scheme 1A ▶; that is, that the product of the folding reaction is the native H3–H4 dimer. The double-jump data, described above, also showed that the development of the population of native oligomers is limited by the first-order folding reaction. However, because of SF-mixing difficulties, it was not possible to demonstrate definitively that folding of the entire population of native oligomers is constrained by the first-order folding reaction. Therefore, a parallel pathway of direct folding to native oligomers in the burst-phase reaction cannot be excluded.
The four-helix bundle tetramer interface was disrupted by the introduction of charged residues into a hydrophobic environment by the H3–C110E mutation. The resulting mutant H3–C110E/WT H4 protein was a dimer, even under stabilizing conditions (Fig. 6 ▶, inset). The observed folding relaxation times of this obligate dimer were similar to those ascribed to the folding of the wild-type H3–H4 dimer (Fig. 7 ▶). Furthermore, unlike the wild-type H3–H4 system, which is an equilibrium of dimeric and tetrameric species, in 0 M TMAO, the H3–C110E/WT H4 dimer unfolds by a single kinetic phase that is very similar to the faster kinetic phase observed for the wild-type dimer species of the H3–H4 dimer–tetramer ensemble.
The stability associated with the H3–H4 dimeric intermediate’s conversion to native H3–H4 dimer can be determined from the kinetic values in 0 M TMAO (Table 1), using the following relationships:
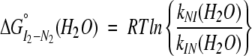 |
(1a) |
 |
(1b) |
The m value associated with a folding/unfolding reaction is generally proportional to the change in solvent-accessible surface area between the folded and unfolded species (Myers et al. 1995). The ΔG° (H2O) and m values for the folding of the I2 dimeric intermediate are 1.8 kcal mole−1 and 1.2 kcal mole−1M−1, respectively. Comparison of these parameters to those determined from equilibrium unfolding data would be informative. However, such a comparison is complicated because the stability of the H3–H4 dimer could only be determined in the presence of 1 M TMAO, because of the instability of the H3–H4 ensemble (Banks and Gloss 2003); yet, in 1 M TMAO, the unfolding phase of the H3–H4 dimer is not observed because the ensemble is predominantly tetrameric. However, the ΔG and m values associated with the folding of the dimeric intermediate are certainly less than that expected for the complete folding of the native dimer. The kinetic ΔG and m values suggest that the folding of the burst-phase dimeric intermediate (1) provides a significant amount of the stability of the native dimer, and (2) results in the burial of a large portion of the surface area excluded from solvent in the native dimer.
Folding and unfolding of the H3–H4 tetramer
The complete assembly of the H3–H4 tetramer requires the association and folding of two dimerization interfaces—the histone fold motif of the H3–H4 dimer and the four-helix bundle of the H3–H3′ tetramer interface. The histone fold is the larger, more stable interface, with ~76% more buried surface area than the four-helix bundle interface (Banks and Gloss 2003). The formation of the tetramer can be monitored by a decrease in FL upon dimer association. SF-FL kinetics demonstrate that the tetramerization interface is formed after complete folding of the H3–H4 dimer, through a rapid association reaction. As this reaction is complete within 5 msec at dimer concentrations of 2 μM, the H3–H4 dimers must associate with a rate constant ≥107 M−1sec−1 under strongly refolding conditions.
The stability difference between the dimeric and tetrameric species is relatively small, ~2.6 kcal mole−1 in 1 M TMAO at 4 μM monomer. Therefore, the dimer and the tetramer exist in a dynamic equilibrium with each other; their relative populations have been estimated from the relative amplitudes of the fast (dimeric) and slow (tetrameric) unfolding reactions (Banks and Gloss 2003). The unfolding of the tetrameric component of the ensemble is slower than the unfolding of the dimeric component because of the pre-equilibrium that exists between the dimer and slightly more stable tetramer (illustrated in Scheme 1 ▶). However, the major rate-determining step in the unfolding of the H3–H4 species is the unfolding of the H3–H4 dimer. Hence, the m‡ values for the unfolding of the dimer and tetrameric species in 0 M TMAO are quite similar (Table 1), as they represent traversing a similar rate-determining barrier. The relative unfolding rates for the dimer and tetramer, over a similar rate-determining barrier, can be used to estimate the difference in free energy between the two oligomeric species; at 4 μM monomer (Table 1), the predicted stability difference is ~2.7 kcal mole−1, in agreement with the expected equilbrium value at this monomer concentration.
The GdnHCl dependence of the folding and unfolding reactions further support the conclusions that (1) the slow unfolding species represents the tetrameric ensemble, and (2) that the association of two dimers to form the tetramer is rapid, relative to the rate-determining step of dimer folding. In 0 M TMAO, the observed folding relaxation time converges with the faster unfolding phase in the transition region (Fig. 2A ▶). As discussed above, this convergence demonstrates that the reactions interconvert between the same initial and final species. Furthermore, the folding and unfolding relaxation times of the obligatory dimeric H3–C110E/H4 agree very well with these two reactions that have been ascribed to the wild-type H3–H4 dimer folding/unfolding (Fig. 7 ▶). No observable kinetic folding phase converges with the slower unfolding reaction. In 1 M TMAO, only the slower unfolding phase is observed, as the equilibrium ensemble is predominantly tetrameric (Banks and Gloss 2003). In the transition region, there is no convergence of the unfolding phase (ascribed to the tetramer) with the observed folding phase (attributed to the conversion of the dimeric intermediate to the native H3–H4 dimer).
Reaction coordinate diagram for the folding of the H3–H4 ensemble
A summary of the mechanism and data can be easily visualized in a reaction coordinate diagram. Using the mechanism of Scheme 1A ▶, the relative energies of the various species and transition state ensembles traversed in the folding of the (H3–H4)2 tetramer are shown in the reaction coordinate diagram of Scheme 1B ▶, for a standard state of 1 μM tetramer (4 μM monomer), a condition used in many of the experiments described in this report.
The stability of the (H3–H4)2 tetramer was arbitrarily set to 0 kcal mole−1. The relative free energy of the unfolded monomers and H3–H4 dimer were determined from equilibrium studies (Banks and Gloss 2003). The stability of the dimeric H3–H4 I2 intermediate was estimated to be 1.8 kcal mole−1 less than that of the native H3–H4 dimer, based on the ΔG° (H2O) values estimated from the observed folding kinetics and the faster unfolding kinetic phase in 0 M TMAO. Transition state energies were estimated from the relationship: kobs = ka • e(−ΔG‡/RT), based on the Eyring equation. The values of ka, the exponential prefactors, were estimated from the Kramers formalism, as described previously (Gloss and Matthews 1998a). The transition state energies between 4U and 2I2 and between the H3–H4 dimer and (H3–H4)2 tetramer were estimated from minimal association rates of 107 M−1sec−1 and a diffusion limited rate constant, ka of 109 M−1sec−1 (Janin 1997; Gloss and Matthews 1998a). The free energy of the transition state between the dimeric intermediate and the native H3–H4 dimer was estimated using a ka of 5 × 108 sec−1, derived from model peptide studies (Thompson et al. 1997; Gloss and Matthews 1998a; Munoz et al. 1998).
The reaction coordinate in Scheme 1B ▶ has been drawn to illustrate the folding reactions of the H3 and H4 monomers. However, it serves to illustrate several key features of both the folding and unfolding reactions: (1) The largest kinetic barrier to folding and unfolding is the transition state between the dimeric intermediate and the native H3–H4 dimer; (2) the burst phase dimeric intermediate exhibits much of the stability of the native dimer; (3) there is a pre-equilibrium between the native dimers and the native tetramer, which serves to decrease the unfolding rate of the tetrameric species.
H3–H4 folding mechanism compared to other DNA-binding dimers
Small, monomeric, single-domain protein folding model systems that can fold by a two-state kinetic mechanism (see Introduction) have provoked questions about the role of transient kinetic intermediates in protein folding. Do the kinetic intermediates observed in the folding of larger proteins assist and accelerate the folding reaction or do they represent kinetic traps that frustrate the folding protein and hinder rapid, productive conversion to the native species? In the folding of oligomeric species of diverse structural content and topology, monomeric and dimeric intermediates have been observed (see Introduction). It is important to extend our understanding of the role of intermediates to oligomeric systems, where efficient folding requires the assembly of not one, but multiple, polypeptide chains. Studies of the histone fold help to further address the issue of kinetic folding intermediates in a class of oligomeric proteins: those with intertwined, segment-swapped, α-helical interfaces, and specifically, DNA-binding proteins.
The histone fold, contained in the H3–H4 dimer, is a segment-swapped, α-helical interface in which the monomers are intertwined around each other. Domain swapping is an oligomerization mechanism, first described by Eisenberg et al. (Bennett et al. 1995; Liu and Eisenberg 2002), in which a domain that can fold within a monomer is swapped, in an oligomeric structure, with the identical domain from another monomer, via a flexible hinge region. Nussinov et al. (Xu et al. 1998) suggested the term segment-swapped for those structures in which the swapped structural elements do not form a compact domain, and expanded the concept to obligatorily swapped structures, for which closed monomers are unstable and have not been observed. The folding mechanisms of two other DNA-binding dimers with segment-swapped, α-helical interfaces have been reported: the E. coli homodimeric proteins Trp repressor and Factor for Inversion Stimulation (FIS). There is a striking similarity in the kinetic folding mechanisms of these proteins and the heterodimeric histone fold, in that all three transiently populate a burst-phase, partially folded, dimeric intermediate.
The folding landscape of full-length Trp repressor is complicated and rough (Gloss et al. 2001 and references therein), with three parallel channels and monomeric and dimeric intermediates. However, the [2–66]2 core dimerization domain of Trp repressor folds more simply, through a single channel, with a rapid association reaction to form a dimeric intermediate, followed by folding to the native dimer (Gloss and Matthews 1998a,b). FIS also folds to a highly helical dimeric intermediate in the stopped-flow dead time, and then reaches the native state through a subsequent first-order folding reaction (Topping et al. 2003). In short, the mechanism shown in Scheme 1A ▶ for the folding to the H3–H4 dimer also describes the folding of FIS and the Trp repressor dimerization domain. For the latter two proteins, it has been shown that the dimeric intermediate is an obligatory, on-pathway kinetic species. These three examples suggest that dimeric intermediates may be a common feature in the folding landscape of segment-swapped, α-helical interfaces, despite different topologies.
Materials and methods
Materials
Ultrapure GdnHCl was purchased from ICN Biomedicals. TMAO was purchased from Sigma. TMAO was deionized and the concentration determined as described previously (Banks and Gloss 2003). All other chemicals were of reagent grade. The H3–C110E mutant was constructed by four-way PCR methods (Ho et al. 1989). The PCR product was subcloned into a pET3d vector, and the inserted gene was sequenced to verify the presence of the desired mutation and lack of any extraneous mutations. All histones (wild-type H3, H4, and C110E-H3) were produced recombinantly in E. coli; overexpression, purification, and reconstitution methods have been previously published (Luger et al. 1997b; Banks and Gloss 2003). Extinction coefficients were determined at 280 nm by the method of Gill and von Hippel (1989), yielding values of 19.3 mM−1 cm−1 and 8.7 mM−1 cm−1 for the wild-type tetramer and C110E dimer, respectively.
Methods
CD and fluorescence spectroscopy
Experimental conditions were 25°C in a buffer of 200 mM KCl, 20 mM potassium phosphate at pH 7.2, 1 mM EDTA, 3 mM β-ME, in 0 or 1 M TMAO. Stopped-flow data were collected on an AVIV 202SF spectrophotometer interfaced with an AVIV stopped-flow tower. SF-CD data were collected at 222 nm. SF-FL data were collected with an excitation wavelength of 280 nm, with emission detected at 90° to the incoming light, after a 295-nm cutoff filter. Rapid, efficient mixing was achieved with a two-syringe delta mixer with dead times of 5 to 8 msec, under the push conditions of ~2 ml/sec. Folding data were collected for 2.5 sec and unfolding data were collected for 42 sec. To enhance the signal-to-noise ratio, 30–40 shots were averaged for SF-CD and 15–20 shots averaged for SF-FL data for each kinetic trace.
Kinetic data analysis
Individual kinetic traces were locally fit with KaleidaGraph 3.5 for Windows (Synergy Software) to an equation of the exponential form
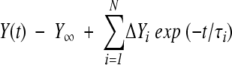 |
(2) |
where Y∞ is the final signal of the sample at equilibrium, ΔYi is the change in signal associated with the kinetic phase, and τi is its relaxation time. SF-CD and SF-FL refolding data were fit to a single exponential, in the presence and absence of TMAO, at all protein and final GdnHCl concentrations. At all final [GdnHCl], SF-CD and SF-FL unfolding data were fit to a sum of two exponentials in 0 M TMAO, and to a single exponential in 1 M TMAO. The program Savuka 5.1 (described elsewhere, Zitzewitz et al. 1995; Gualfetti et al. 1999a,b) was used to fit the GdnHCl dependence of the folding and unfolding relaxation times of multiple SF-CD and SF-FL traces to equation 3:
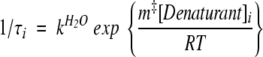 |
(3) |
where kH2O is the rate constant in the absence of GdnHCl and m‡ reflects the sensitivity of the reaction to [GdnHCl].
Equilibrium methods for H3–C110E/WT-H4
The dimeric state of the C110E-containing H3–H4 protein was confirmed by HPLC-size-exclusion chromatography. Buffer conditions were: 1 M KCl, 20 mM potassium phosphate at pH 7.2, 1 mM EDTA, 3 mM β-ME. Wild-type H3–H4 and H2A-H2B oligomers and the H3–C110E/WT H4 proteins were chromatographed at room temperature on a Phenomenex BioSep-SEC-S 3000 column. The elution of the H2A-H2B dimer and the H3–H4 tetramer has been compared previously to the elution of standards: bovine serum albumin, ovalbumin, chymotrypsinogen, and ribonuclease A (Banks and Gloss 2003).
Equilibrium unfolding transitions for the H3–C110E/H4 dimer were collected as described previously for the wild-type H3–H4 system in 1 M TMAO (Banks and Gloss 2003). CD spectra from 260 to 220 nm were collected at each [GdnHCl] in an automated titration. The spectral data were analyzed using singular value decomposition (Henry and Hofrichter 1992). FL titrations were monitored at 305 nm. CD and FL data were globally fitted, using a two-state dimer unfolding model, such as described elsewhere (Gloss and Placek 2002).
Acknowledgments
This work was supported by grants to L.M.G. from the American Cancer Society (RPG-00-085-01-GMC) and the National Science
Foundation (MCB-9983831). D.D.B. was partially supported by an NIH Biotechnology training grant (GM08336-13). The pET overexpression vectors for wild-type histones were kindly provided by Karolin Luger and Timothy Richmond of the Institute for Molecular Biology and Biophysics at the ETHZ, Zurich, Switzerland (K.L. is currently at Colorado State University). Brian Thome and Ananthi Asirvatham helped in the generation of mutants in the H3 interface to disrupt tetramer formation.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Abbreviations
β-ME, 2-mercaptoethanol
BP, burst phase
CD, circular dichroism
CM, the denaturant concentration at which the apparent fraction of unfolded monomer constitutes 50% of the population
FL, fluorescence
GdnHCl, guanidinium hydrochloride
HPLC-SEC, high pressure liquid chromatography size-exclusion chromatography
m value and m‡ value, the denaturant concentration dependence of the equilibrium constant and on the folding and unfolding rates, respectively
MRE, mean residue ellipticity
SF, stopped-flow
TMAO, trimethylamine-N-oxide
WT, wild type.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.03535504.
References
- Arents, G. and Moudrianakis, E.N. 1995. The histone fold: A ubiquitous architectural motif utilized in DNA compaction and protein dimerization. Proc. Natl. Acad. Sci. 92 11170–11174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arents, G., Burlingame, R.W., Wang, B.C., Love, W.E., and Moudrianakis, E.N. 1991. The nucleosomal core histone octamer at 3.1 Å resolution: A tripartite protein assembly and a left-handed superhelix. Proc. Natl. Acad. Sci. 88 10148–10152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banks, D.D. and Gloss, L.M. 2003. Equilibrium folding of the core histones: The H3–H4 tetramer is less stable than the H2A–H2B dimer. Biochemistry 42 6827–6839. [DOI] [PubMed] [Google Scholar]
- Baxevanis, A.D., Godfrey, J.E., and Moudrianakis, E.N. 1991. Associative behavior of the histone (H3–H4)2 tetramer: Dependence on ionic environment. Biochemistry 30 8817–8823. [DOI] [PubMed] [Google Scholar]
- Baxevanis, A.D., Arents, G., Moudrianakis, E.N., and Landsman, D. 1995. A variety of DNA-binding and multimeric proteins contain the histone fold motif. Nucleic Acids Res. 23 2685–2691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett, M.J., Schlunegger, M.P., and Eisenberg, D. 1995. 3D domain swapping: A mechanism for oligomer assembly. Protein Sci. 4 2455–2468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birck, C., Poch, O., Romier, C., Ruff, M., Mengus, G., Lavigne, A.C., Davidson, I., and Moras, D. 1998. Human TAFII28 and TAFII18 interact through a histone fold encoded by atypical evolutionary conserved motifs also found in the SPT3 family. Cell 94 239–249. [DOI] [PubMed] [Google Scholar]
- Clark, A.C., Raso, S.W., Sinclair, J.F., Ziegler, M.M., Chaffotte, A.F., and Baldwin, T.O. 1997. Kinetic mechanism of luciferase subunit folding and assembly. Biochemistry 36 1891–1899. [DOI] [PubMed] [Google Scholar]
- Daggett, V. and Fersht, A. 2003. The present view of the mechanism of protein folding. Nat. Rev. Mol. Cell Biol. 4 497–502. [DOI] [PubMed] [Google Scholar]
- Decanniere, K., Babu, A.M., Sandman, K., Reeve, J.N., and Heinemann, U. 2000. Crystal structures of recombinant histones HMfA and HMfB from the hyperthermophilic archaeon Methanothermus fervidus. J. Mol. Biol. 303 35–47. [DOI] [PubMed] [Google Scholar]
- Gill, S.C. and von Hippel, P.H. 1989. Calculation of protein extinction coefficients from amino acid sequence data. Anal. Biochem. 182 319–326. [DOI] [PubMed] [Google Scholar]
- Gittelman, M.S. and Matthews, C.R. 1990. Folding and stability of trp aporepressor from Escherichia coli. Biochemistry 29 7011–7020. [DOI] [PubMed] [Google Scholar]
- Gloss, L.M. and Matthews, C.R. 1998a. The barriers in the biomolecular and unimolecular folding reaction of the dimeric core domain of Escherichia coli Trp repressor are dominated by enthalpic contributions. Biochemistry 37 16000–16010. [DOI] [PubMed] [Google Scholar]
- ———. 1998b. Mechanism of folding of the dimeric core domain of Escherichia coli Trp repressor: A nearly diffusion-limited reaction leads to the formation of an on-pathway dimeric intermediate. Biochemistry 37 15990–15999. [DOI] [PubMed] [Google Scholar]
- Gloss, L.M. and Placek, B.J. 2002. The effect of salts on the stability of the H2A–H2B histone dimer. Biochemistry [DOI] [PubMed]
- Gloss, L.M., Simler, B.R., and Matthews, C.R. 2001. Rough energy landscapes in protein folding: Dimeric E. coli Trp repressor folds through three parallel channels. J. Mol. Biol. 312 1121–1134. [DOI] [PubMed] [Google Scholar]
- Gualfetti, P.J., Bilsel, O., and Matthews, C.R. 1999a. The progressive development of structure and stability during the equilibrium folding of the α subunit of tryptophan synthase from Escherichia coli. Protein Sci. 8 1623–1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gualfetti, P.J., Iwakura, M., Lee, J.C., Kihara, H., Bilsel, O., Zitzewitz, J.A., and Matthews, C.R. 1999b. Apparent radii of the native, stable intermediates and unfolded conformers of the α-subunit of tryptophan synthase from E. coli, a TIM barrel protein. Biochemistry 38 13367–13378. [DOI] [PubMed] [Google Scholar]
- Henry, E.R. and Hofrichter, J. 1992. Singular value decomposition: Application of analysis of experimental data. Methods Enzymol. 210 129–192. [Google Scholar]
- Ho, S.N., Hunt, H.D., Horton, R.M., Pullen, J.K., and Pease, L.R. 1989. Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene 77 51–59. [DOI] [PubMed] [Google Scholar]
- Inlow, J.K. and Baldwin, T.O. 2002. Mutational analysis of the subunit interface of Vibrio harveyi bacterial luciferase. Biochemistry 41 3906–3915. [DOI] [PubMed] [Google Scholar]
- Jackson, S.E. 1998. How do small single-domain proteins fold? Fold Des. 3 R81–91. [DOI] [PubMed] [Google Scholar]
- Jaenicke, R. 1987. Folding and association of proteins. Prog. Biophys. Mol. Biol. 49 117–237. [DOI] [PubMed] [Google Scholar]
- Jaenicke, R. and Lilie, H. 2000. Folding and association of oligomeric and multimeric proteins. Adv. Protein Chem. 53 329–401. [DOI] [PubMed] [Google Scholar]
- Janin, J. 1997. The kinetics of protein–protein recognition. Proteins 28 153–161. [DOI] [PubMed] [Google Scholar]
- Karantza, V., Baxevanis, A.D., Freire, E., and Moudrianakis, E.N. 1995. Thermodynamic studies of the core histones: Ionic strength and pH dependence of H2A-H2B dimer stability. Biochemistry 34 5988–5996. [DOI] [PubMed] [Google Scholar]
- Karantza, V., Freire, E., and Moudrianakis, E.N. 1996. Thermodynamic studies of the core histones: pH and ionic strength effects on the stability of the (H3–H4)/(H3–H4)2 system. Biochemistry 35 2037–2046. [DOI] [PubMed] [Google Scholar]
- ———. 2001. Thermodynamic studies of the core histones: Stability of the octamer subunits is not altered by removal of their terminal domains. Biochemistry 40 13114–13123. [DOI] [PubMed] [Google Scholar]
- Kim, P.S. and Baldwin, R.L. 1990. Intermediates in the folding reactions of small proteins. Annu. Rev. Biochem. 59 631–660. [DOI] [PubMed] [Google Scholar]
- Kim, D.H., Jang, D.S., Nam, G.H., and Choi, K.Y. 2001a. Folding mechanism of ketosteroid isomerase from Comamonas testosteroni. Biochemistry 40 5011–5017. [DOI] [PubMed] [Google Scholar]
- Kim, D.H., Nam, G.H., Jang, D.S., Yun, S., Choi, G., Lee, H.C., and Choi, K.Y. 2001b. Roles of dimerization in folding and stability of ketosteroid isomerase from Pseudomonas putida biotype B. Protein Sci. 10 741–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraulis, P.J. 1991. MOLSCRIPT: A program to produce both detailed and schematic plots of protein structures. J. Appl. Crystallogr. 24 946–950. [Google Scholar]
- Liu, Y. and Eisenberg, D. 2002. 3D domain swapping: As domains continue to swap. Protein Sci. 11 1285–1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luger, K., Mader, A.W., Richmond, R.K., Sargent, D.F., and Richmond, T.J. 1997a. Crystal structure of the nucleosome core particle at 2.8 Å resolution. Nature 389 251–260. [DOI] [PubMed] [Google Scholar]
- Luger, K., Rechsteiner, T.J., Flaus, A.J., Waye, M.M., and Richmond, T.J. 1997b. Characterization of nucleosome core particles containing histone proteins made in bacteria. J. Mol. Biol. 272 301–311. [DOI] [PubMed] [Google Scholar]
- Mann, C.J., Xiao, S., and Matthews, C.R. 1995. Characterization of the slow folding reactions of trp aporepressor from Escherichia coli by mutational analysis of prolines and catalysis by a peptidylprolyl isomerase. Biochemistry 34 14573–14580. [DOI] [PubMed] [Google Scholar]
- Matthews, C.R. 1993. Pathways of protein folding. Annu. Rev. Biochem. 62 653–683. [DOI] [PubMed] [Google Scholar]
- Milla, M.E. and Sauer, R.T. 1994. P22 Arc repressor: Folding kinetics of a single-domain, dimeric protein. Biochemistry 33 1125–1133. [DOI] [PubMed] [Google Scholar]
- Munoz, V., Henry, E.R., Hofrichter, J., and Eaton, W.A. 1998. A statistical mechanical model for β-hairpin kinetics. Proc. Natl. Acad. Sci. 95 5872–5879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers, J.K., Pace, C.N., and Scholtz, J.M. 1995. Denaturant m values and heat capacity changes: Relation to changes in accessble surface areas of protein folding. Protein Sci. 4 2138–2148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noland, B.W., Dangott, L.J., and Baldwin, T.O. 1999. Folding, stability, and physical properties of the α subunit of bacterial luciferase. Biochemistry 38 16136–16145. [DOI] [PubMed] [Google Scholar]
- Placek, B.J. and Gloss, L.M. 2002. The N-terminal tails of the H2A–H2B Histones affect dimer structure and stability. Biochemistry 41 14960–14968. [DOI] [PubMed] [Google Scholar]
- Plaxco, K.W., Simons, K.T., Ruczinski, I., and Baker, D. 2000. Topology, stability, sequence, and length: Defining the determinants of two-state protein folding kinetics. Biochemistry 39 11177–11183. [DOI] [PubMed] [Google Scholar]
- Seckler, R. 2000. Assmbly of multi-subunit structures. In Mechanisms of protein folding, 2nd ed. (ed. R.H. Pain), pp. 279–308. Oxford University Press, New York.
- Srivastava, A.K. and Sauer, R.T. 2000. Evidence for partial secondary structure formation in the transition state for arc repressor refolding and dimerization. Biochemistry 39 8308–8314. [DOI] [PubMed] [Google Scholar]
- Starich, M.R., Sandman, K., Reeve, J.N., and Summers, M.F. 1996. NMR structure of HMfB from the hyperthermophile, Methanothermus fervidus, confirms that this Archaeal protein is a histone. J. Mol. Biol. 255 187–203. [DOI] [PubMed] [Google Scholar]
- Thompson, P.A., Eaton, W.A., and Hofrichter, J. 1997. Laser temperature jump study of the helixcoil kinetics of an alanine peptide interpreted with a ‘kinetic zipper’ model. Biochemistry 36 9200–9210. [DOI] [PubMed] [Google Scholar]
- Topping, T.B., Hoch, D.A., and Gloss, L.M. 2003. Folding mechanism of FIS, the intertwined, dimeric factor for inversion stimulation. J. Mol. Biol. 335 1065–1081. [DOI] [PubMed] [Google Scholar]
- Wallace, L.A. and Dirr, H.W. 1999. Folding and assembly of dimeric human glutathione transferase A1-1. Biochemistry 38 16686–16694. [DOI] [PubMed] [Google Scholar]
- Wallace, L.A. and Matthews, C.R. 2002. Sequential vs. parallel protein-folding mechanisms: Experimental tests for complex folding reactions. Biophys. Chem. 101–102 113–131. [DOI] [PubMed] [Google Scholar]
- Wallace, L.A., Sluis-Cremer, N., and Dirr, H.W. 1998. Equilibrium and kinetic unfolding properties of dimeric human glutathione transferase A1-1. Biochemistry 37 5320–5328. [DOI] [PubMed] [Google Scholar]
- Xie, X., Kokubo, T., Cohen, S.L., Mirza, U., Hoffmann, A., Chait, B.T., Roeder, R.G., Nakatani, Y., and Burley, S.K. 1996. Structural similarity between TAFs and the heterotetrameric core of the histone octamer. Nature 380 316–322. [DOI] [PubMed] [Google Scholar]
- Xu, D., Tsai, C.J., and Nussinov, R. 1998. Mechanism and evolution of protein dimerization. Protein Sci. 7 533–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu, W., Sandman, K., Lee, G.E., Reeve, J.N., and Summers, M.F. 1998. NMR structure and comparison of the archaeal histone HFoB from the mesophile Methanobacterium formicicum with HMfB from the hyperthermophile Methanothermus fervidus. Biochemistry 37 10573–10580. [DOI] [PubMed] [Google Scholar]
- Zitzewitz, J.A., Bilsel, O., Luo, J., Jones, B.E., and Matthews, C.R. 1995. Probing the folding mechanism of a leucine zipper peptide by stopped-flow circular dichroism spectroscopy. Biochemistry 34 12812–12819. [DOI] [PubMed] [Google Scholar]
- Zitzewitz, J.A., Ibarra-Molero, B., Fishel, D.R., Terry, K.L., and Matthews, C.R. 2000. Preformed secondary structure drives the association reaction of GCN4-p1, a model coiled-coil system. J. Mol. Biol. 296 1105–1116. [DOI] [PubMed] [Google Scholar]