

Abstract
Xyl1 from Streptomyces sp. S38 belongs to the low molecular mass family 11 of endo-β-1,4-xylanases. Its three-dimensional structure has been solved at 2.0 Å and its optimum temperature and pH for enzymatic activity are 60°C and 6.0, respectively. Aspergillus kawachii xylanase XynC belongs to the same family but is an acidophilic enzyme with an optimum pH of 2.0. Structural comparison of Xyl1 and XynC showed differences in residues surrounding the two glutamic acid side chains involved in the catalysis that could be responsible for the acidophilic adaptation of XynC. Mutations W20Y, N48D, A134E, and Y193W were introduced by site-directed mutagenesis and combined in multiple mutants. Trp 20 and Tyr 193 are involved in substrate binding. The Y193W mutation inactivated Xyl1 whereas W20Y decreased the optimum pH of Xyl1 to 5.0 and slightly increased its specific activity. The N48D mutation also decreased the optimum pH of Xyl1 by one unit. The A134E substitution did not induce any change, but when combined with N48D, a synergistic effect was observed with a 1.4 unit decrease in the optimum pH. Modeling showed that the orientations of residue 193 and of the fully conserved Arg 131 are different in acidophilic and “alkaline” xylanases whereas the introduced Tyr 20 probably modifies the pKa of the acid–base catalyst via residue Asn 48. Docking of a substrate analog in the catalytic site highlighted striking differences between Xyl1 and XynC in substrate binding. Hydrophobicity calculations showed a correlation between acidophilic adaptation and a decreased hydrophobicity around the two glutamic acid side chains involved in catalysis.
Keywords: endo-β-1,4-xylanase; acidophilicity; site-directed mutagenesis; structural analysis; hydrophobicity; docking
Xylan, the major constituent of hemicellulose, is a heterogeneous polysaccharide consisting of a β-1,4-linked D-xylose backbone substituted to varying degrees with O-acetyl, α-L-arabinofuranosyl, 4-O-methylglucuronic acid groups or α-1,2-linked glucuronic acids (Singh et al. 2003). Its complete breakdown requires a set of enzymes among which endo-β-1,4-xylanases (EC 3.2.1.8) are crucial for depolymerization into xylo-oligomers of various lengths. Xylanases are widely used in many industrial applications such as pulp biobleaching in the paper industry, baking, saccharification of lignocellulosic biomass, wine making, and fruit juice clarification (Sunna and Antranikian 1997; Wouters et al. 2001). On the basis of amino acid sequence similarities and hydrophobic cluster analysis, xylanases have been classified into families 10 and 11 of glycoside hydrolases although psychrophilic xylanases have been recently discovered and classified into family 8 of glycoside hydrolases (Wong et al. 1988; Gilkes et al. 1991; Henrissat and Davies 1997; Collins et al. 2002). Family 10 xylanases and some cellulases exhibit molecular masses higher than 30 kD and (α/β)8 barrel folds whereas the Mr values of family 11 are generally lower and their structures consist of a single catalytic domain with a sandwichlike fold. This fold resembles a partly closed right hand composed of two β-sheets and one α helix, which form a large cleft where 3 to 7 xylose subunits can be accommodated (Torronen and Rouvinen 1995; Gruber et al. 1998). Family 11 members are highly similar; they show closely related three-dimensional structures and catalytic site geometries (Torronen et al. 1993; Torronen and Rouvinen 1997). Catalysis occurs by a double displacement mechanism whereby the anomeric configuration of the glycosidic oxygen is retained. It is thought to involve two conserved glutamic acids, functioning, respectively, as nucleophile and as general acid–base catalyst. However, the optimum pH is highly variable within this family and xylanase activity can be observed at pH values ranging from 2.0 to 11.0. Acidophilic and “alkaline” xylanases are active in pH ranges 2.0–6.0 and 4.0–11.0, respectively (Torronen and Rouvinen 1997; Joshi et al. 2000). It is of interest to understand these features in relation to industrial applications where endo-β-1,4-xylanases are widely used under extreme pH and temperature conditions. Recent studies have partly clarified the basis of acidophilicity, and it appears that the pH profile is influenced by the nature of residues surrounding the catalytic glutamic acids whose pKas are finely tuned by a complex hydrogen bond network (Torronen and Rouvinen 1997). Indeed, comparison of primary and tertiary structures have shown that, in the catalytic site of acidophilic endo-β-(1,4)-xylanases, an aspartic acid is hydrogen bonded to the acid/base catalyst whereas it is replaced by an asparagine in the alkaline xylanases (Torronen and Rouvinen 1995). Moreover, site-directed mutagenesis studies have shown the importance of neighboring residues in the finetuning of the pKas of these glutamates, in particular, that of the acid–base catalyst (Krengel and Dijkstra 1996; Fushinobu et al. 1998; Joshi et al. 2001). Interestingly, conformational changes induced by either substrate binding or pH modification have been observed in the catalytic site (Torronen and Rouvinen 1995). Indeed, the acid–base catalyst can adopt two positions that might correspond to different pKa values so that it can accept or donate a proton (Torronen et al. 1994). Moreover, crystal structures and molecular dynamic simulations of the catalytic pathway have suggested an opening–closing movement of the active site proceeding by way of three different conformations (Muilu et al. 1998).
Xyl1 from Streptomyces sp. S38 is a family 11 endo-β-1,4-xylanase. It is one of the most active xylanases presently described (Georis et al. 2000). Its optimum temperature is 60°C and its optimum pH about 6.0. It exhibits interesting properties for bleaching and delignification and its structure has been solved at 2.0 Å (Wouters et al. 2001). In an attempt to better understand the adaptation of endo-β-1,4-xylanases to acidic environments, the Xyl1 structure was compared to those of two acidophilic xylanases, and features potentially responsible for acidophilicity were identified and introduced in Xyl1 by site-directed mutagenesis.
Results
Comparison of Xyl1 with acidophilic endo-β-1,4-xylanases
Alignment of the amino acid sequence of Xyl1 with those of other family 11 endo-β-1,4-xylanases highlighted two groups, one containing so-called alkaline xylanases presenting basic pIs and the second acidophilic xylanases with acidic pIs and lower optimum pH values (Fig. 1 ▶). Xyl1 shares 51.5% and 45.9% identity, respectively, with the acidophilic Aspergillus kawachii XynC and Trichoderma reesei Xyn1 enzymes. Comparison of the three-dimensional structures of the enzymes and sequence alignment highlighted the following interesting differences:
Figure 1.

Sequence alignment of family 11 xylanases. The three acidophilic enzymes are from Aspergillus kawachii (XynC; Ito et al. 1992), Aspergillus niger (Xyl1; Krengel et al. 1996), and Trichoderma reesei (XYN1, Torronen et al. 1992). The so-called “alkaline” enzymes are from Trichoderma reesei (XYN2; Torronen et al. 1992), Trichoderma harzianum (Wong and Saddler 1992), Aspergillus niger (XynNB; Krengel and Dijkstra 1996), Streptomyces sp. S38 (Xyl1; Georis et al. 2000), Streptomyces lividans (XynB; Kluepfel et al. 1990), Streptomyces EC3 (xylanase C; Mazy-Servais et al. 1996), Bacillus D3 (Harris et al. 1997), and Bacillus circulans (XynA; Yang et al. 1989). The residues referred to in the text are in bold, the catalytic Glu residues underlined, and the mutated residues in bold and underlined. The individual numbering of each enzyme is shown. In the text, the standard numbering resulting from the alignment is used consistently. n.d.: Not determined.
At the edge of the cleft of Xyl1, residues 48 and 134 are Asn and Ala, respectively (Fig. 2 ▶). Asn48 is conserved in the alkaline xylanase group and is assumed to be, in part, responsible for the pH adaptation (Fig. 1 ▶; Torronen and Rouvinen 1995; Fushinobu et al. 1998). As seen in Table 1, Asn 48 is located at a bonding distance of 3.6 Å from the acid/base catalyst Glu 191 (distance between heavy atoms; Table 1; Wouters et al. 2001). The corresponding residue in acidophilic xylanases is an Asp making a shorter hydrogen bond with the acid–base catalyst; the distances between AspOD1 and the acid/base catalyst Glu(A/B)OE1 are 2.8 Å and 2.9 Å in XynC and Xyn1, respectively (Fig. 1 ▶; Table 1). In the acidophilic xylanases, the residue corresponding to Ala 134 is a Glu located in the thumb at the bottom and pointing into the active site. Together with the Asp 48 residue at the lower boundary of the cleft, Glu 134 confers a significantly more negative environment along the edge of the acidophilic enzyme cleft (Fushinobu et al. 1998).
Arg 131, fully conserved among family 11 xylanases, points towards the thumb in Xyl1, whereas it protrudes toward the cord in acidophilic enzymes (Fig. 2 ▶).
The compared structures present aromatic residues nicely lined up inside the active site and at the edge of the cleft. In particular, Tyr 16, Tyr 83, Tyr 95, and Tyr 185 are conserved among all family 11 xylanases, whereas Trp 148 is only conserved in alkaline xylanases and is usually replaced by Phe or Ile in the acidophilic group (Fig. 1 ▶). Tyr 127 is substituted by Trp in Xyn1 and by a Cys forming a disulfide bridge with Cys 106 in XynC. Interestingly, in acidophilic xylanases, Trp 20 and Tyr 193 are replaced by Tyr and Trp, respectively. Tyr 20 in Xyn1 is involved in a stacking interaction with the xylose ring at subsite -2 (Torronen and Rouvinen 1997). In the same way, Trp 193 in Xyn1 is assumed to determine subsite +2 (Torronen and Rouvinen 1997). This difference between acidophilic and alkaline xylanases is conserved among all members of the two groups (Fig. 1 ▶).
Two salt bridges are found in Xyn1 and three in Xyl1 and XynC (Table 2). One (A) is common to the three enzymes and is the only one shared by Xyl1 and the acidophilic enzymes. Salt bridge B, located in the thumb is only found in this latter group. It cannot be formed in Xyl1 because of the E134A substitution. Salt bridge C, only found in XynC, is replaced by a hydrogen bond in Xyl1. Salt bridges D and E are specific to Xyl1.
Figure 2.

Docking of a chain of eight xylose residues in the active site of XynC and Xyl1. Six of the eight xylose residues were accommodated in the catalytic sites of Xyl1 and XynC. The geometry of the complex was optimized by energy minimization.
Table 1.
Some important interatomic distances in the crystallographic (XynA, Xyl1-WT, XYN1, and XynC) and modeled (Xyl1 mutants) structures of the proteins
| XynA | Xyl1 | A134E | W20Y/N48D/A134E | N48D | W20Y | N48D/A134E | XYN1 | XynC | |
| Optimum pH | 6 | 6 | 6 | 5.3 | 5 | 5 | 4.7 | 3.5 | 2 |
| N/D48OD1–Glu191OE1 | 3.59 | 3.6 | 3.53 | 3.45 | 3.03 | 2.95 | 3.47 | 2.9 | 2.8 |
| Glu191OE2–(Wat1)O | 2.97 | 2.99 | 2.99 | 2.85 | 2.71 | 2.7 | 2.65 | 2.65 | 2.59 |
| (Wat1)O–Tyr95OH | 2.96 | 2.95 | 2.95 | 2.7 | 2.85 | 2.76 | 2.71 | 2.68 | 2.67 |
| Tyr95OH–Glu93OE1 | 2.77 | 2.75 | 2.78 | 2.76 | 2.82 | 2.82 | 2.77 | 2.75 | 2.71 |
| Glu93OE2–(Wat2)O | 2.84 | 2.82 | 3.1 | 3.38 | 3.41 | 2.82 | 3.39 | 3.32 | 3.37 |
| Glu93OE2–Tyr83OH | 2.76 | 2.75 | 2.76 | 2.74 | 2.77 | 2.75 | 2.73 | 2.74 | 2.76 |
| Glu134OE1–(Wat2)O | — | — | 2.6 | 2.74 | — | — | 2.75 | 2.75 | 2.77 |
| Glu191OE2–(Wat1)O | 8.7 | 8.69 | 8.72 | 8.31 | 8.33 | 8.28 | 8.13 | 8.08 | 7.97 |
| Arg131NE–Glu191OE1 | 7.16 | 8.99 | 8.20 | 8.27 | 8.85 | 8.69 | 8.26 | 8.08 | 10.12 |
| Arg131NH1–N/D48OD1 | 4.51 | 6.98 | 6.28 | 6.23 | 6.56 | 6.50 | 6.14 | 8.08 | 10.45 |
XynA: Bacillus circulans; XYN1: Trichoderma reesei; XynC: Aspergillus kawachii; Xyl1: Streptomyces sp. S38; Wat1 and Wat2: water molecules.
Table 2.
Side-chain interactions in the wild-type enzymes
| Salt bridge | XYN1 | XynC | Xyl1 | Std. numbering |
| A | E80-R132 | E84-R138 | D92-R144 | 98–155 |
| B | R109-E112 | R115-E118 | R121/A124 | 131–134 |
| C | N107/R109 | D113-R115 | T119/R121 | 129–131 |
| D | Q116/- | T116/- | E128-K131 | 138–141 |
| E | -/T103 | N90/Q109 | R98-D115 | 104–125 |
Salt bridges in the wild-type enzymes are in bold. The corresponding residues that cannot form salt bridges are shown in italic. The individual numbering of each enzyme is used, but the last column shows the standard numbering of Figure 1 ▶.
Site-directed mutagenesis
On the basis of these observations, four residues that could be responsible for the acidophilic adaptation were chosen and mutated in the wild-type Xyl1 enzyme. Four variants were constructed with mutations W20Y, N48D, A134E, and Y193W and three combinations W20Y/Y193W, N48D/ A134E, and W20Y/N48D/A134E, made in order to study the possible interdependence of these mutations and partly reproduce the catalytic site of an acidophilic xylanase. All substitutions were located in the catalytic site, in the neighborhood of the two Glu residues involved in the catalysis.
Preliminary characterization of the recombinants proteins
The modified proteins were expressed in Streptomyces parvulus as previously described (Georis et al. 2000). All recombinant strains exhibited xylanolytic activity when plated on 1% oats spelt plates with the exceptions of those producing the Y193W and W20Y/Y193W mutants. Secretion of the two proteins was verified by Western blot, but no activity was detected in the culture supernatants.
Thermophilicity and thermostability
Thermophilicity of the active recombinants proteins N48D, A134E, W20Y, N48D/A134E, and W20Y/N48D/A134E was evaluated and the temperature dependence of their activities established. All the purified proteins exhibited maximal activity at 60°C, similar to the wild-type enzyme (not shown).
Thermal unfolding curves were established at pH 6.0 between 35°C and 85°C. The curves were obtained by following the decrease of fluorescence at 323 nm induced by a temperature gradient of 1 °C/min. Thermal denaturation of the wild-type enzyme Xyl1 is irreversible and its apparent Tm was previously evaluated at 61.1 ± 0.1 °C under the same conditions (Georis et al. 2000). The apparent Tm value of W20Y was higher whereas those of N48D, A124E, N48D/A134E and W20Y/N48D/A134E were slightly lower (Table 3).
Table 3.
Properties of the purified wild-type and mutant proteins
| Mutation(s) | Optimum pH | Specific activity (IU/mg) | Tm (°C) |
| Wild type | 6 | 2100 ± 200 | 61.1 ± 0.1 |
| N48D | 5 | 1180 ± 90 | 60.9 ± 0.1 |
| W20Y | 5 | 2240 ± 200 | 63.7 ± 0.2 |
| A134E | 6 | 530 ± 60 | 61 ± 0.1 |
| N48D/A134E | 4.7 | 850 ± 70 | 59.1 ± 0.3 |
| W20Y/N48D/A134E | 5.3 | 580 ± 40 | 59.2 ± 0.2 |
Specific activities (IU/mg) of the purified recombinant and wild-type proteins and Tm values obtained by fluorescence (°C).
Acid stability
The wild-type enzyme and the mutants were tested for their stability by measuring their residual activities at optimum pH after incubation at pH 1.0, 3.0, and 6.0 at 20°C. Whatever the pH, 100% activity was retained after a 24-h incubation. Moreover, far- and near-UV CD spectra were recorded between 195 and 260 nm from pH 1.0 to 6.0 for the wild-type enzyme and the mutants after 10 min and 24 h of incubation at 20°C. No change in the spectra was observed, suggesting that all the proteins were quite stable and that the low pH did not induce major changes in the native structure (not shown).
pH profiles of the purified proteins
The activity of the five purified proteins, W20Y, N48D, A134E, N48D/A134E, and W20Y/N48D/A134E, was assayed at pH values between 2.0 and 9.0. The wild-type enzyme optimum pH was close to 6.0 (Fig. 3 ▶). Its activity was reduced by 95% at pH 3.0 and was negligible at pH 2.0. N48D and W20Y were optimally active at pH 5.0. Moreover, they showed, respectively, 33% and 30% of their maximum respective activities at pH 3.0 (Fig. 3 ▶). The A134E mutant showed an activity profile similar to that of the wild-type enzyme although slightly narrower. Interestingly, the double mutant N48D/A134E was optimally active at pH 4.7 and retained 37% of the maximum activity at pH 3.0, suggesting that the A134E mutation lowers the optimum pH of this mutant but not that of the wild-type enzyme. However, the triple mutant showed an optimum pH of about 5.0 like N48D and W20Y, and thus it seems that the effects of some of the mutations are not additive.
Figure 3.

pH dependence of the activity of the wild-type and mutant enzymes. The solid lines were obtained by fitting the experimental data to the following equation:
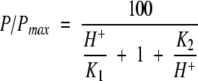 |
P represents the activity of the mutant and wild-type enzymes measured at various pH, and Pmax, the maximum activity observed for the wild-type enzyme (pH 6.0–6.5). Activities measured with 1% and 2% suspensions of oat spelt xylan were nearly identical at all pH values, indicating that P/Pmax mainly reflected kcat.
The specific activity of the wild-type enzyme was evaluated at 2100 ± 200 IU/mg at 60°C (Georis et al. 1999). With the exception of the W20Y protein, which presented a slightly higher specific activity, all mutants showed decreased specific activities at their respective optimum pH values (Table 3).
Modeling of the mutants
All distances referred to in the text are summarized in Table 1.
In the wild-type enzyme, the Trp 20 side chain does not interact with other residues. Modeling of the W20Y mutant is relatively straightforward, leading to the formation of a Tyr20OH–Asn48OD1 hydrogen bond, which itself induces a shortening of the Asn48ND2–Glu191OE1 distance from 3.6 Å to 2.95 Å.
The replacement of Ala 134 by Glu can give rise to several conformations of the latter side chain. All starting conformations lead to models in which Arg 131 is strongly hydrogen bonded to Glu 134 and Wat2 is also hydrogen bonded to the same residue. However, the formation of the salt bridge only slightly reorients the Arg side chain with respect to its position in the wild-type Xyl1. The Arg131NH1–Asp48OD1 and Arg131NE–Glu191OE1 distances decrease from 6.98 Å to 6.28 Å and from 8.99 Å to 8.20 Å, respectively.
The distances between Wat1 and Tyr 95 on the one hand and Glu 191 on the other are similar to those found in the wild-type enzyme. By contrast, a shortening of the Tyr95OH–(Wat1)O–Glu191OE2 distances is also observed (2.95 Å/2.85 Å and 2.99 Å/2.71 Å) in the N48D model, and this is even more obvious in the cases of the N48D/A134E and W20Y/N48D/A134E mutants.
The side chain of Tyr 193 is not involved in hydrogen bonding interactions with other residues of the wild-type enzyme, and is located in a position that does not impair binding of the substrate. The Y193W mutation yields two possible stable conformations of the residue: In the first one, a hydrogen bond is formed between Trp193NE1 and Glu191OE2 (3.27 Å) whereas in another one, 10 kcal/mole less stable, a Trp193NE1–Gln194OE2 hydrogen bond (3.25 Å) is observed. In both cases, the Trp side chain protrudes into the active site, its orientation being somewhat constrained by the Asn 76 side chain.
Hydrophobicity calculations
The method of Mehler and Guarnieri (1999; see Materials and Methods) was used to evaluate the environment of Glu 191 and Glu 93 because the local environment should be a key determinant of pKa values of these residues. This was done using equation 1 and taking account of all heavy atoms within a sphere of 4.25 Å radius around the two catalytically important glutamic acids. In Xyl1 and in the mutants, Asn/ Asp 48, Val 50, Tyr 79, Thr 81, Tyr 83, Trp 85, Tyr 95, Phe 144, and Gln 146 were concerned. Analysis of the local environments shows that the nearest neighbors of the acid–base catalyst are less hydrophilic than those of the nucleophile (Table 4). Interestingly, in XynC, Trp 193 influences the hydrophobicity around the Glu residues involved in catalysis whereas it does not in Xyl1. Moreover, due to mutations around the Glu residues and/or a slight reorganization of the active-site structure, the hydrophobicity values slightly change in the mutants.
Table 4.
Hydrophobicities of the microenvironments of the two catalytic glutamic acid residues
| Enzyme | Glu 93 | Glu 191 | Total |
| Xyl1 | −1.85 | −0.72 | −2.57 |
| W20Y | −2.17 | −2.06 | −4.23 |
| N48D | −1.85 | −2.16 | −4.01 |
| A134E | −1.85 | −0.72 | −2.57 |
| N48D/A134E | −2.48 | −2.48 | −4.96 |
| W20Y/N48D/A134E | −2.70 | −2.48 | −5.18 |
| XynC | −3.77 | −1.85 | −5.62 |
Values were obtained with equation 1 (see Materials and Methods). The more negative the value, the less hydrophobic the microenvironment of the two catalytic glutamic acid residues.
Enzyme–substrate interactions
Modeling of the substrate in the catalytic site of XynC and Xyl1 was performed and residues involved in the interactions compared (Fig. 2 ▶). In XynC, subsites are defined as −2, −1, +1, +2, and +3 whereas in Xyl1, one additional site (−3) is thought to be involved in the binding of the substrate (Georis et al. 2000). Interactions with subsite +1 are strictly conserved in the two xylanases and probably among all the aligned family 11 enzymes. A chain containing eight xylose rings was docked into the active site of Xyl1, on the basis of the results of the crystal structure of Bacillus agaradhaerens XYN11 (Sabini et al. 2001), and the geometry of the complex optimized by energy minimization. A similar analysis was performed for XynC.
In both structures, the xylose ring in subsite +1 interacts with Arg 131 and Tyr 95. In turn, the xylose in subsite −1 interacts with the carbonyl group of the conserved Pro 135, whereas the OH group of Tyr 185 is hydrogen bonded to xylose at subsite −2.
However, some interactions are different: At subsite −1 the xylose residue is hydrogen bonded to the guanidinium group of Arg 131 in Xyl1, an interaction that is not found in the case of XynC. In addition, some hydrogen bond interactions between xylose at −1 (Tyr 20) and −2 (Gln 18) are not observed with Xyl1, due to the following differences: Gln 18 in XynC is replaced by Ser in Xyl1 and XynC Tyr 20 is a Trp in Xyl1. In this latter case, the stacking interaction between the ring of residue 20 and the xylose moiety at subsite −1 is, however, conserved.
Discussion
Xyl1 from Streptomyces sp. S38 belongs to family 11 of endo-β-1,4-xylanases. Comparison of its structure with those of acidophilic xylanases of the same family revealed that acidophilic adaptation could be partly explained by replacements of residues surrounding the two glutamic acids involved in the catalysis.
The environment of the nucleophile Glu 93 is more conserved than that of the acid–base catalyst Glu 191. As previously described for Bacillus circulans XynA, the N48D mutation improved the acidophilic performance of Xyl1 by lowering its optimum pH (6.0 to 5.0) but at the cost of a decrease of nearly 50% in specific activity, whereas in the case of B. circulans enzyme, this mutation yielded an enzyme slightly more active than the wild type (Joshi et al. 2000). However, the decreased specific activity observed here might be due to the utilization of a different substrate. This result agrees with observations made by Fushinobu et al. (1998) on the mutational analysis of the Aspergillus kawachii XynC in which the inverse (D → N) mutation drastically increased the optimum activity pH from 2.0 to 5.0.
In the wild-type Xyl1 enzyme, residue Asn 48 is connected to the acid–base catalyst Glu 191 and makes a 0.6 Å longer bond than the corresponding hydrogen bond between Asp 48 and Glu 191 in the acidophilic XynC (3.6 Å versus 3 Å). Indeed, as previously suggested, Asn 48 and Glu 191 may function together as the general acid–base catalyst using a “reverse protonation” mechanism (Joshi et al. 2000). In this context, the Q146E mutation performed in the B. circulans XynA lowered its optimum pH by 0.6 units. Residue 146 is situated near the nucleophile Glu 93 and the introduction of a carboxylic group in position 146 lowers the pKa of Glu 93. These mutations highlight the importance of the close neighboring residues in determining the pH activity range.
Xylanases of the acidophilic group present a glutamic acid in the thumb (Glu 134). In XynC, this residue is assumed to contribute to the negative charge at the edge of the cleft and to partly determine its low optimum pH (Fushinobu et al. 1998). The neighboring Arg 131 is highly conserved in family 11 xylanases, underlining its importance (Sapag et al. 2002). In Xyl1, this residue is assumed to be close enough to maintain the low pKa of the nucleophile Glu 93 (Wouters et al. 2001). Interestingly, Joshi et al. showed that the R131N mutation in B. circulans XynA resulted in a dramatic activity loss accompanied by an increase of the pKas of the two glutamates but especially of the acid–base catalyst Glu 191, whereas mutation R131A in Thermomyces lanuginosus xylanase YNA was predicted to increase the pKa of the nucleophile without affecting the acid–base catalyst (Gruber et al. 1998; Joshi et al. 2001). The environment of residue Arg 131 is different in Xyl1 and in XynC and leads to a different orientation of its lateral chain. In Xyl1, this residue points toward the thumb, whereas, in the acidophilic xylanase, it protrudes towards the cord. In XynC, this residue is connected to Asp 129 and Glu 134 by two salt bridges. In Xyl1, none of these salt bridges can be formed because residue 129 and 134 are Thr and Ala, respectively. Moreover, Arg 131 fills the void space available due to the low steric hindrance of the shorter lateral chain of Ala 134. Docking experiments showed that, in Xyl1, this residue binds the substrate at subsites −1 and +1, whereas in XynC it only binds the xylose ring at subsite +1. The A134E mutation in the catalytic site of Xyl1 had no effect on the optimum pH of Xyl1 but decreased its specific activity by 75%. This substitution allows the putative formation of the salt bridge Arg131ND1–Glu134OE1 as found in XynC but not the formation of the second one. Thus, because of the lack of the second salt bridge, complete reorientation of Arg 131 in the mutant A134E probably could not occur as in XynC and the binding to the xylose ring in subsite −1 remains the same as in the wild-type Xyl1. In fact, this residue makes a bond network with neighboring residues and the substrate that is much of interest and probably is an important key in the acidophilic adaptation of xylanases. However, combination of mutations A134E and N48D led to a larger decrease of the optimum pH (1.4 pH unit) than when those mutations were separately introduced or with the additional W20Y substitution. This synergistic effect still remains obscure and it is not understood why the triple mutant does not present the largest decrease in optimum pH. Solving the three-dimensional structures of mutants will be necessary to understand and explain the exact effects of these mutations.
In the active site cleft of XynC, five subsites (+3, +2, +1, −1, and −2) have been identified where Tyr 20 and Trp 193 are located at subsites −2 and +1, respectively (Fushinobu et al. 1998). In this way, the structure of the inactive mutant E191C of B. circulans XynA complexed with xylotetraose revealed a stacking interaction of Trp 20 with the xylose ring at subsite −2 (Wakarchuk et al. 1994). Moreover, in Trichoderma reesei XYN1 and XYN2 xylanases, residue 20 is assumed to be part of subsites −2 and residue 193 of subsites +1 and +2, respectively (Torronen and Rouvinen 1995,Torronen and Rouvinen 1997). The W20Y mutation lowered the optimum pH of Xyl1 by one unit pH and slightly increased its specific activity, whereas Y193W inactivated the enzyme. We assume that the introduced Tyr 20 acts in the same way as Asp 48 to decrease the optimum pH of Xyl1. Actually, modeling showed that Tyr 20 attracts residue Asn 48 in a rocking motion by formation of a hydrogen bond between Tyr20OH and Asn48OD1 and induces a shortening of the distance between Glu191OE1 and Asn48ND2 from 3.6 Å to 2.95 Å. Interestingly, the orientation of residue 193 is different in acidophilic and in alkaline xylanases. In Xyl1, Tyr 193 protrudes into the solvent, whereas, in XynC, Trp 193 bends into the cleft. The Y193W substitution in Xyl1 may sterically affect neighboring residues, in particular residue Asn 48, which is very close, but also Asn 76, which may hinder the correct positioning of the Trp ring and thus probably trigger a reorganization of the hydrogen bond network. It remains noteworthy that, in XynC, Trp 193 influences the hydrophobicity around the Glu residues involved in catalysis and may probably influence the pKa of the acid/base catalyst. With the exception of the triple mutant W20Y/ N48D/A134E, calculations have shown that hydrophobicity around the nucleophile and the acid–base catalyst decreased with increasing acidophilic activity. This observation was also made in the case of the alkalophilic variants of the Neocallimastix patriciarum xylanase (Chen et al. 2001), where mutations increased hydrophobicity and alkalophilicity of the enzyme.
Materials and methods
Bacterial hosts strains and plasmids
Streptomyces parvulus IMET41380 and the Streptomyces plasmid vector pIJ486 were gifts from Drs. D.A. Hopwood and T. Kieser (John Innes Institute, Norwich, UK). pDML 1011 and pDML1012 were used as previously described (Georis et al. 1999). Escherichia coli DH5α was used as host strain. The E. coli–Streptomyces shuttle plasmid resulted from the subcloning of pDML1011 derivatives (obtained by site-directed mutagenesis) in the unique HindIII site of the pIJ486.
Media, antibiotics, and culture conditions
Streptomyces liquid cultures were grown in YEME (Hopwood et al. 1985) and M13 media (Morosoli et al. 1986) on rotatory shakers at 220 rpm and 28°C. R2YE or minimal agar medium (Hopwood et al. 1985) were used for plating. The E. coli strain was grown at 37°C in Luria–Bertani broth. Thiostrepton (Squibb) was used at final concentrations of 25 μg/mL in liquid and solid media, and 50 μg/mL in overlays. Ampicillin (Sigma Chemical) was used at 100 μg/mL.
After transformation by shuttle plasmids carrying different mutations, Xyl1 recombinant proteins were produced as follows: 25 mL of 48-h cultures of S. parvulus in Tryptic Soy Broth (TSB) were used to inoculate 1 L of M13 medium with 1% xylose as carbon source and 25 μg/mL of thiostrepton (Squibb). Cultures were grown in 1-L Erlenmeyer flasks containing a stainless steel spring to improve oxygen transfer and 250 mL of medium. They were incubated for 72 h at 28°C and 220 rpm on a rotatory shaker. The production of xylanase was monitored at 8-h intervals.
Cloning, mutagenesis, and DNA sequencing
Site-directed mutagenesis was performed on pDML1012 (Georis et al. 2000) by using the Quick-Change Site Directed Mutagenesis Kit from Stratagene according to the protocol recommended by the manufacturer and the following primers (Pharmacia LKB): W20Y (up): GGCTACTACTACTCGTTCTACACCGACGGCG GCG; W20Y (down): CGCCGCCGTCGGTGTAGAACGAGTA GTAGTAGCC; N48D (up): GCGGCGACTTCGTCGCAGGCAA GGG; N48D (down): CCCTTGCCTGCGACGAAGTCGCC GC; A134E (up): ACGCGGGTCAACGAGCCGTCCGTCGA AG; A134E (down): CTTCGACGGACGGCTCGTTGACCCGC GT; Y193W (up): GGCGACGGAGGGCTGGCAGAGCAGT GGGAGC; Y193W (down): GCTCCCACTGCTCTGCCAGCCC TCCGTCGCC. The polymerase chain reaction (PCR) conditions were 5 min at 95 °C, 15 times (30 s at 95 °C; 1 min at 55 °C; 8 min at 72 °C) and 3 min at 72 °C. pDML 1012 derivatives were isolated using GFX PCR DNA and Gel Band Purification Kit (Amersham Pharmacia Biotech Inc.) and nucleotide sequences were determined by double-strand sequencing using the dideoxy-nucleotide method and the Thermosequenase fluorescent DNA sequencing kit (Amersham) with universal and reverse primers. Experiments were performed on an Alf automated fluorescent DNA sequencer (Pharmacia).
pH-dependence of xylanase activity
Endo-β-1,4-xylanase (EC 3.2.1.8) activity was measured by the dinitrosalicylic acid method (DNS; Miller 1959). Aliquots (40 μL) of diluted enzyme were mixed with 360 μL of a 1% suspension of oat spelt xylan (Sigma Chemical Co.) at different pH and incubated for 10 min at 50 °C. Buffers were 0.05 M HCl/KCl (pH 1.0 to 2.0), 0.05 M sodium citrate (pH 2.5 to 6.0), 0.05 M sodium phosphate (pH 6.0 to 7.0) and 0.05 M Tris/HCl (pH 7.5 to 9.0). One international unit (IU) is defined as the amount of enzyme that releases 1 μmole of reducing sugar per minute.
Proteins purification and analysis
Culture supernatants (1 L) were harvested by continuous centrifugation at 10,000g for 20 min. They were microfiltered on 0.4 μm Millipore filters (Millipore) and diluted 10-fold with water. After adjustment to pH 5.0, 300 g of moist carboxymethylcellulose (CMC) equilibrated in 10 mM sodium citrate buffer (pH 5.0) was added. The exchanger was washed with 3 L of the same buffer and harvested by decanting. Elution of recombinant enzymes was performed batchwise with 1 L of 10 mM citrate buffer (pH 5.0) containing 1.5 M NaCl. Solutions were concentrated to 35 mL, dialyzed against 10 mM citrate buffer (pH 5.0) and loaded onto a Sulfopropyl Sepharose Fast Flow column (XK 26/40, 150 mL; Amersham Pharmacia Biotech). The enzyme was eluted with a linear NaCl gradient (450 mL) from 0 to 0.2 M. Fractions exhibiting xylanase activity were pooled, concentrated to 5 mL, and applied onto a molecular sieve column of Sephacryl S100 HR (2.6 × 95 cm, 500 mL; Amersham Pharmacia Biotech) equilibrated in 20 mM phosphate buffer (pH 7.0). The fractions containing xylanolytic activity were collected and concentrated to 0.5 mg/mL. Mr values were estimated by SDS-polyacrylamide gel electrophoresis (Laemmli 1970).
Fluorescence study and circular dichroism spectroscopy
Tryptophan fluorescence was monitored at a protein concentration of 25 μg/mL using a SLM-AMINCO model 8100/2 spectrofluorimeter (Spectronic Instruments) with excitation and emission wavelengths of 284 and 333 nm, respectively. Bandwiths were 1 nm for excitation and 4 nm for emission. Thermal denaturation of the wild-type enzyme and the mutants was recorded by following the fluorescence at 323 nm at a constant heating rate of 1°C/min from 35 to 85°C and pH 6.0. The temperature in the thermostated cell was monitored with a differential thermometer and denaturation curves were established as described by Pace (1986).
Far- and near-UV CD spectra were obtained using a JASCO model J-810 spectropolarimeter by scanning from 195 to 260 nm and 250 to 330 nm, respectively, with 0.2 nm steps and an averaging time of 6 s/nm, at 25 °C. All values were corrected for the contribution of the solvent, and quartz cells of 0.1 and 1 cm were used for far- and near-UV, respectively, at a protein concentration of 0.2 mg/mL.
Amino acid numbering
The standard numbering used in the text results from the alignment numbering in Figure 1 ▶.
Modeling of protein structures
The crystallographic structure of the Streptomyces sp. S38 xylanase Xyl1 (Wouters et al. 2001) was used. Models of the mutant proteins were obtained from the X-ray structure of the wild-type protein, and the local conformational space of the mutated amino acids was searched by a minimum perturbation approach (Shih et al. 1985). The protein structures were manipulated with InsightII (Accelerys Inc.). The geometries of the B. circulans XynA (PDB code 1BVV, from which the inhibitor molecule was removed), T. reesei XYN1 (PDB code 1XYN) and A. kawachii XynC (PDB code 1BK1) xylanases were optimized by energy minimization. As the PDB structure file of XynC did not contain the coordinates of water molecules, a Monte Carlo water bath was generated around the molecule, and their atomic positions were refined together with the enzyme coordinates. The minimization scheme was the same as in the case of the Xyl1 models.
Hydrophobicity
According to Mehler and Guarnieri (1999), a quantitative measure of the hydrophobicity of the microenvironment is needed because the response of the residue to changes in the local environment is the quantity of interest. On the basis of these considerations, the Rekker Fragmental Hydrophobic Constants were selected to evaluate the hydrophobicities of the microenvironments in the present calculations (Rekker 1977,Rekker 1979). The microenvironment of any group is defined as the region that lies within a sphere of radius 4.25 Å from any (nonhydrogen) atom belonging to the group. This radius was chosen because it essentially includes the first shell around each atom, but does not extend to atoms beyond this. Atoms satisfying this distance criterion are included in the calculation of the total hydrophobicity around the given glutamic acid group, according to the equation 1 (Mehler and Guarnieri 1999):
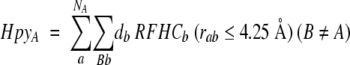 |
(1) |
where RFHCb is the contribution of atom b to the fragment’s fragmental hydrophobic constant, NA is the number of atoms in group A, and db = 0 or 1 if atom b has been counted or not, respectively.
Modeling of the substrate
The structure of a xylane backbone fragment containing eight sugars was built using the biopolymer module of the INSIGHTII (MSI). The crystal structure of the B. circulans xylanase complexed with xylotetraose (PDB code 1BCX; Wakarchuk et al. 1994) and 2-fluoro-xylose (PDB code 1BVV; Sidhu et al. 1999), as well as that of B. agaradhaerens xylanase complexed with xylanopyranoside (PDB code 1QH7; Sabini et al. 1999) were used as references for positioning the substrate in the active cleft of the Streptomyces sp. S38 Xyl1 enzyme. The structures of the four xylanases were superimposed by minimizing the root mean square deviation between the carbons of 12 residues bordering the active site. The two central units of the substrate were superimposed on the mean position of the two xylose residues in the complexes. The six other xyloses (three in the direction of the nonreducing end and three in the direction of the reducing end) were manually oriented to cover hydrophobic areas in the binding cleft and to allow possible hydogen-bonding interactions. The geometries of modeled enzyme–substrate complexes were optimized using the AMBER all atom force field (Pearlman et al. 1995) first by steepest descent energy minimization of atoms with forces above 500 kcal mole−1 Å−1 and then by conjugate gradient energy minimization until the root mean square gradient was less than 0.1 kcal mole−1 Å−1.
Acknowledgments
This work was supported in part by Ministry of Walloon Region, Department of Technology Research and Energy (conventions Bioval 1 no. 981/3860 and no. 981/3709) and by the Fonds pour la Formation à la Recherche dans l’Industrie et l’Agriculture (FRIA, Brussels, Belgium). F.d.L.E. is a fellow of FRIA. The purchase of the Jasco 810 equipment was supported in part by a grant from the Fonds de la Recherche Fondamentale et Collective (F.N.R.S., contract no. 2.4545.01). We thank Drs. J. Georis, M. Galleni, A. Matagne, and F. Bouillenne for their helpful advice.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.03556104.
References
- Chen, Y.L., Tang, T.Y., and Cheng, K.J. 2001. Directed evolution to produce an alkalophilic variant from a Neocallimastix patriciarum xylanase. Can. J. Microbiol. 47 1088–1094. [DOI] [PubMed] [Google Scholar]
- Collins, T., Meuwis, M.A., Stals, I., Claeyssens, M., Feller, G., and Gerday, C. 2002. A novel family 8 xylanase, functional and physicochemical characterization. J. Biol. Chem. 277 35133–35139. [DOI] [PubMed] [Google Scholar]
- Fushinobu, S., Ito, K., Konno, M., Wakagi, T., and Matsuzawa, H. 1998. Crystallographic and mutational analyses of an extremely acidophilic and acid-stable xylanase: Biased distribution of acidic residues and importance of Asp37 for catalysis at low pH. Protein Eng. 11 1121–1128. [DOI] [PubMed] [Google Scholar]
- Georis, J., Giannotta, F., Lamotte-Brasseur, J., Devreese, B., Van Beeumen, J., Granier, B., and Frere, J.M. 1999. Sequence, overproduction and purification of the family 11 endo-β-1,4-xylanase encoded by the xyl1 gene of Streptomyces sp. S38. Gene 237 123–133. [DOI] [PubMed] [Google Scholar]
- Georis, J., de Lemos Esteves, F., Lamotte-Brasseur, J., Bougnet, V., Devreese, B., Giannotta, F., Granier, B., and Frere, J.M. 2000. An additional aromatic interaction improves the thermostability and thermophilicity of a mesophilic family 11 xylanase: Structural basis and molecular study. Protein Sci. 9 466–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilkes, N.R., Henrissat, B., Kilburn, D.G., Miller Jr., R.C., and Warren, R.A. 1991. Domains in microbial β-1, 4-glycanases: Sequence conservation, function, and enzyme families. Microbiol. Rev. 55 303–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruber, K., Klintschar, G., Hayn, M., Schlacher, A., Steiner, W., and Kratky, C. 1998. Thermophilic xylanase from Thermomyces lanuginosus: High-resolution X-ray structure and modeling studies. Biochemistry 37 13475–13485. [DOI] [PubMed] [Google Scholar]
- Harris, G.W., Pickersgill, R.W., Connerton, I., Debeire, P., Touzel, J.P., Breton, C., and Perez, S. 1997. Structural basis of the properties of an industrially relevant thermophilic xylanase. Proteins 29 77–86. [PubMed] [Google Scholar]
- Henrissat, B. and Davies, G. 1997. Structural and sequence-based classification of glycoside hydrolases. Curr. Opin. Struct. Biol. 7 637–644. [DOI] [PubMed] [Google Scholar]
- Hopwood, D.A., Bidd, M.J., Chater, K.F., Kieser, T., Bruton, C.J., Kieser, H.M., Lydiate, D.J., Smith, C.P., Ward, J.M., and Shrempf, H. 1985. Genetic manipulation of Streptomyces. A laboratory manual. The John Innes Foundation, Norwich, UK.
- Ito, K., Iwashita, K., and Iwano, K. 1992. Cloning and sequencing of the xynC gene encoding acid xylanase of Aspergillus kawachii. Biosci. Biotechnol. Biochem. 56 1338–1340. [DOI] [PubMed] [Google Scholar]
- Joshi, M.D., Sidhu, G., Pot, I., Brayer, G.D., Withers, S.G., and McIntosh, L.P. 2000. Hydrogen bonding and catalysis: A novel explanation for how a single amino acid substitution can change the pH optimum of a glycosidase. J. Mol. Biol. 299 255–279. [DOI] [PubMed] [Google Scholar]
- Joshi, M.D., Sidhu, G., Nielsen, J.E., Brayer, G.D., Withers, S.G., and McIntosh, L.P. 2001. Dissecting the electrostatic interactions and pH-dependent activity of a family 11 glycosidase. Biochemistry 40 10115–10139. [PubMed] [Google Scholar]
- Kluepfel, D., Vats-Mehta, S., Aumont, F., Shareck, F., and Morosoli, R. 1990. Purification and characterization of a new xylanase (xylanase B) produced by Streptomyces lividans 66. Biochem. J. 267 45–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krengel, U., and Dijkstra, B.W. 1996. Three-dimensional structure of endo-1,4-β-xylanase I from Aspergillus niger: Molecular basis for its low pH optimum. J. Mol. Biol. 263 70–78. [DOI] [PubMed] [Google Scholar]
- Laemmli, U.K. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227 680–685. [DOI] [PubMed] [Google Scholar]
- Mazy-Servais, C., Moreau, A., Gerard, C., and Dusart, J. 1996. Cloning and nucleotide sequence of a xylanase-encoding gene from Streptomyces sp. strain EC3. DNA Seq. 6 147–158. [DOI] [PubMed] [Google Scholar]
- Mehler, E.L. and Guarnieri, F. 1999. A self-consistent, microenvironment modulated screened coulomb potential approximation to calculate pH-dependent electrostatic effects in proteins. Biophys. J. 77 3–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller, G.L. 1959. Use of dinitrosalicylic acid reagent for determination of reducing sugar. Anal. Chem. 31 426–428. [Google Scholar]
- Morosoli, R., Bertrand, J.L., Mondou, F., Shareck, F., and Kluepfel, D. 1986. Purification and properties of a xylanase from Streptomyces lividans. Biochem. J. 239 587–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muilu, J., Torronen, A., Perakyla, M., and Rouvinen, J. 1998. Functional conformational changes of endo-1,4-xylanase II from Trichoderma reesei: A molecular dynamics study. Proteins 31 434–444. [PubMed] [Google Scholar]
- Pace, C.N. 1986. Determination and analysis of urea and guanidine hydrochloride denaturation curves. Methods Enzymol. 131 266–280. [DOI] [PubMed] [Google Scholar]
- Pearlman, D., Case, D.A., Caldwell, J.A., Ross, W.S., Cheathman, T.E., Ferguson, D.M., Seibel, G.L., Singh, U.C., Weiner, P.K., and Kollman, P.A. 1995. AMBER 4.1. University of California, San Francisco, CA.
- Rekker, R.F. 1977. The hydrophobic fragmental constant. Elsevier, Amsterdam.
- ———. 1979. The hydrophobic fragmental constant: An extension to a 1000 data point set. Eur. J. Med. Chem. 14 479–488. [Google Scholar]
- Sabini, E., Sulzenbacher, G., Dauter, M., Dauter, Z., Jorgensen, P.L., Schulein, M., Dupont, C., Davies, G.J., and Wilson, K.S. 1999. Catalysis and specificity in enzymatic glycoside hydrolysis: A 2,5B conformation for the glycosyl-enzyme intermediate revealed by the structure of the Bacillus agaradhaerens family 11 xylanase. Chem. Biol. 6 483–492. [DOI] [PubMed] [Google Scholar]
- Sabini, E., Wilson, K.S., Danielsen, S., Schulein, M., and Davies, G.J. 2001. Oligosaccharide binding to family 11 xylanases: Both covalent intermediate and mutant product complexes display (2,5)B conformations at the active centre. Acta Crystallogr. D Biol. Crystallogr. 57 1344–1347. [DOI] [PubMed] [Google Scholar]
- Sapag, A., Wouters, J., Lambert, C., de Ioannes, P., Eyzaguirre, J., and Depiereux, E. 2002. The endoxylanases from family 11: Computer analysis of protein sequences reveals important structural and phylogenetic relationships. J. Biotechnol. 95 109–131. [DOI] [PubMed] [Google Scholar]
- Shih, H.H., Brady, J., and Karplus, M. 1985. Structure of proteins with single-site mutations: A minimum perturbation approach. Proc. Natl. Acad. Sci. 82 1697–1700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidhu, G., Withers, S.G., Nguyen, N.T., McIntosh, L.P., Ziser, L., and Brayer, G.D. 1999. Sugar ring distortion in the glycosyl-enzyme intermediate of a family G/11 xylanase. Biochemistry 38 5346–5354. [DOI] [PubMed] [Google Scholar]
- Singh, S., Madlala, A.M., and Prior, B.A. 2003. Thermomyces lanuginosus: Properties of strains and their hemicellulases. FEMS Microbiol. Rev. 27 3–16. [DOI] [PubMed] [Google Scholar]
- Sunna, A. and Antranikian, G. 1997. Xylanolytic enzymes from fungi and bacteria. Crit. Rev. Biotechnol. 17 39–67. [DOI] [PubMed] [Google Scholar]
- Torronen, A. and Rouvinen, J. 1995. Structural comparison of two major endo-1,4-xylanases from Trichoderma reesei. Biochemistry 34 847–856. [DOI] [PubMed] [Google Scholar]
- ———. 1997. Structural and functional properties of low molecular weight endo-1,4-β-xylanases. J. Biotechnol. 57 137–149. [DOI] [PubMed] [Google Scholar]
- Torronen, A., Mach, R.L., Messner, R., Gonzalez, R., Kalkkinen, N., Harkki, A., and Kubicek, C.P. 1992. The two major xylanases from Trichoderma reesei: Characterization of both enzymes and genes. Biotechnology 10 1461–1465. [DOI] [PubMed] [Google Scholar]
- Torronen, A., Kubicek, C.P., and Henrissat, B. 1993. Amino acid sequence similarities between low molecular weight endo-1,4-β-xylanases and family H cellulases revealed by clustering analysis. FEBS Lett. 321 135–139. [DOI] [PubMed] [Google Scholar]
- Torronen, A., Harkki, A., and Rouvinen, J. 1994. Three-dimensional structure of endo-1,4-β-xylanase II from Trichoderma reesei: Two conformational states in the active site. EMBO J. 13 2493–2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakarchuk, W.W., Campbell, R.L., Sung, W.L., Davoodi, J., and Yaguchi, M. 1994. Mutational and crystallographic analyses of the active site residues of the Bacillus circulans xylanase. Protein Sci. 3 467–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong, K.K.Y. and Saddler, J.N. 1992. Trichoderma xylanases, their properties and application. Crit. Rev. Biotechnol. 12 413–435. [Google Scholar]
- Wong, K.K., Tan, L.U., and Saddler, J.N. 1988. Multiplicity of β-1,4-xylanase in microorganisms: Functions and applications. Microbiol. Rev. 52 305–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wouters, J., Georis, J., Engher, D., Vandenhaute, J., Dusart, J., Frere, J.M., Depiereux, E., and Charlier, P. 2001. Crystallographic analysis of family 11 endo-β-1,4-xylanase Xyl1 from Streptomyces sp. S38. Acta Crystallogr. D Biol. Crystallogr. 57 1813–1819. [DOI] [PubMed] [Google Scholar]
- Yang, R.C., MacKenzie, C.R., Bilous, D., and Narang, S.A. 1989. Hyperexpression of a Bacillus circulans xylanase gene in Escherichia coli and characterization of the gene product. Appl. Environ. Microbiol. 55 1192–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]