

Abstract
It remains poorly understood as to how newly synthesized proteins that are required to act at specific synapses are translocated into only selected subsets of potentiated dendritic spines. Here, we report that F-actin, a major component of the skeletal structure of dendritic spines, may contribute to the regulation of synaptic specificity of protein translocation. We found that the stabilization of F-actin blocked the translocation of GFP-CaMKII and inhibited the diffusion of 3-kDa dextran into spines (in 2–3 weeks cultures). Neuronal activation in hippocampal slices and cultured neurons led to an increase in the activation (decrease in the phosphorylation) of the actin depolymerization factor, cofilin, and a decrease in F-actin. Furthermore, the induction of long-term potentiation by tetanic stimulation induced local transient depolymerization of F-actin both in vivo and in hippocampal slices (8–10 weeks), and this local F-actin depolymerization was blocked by APV, a N-methyl-d-aspartate (NMDA) receptor antagonist. These results suggest that F-actin may play a role in synaptic specificity by allowing protein translocation into only potentiated spines, gated through its depolymerization, which is probably triggered by the activation of NMDA receptors.
Keywords: dendritic spine, F-actin, information storage, LTP, protein translocation, synaptic specificity
Introduction
Dendritic spines are unique subcellular structures of neurons that have been studied for over 100 years (Cajal, 1888). They serve as postsynaptic sites for synaptic connections between neurons. The function of dendritic spines is still under intensive investigation, but spines have been implicated in mechanisms of memory. Changes in synaptic strength, such as long-term potentiation (LTP) and long-term depression (LTD), are believed to mediate memory. Protein synthesis is required for the late phase of LTP (Frey & Morris, 1997), the late phase of LTD (Linden, 1996), and the formation of memory (Davis & Squire, 1984). Furthermore, the induction of LTP (Shi et al., 1999) or the activation of neurons using glutamate and glycine (Shen & Meyer, 1999) results in the translocation of protein into dendritic spines. LTP exhibits activity-dependent synaptic specificity, resulting in potentiation of activated synapses only. Therefore, synaptic specificity of protein translocation into spines may be critical for long-term information storage (Frey & Morris, 1997; Schuman, 1997).
Proposed mechanisms accounting for synaptic specificity of LTP have been focused on active protein delivery, and the possibility of passive gating at dendritic spines has been virtually ignored. Recently, it has been recognized that dendritic spines may not be just an open space allowing free diffusion of all molecules. For example, application of Alexa dyes shows that not every dendritic spine is equally accessible to such diffusible dye molecules (De Simoni et al., 2004). The mechanisms and functional signifiscance of dendritic spine gating remain to be explored. In the present study, we have investigated the role of actin in the gating process in dendritic spines.
Actin is highly concentrated in dendritic spines and serves as a major skeletal protein in the subcellular structure (Fifkova & Delay, 1982; Matus et al., 1982). Whether this highly concentrated actin is needed for unique functions of dendritic spines is still unknown. Intracellular actin is present in monomer form (G-actin) or polymerized filament form (F-actin). Once it reaches a critical concentration, G-actin (with ATP) tends to form F-actin and eventually reaches a steady state. At the steady state, ATP will be hydrolysed into ADP and the length of filament will become stable (Korn et al., 1987). Observation with electron microscopy reveals that at the steady state, F-actin may form massive filament networks (Hartwin, 1992). A number of proteins that regulate actin cytoskeleton have been identified, including cofilin (also named actin depolymerization factor) and profilin (Bamburg et al., 1999; Chen et al., 2000; Pollard et al., 2000; Wear et al., 2000; Condeelis, 2001; Holt & Koffer, 2001). While profilin mediates the formation of actin filaments, cofilin can bind to F-actin in the interface between two subunits and effectively depolymerize the filament.
In addition to its capability of polymerization, actin can bind to many other proteins in a neuron, such as actinin in the postsynaptic density (Wyszynski et al., 1997) and synaptopodin in the spine neck (Deller et al., 2000). Those proteins may form a cross-linking complex that could prevent proteins or other large molecules from being translocated into the spine. Indeed, a meshwork-like structure has been observed in neuronal spines by cryofracture electron microscopy (Landis & Reese, 1983). More recently, massive labeling of F-actin in many dendritic spines was observed using electron microscopy (Capani et al., 2001).
Several lines of evidence support that F-actin plays a role in LTP induction and protein transport. Induction of LTP has been reported to increase F-actin content of dendritic spines in hippocampus in vitro and in vivo (Fukazawa et al., 2003; Okamoto et al., 2004). Furthermore, blocking actin polymerization with latrunculin A leads to a reduction of CaMKII labeling in distal dendrites in cultured hippocampal neurons (Allison et al., 2000) and also a decrease in LTP induction in hippocampal slices (Krucker et al., 2000). These previous studies have been taken as evidence that actin polymerization is necessary for the structural changes that occur with LTP. In contrast, the stabilization of F-actin by phalloidin reduces the level of expression of LTP (Kim & Lisman, 1999), and glutamate and glycine cause loss of F-actin (Shen & Meyer, 1999), suggesting that depolymerization of F-actin may also be important for spine changes induced by neuronal activity. Thus, additional studies are needed to better understand the complex dynamics and role of actin in the induction and maintenance of LTP.
In this report, we examined in more detail the dynamic changes of actin in dendritic spines under different conditions of neuronal activation. We hypothesize that neuronal activation, such as with LTP, leads to a biphasic effect on actin, consisting of an initial transient depolymerization of F-actin followed by a longer-lasting increase in F-actin. At its steady state, F-actin may constitute a complex that blocks the movement of protein into spines. With neuronal activation, the transient depolymerization of F-actin may allow protein translocation into spines, which combined with the subsequent increase in F-actin then results in structural changes associated with LTP. Thus, regulation of actin-mediated gating may provide a mechanism for specificity of protein translocation into spines.
Materials and methods
Electrophysiology in hippocampal slices
Detailed methods for slice electrophysiology have been previously described (Ouyang et al., 1997, 1999). Briefly, adult rats (8–10 weeks old) were anesthetized with halothane and decapitated. Acute hippocampal slices (500 µm) were prepared with a tissue chopper, and rested for at least 2 h in a humidified air chamber. Recording was performed in a chamber perfused with oxygenated artificial cerebrospinal fluid (ACSF; in mm: NaCl, 119; KCl, 2.5; MgSO4, 1.3; CaCl2, 2.5; NaH2PO4, 1.0; NaHCO3, 26.2; d-glucose, 11.0). A glass recording electrode was placed in the stratum radiatum of the CA1 region to measure extracellular field EPSPs. Two tungsten stimulating electrodes were placed approximately 1 mm apart in the stratum radiatum on opposite sides of the recording electrode to activate independent synaptic inputs. After a stable baseline was obtained for 30 min, a series of four tetani (100 Hz for 1 s each) with 30-s intervals were given to one of the two pathways, resulting in LTP only in the tetanized pathway (Ouyang et al., 1997). Subsequent analysis of F-actin and MAP2 levels compared the tetanized vs. non-tetanized sides. For each recorded slice, an adjacent slice from the same hippocampus was placed in the perfusing chamber without stimulating electrodes to serve as a chamber control. The slices were subsequently fixed in ice-cold 4% paraformaldehyde and 0.25% glutaraldehyde for 1 h, and cut into 50-µm sections for immunolabeling.
Electrophysiology for induction of LTP in hippocampus in vivo
Rats (8–10 weeks old) were anaesthetized with 3% halothane and placed in a stereotactic frame for electrode implantation. Aseptic technique was used to make 0.7-mm burr holes for stimulation (4.2 mm lateral to lambda) and recording (4.0 mm posterior, 2.0 mm lateral to bregma). Halothane was reduced to 1% during recording experiments. Concentric bipolar tungsten electrodes (10–15 KOhm impedance, WPI TM33CCINS) mounted on micromanipulators were used for both stimulation and recording. Optimal electrode depth was determined by observing the evoked synaptic response. Square wave stimuli (100–200 µs) were applied to the stimulating electrode from an isolated stimulator (Axon Instruments Isolator-11, Foster City, CA, USA). The stimulus intensity was obtained by applying graded intensities and selecting the intensity that produced a half-maximal amplitude population spike. Medial perforant pathway synaptic responses typically exhibited a monophasic response with a graded amplitude to increasing stimulus intensity, and a graded amplitude population spike on the rising phase of the synaptic response. The population spike exhibited almost complete paired-pulse inhibition at a 20-ms interval. For LTP experiments a stable baseline response was established by recording evoked synaptic responses every 5 min for 20 min. After recording the baseline response, LTP was induced by 2000 total stimuli, delivered in five 1-s-long tetanic (400 Hz) stimulus trains, 2 min apart, as described by Fukazawa et al. (2003). Responses were recorded at 5-min intervals after tetanus for different lengths of time (5, 30 or 60 min), after which the animal was quickly perfusion-fixed for F-actin immunolabeling.
Immunolabeling and imaging
Sections from slices, brains or fixed culture were first treated with Triton X-100, preblocked with buffer containing normal goat serum and then incubated with primary antibodies followed by three washes. The sections were further incubated with secondary antibodies, which were either fluorescein or cy3 conjugated goat anti-mouse or anti-rabbit IgG. Following the extensive wash, sections or cultures were mounted with anti-fade medium. Images were acquired using confocal microscopy and processed by Photoshop, and quantitatively analysed with either McPhase as previously described (Ouyang et al., 1997, 1999) or MetaMorph in later experiments. The number of spines and length of dendrites were measured with Neuron Zoom.
Western blotting
To study changes in phosphorylation of cofilin at Ser-3, we used similar quantitative immunoblotting methods as previously described (Ouyang et al., 1999). Briefly, whole-cell lysates of cultures or homogenates of hippocampal slices were prepared in sodium dodecyl sulphate–polyacrylamide gel electrophoresis (SDS–PAGE) sample buffer (containing 3% SDS, 2% β-mercaptoethanol and 5% glycerol in 60 mm Tris buffer, pH 6.7), boiled for 5 min, and stored at −20 °C. After the protein concentration was determined by using a modified Lowry method, an equal amount of protein was resuspended in the sample buffer, resolved in 7% SDS–PAGE and transferred to nitrocellular membranes. Blots were incubated with antibody for p-cofilin Ser-3 1 : 1000 (Cell Signaling, MA, USA). Bound antibodies were detected by the enhanced chemiluminescent method (Pierce). Computer-aided imaging analysis was conducted with MetaMorph. The ratio of changes was calculated and graphed with Excel.
Electron microscopic observation using photo-oxidation of eosin-phalloidin
Hippocampal slices were fixed 30 min after induction of LTP by tetanic stimulation with 4% paraformaldehyde and 2% glutaraldehyde for 1 h on ice. Slices were then cut into sections with a vibratome and processed as previously described (Capani et al., 2001). After additional washes in sodium cacodylate buffer, tissue sections labeled with eosin-phalloidin were mounted on glass-welled tissue culture dishes (MatTek) pretreated with Cell Tak adhesive (Collaborative Research). Slices were fixed again for 2–5 min with 2% glutaraldehyde in 0.1 m cacodylate buffer, rinsed in a buffer for several minutes, and placed in cacodylate buffer +ed with 50 mm glycine and potassium cyanide for an additional 5 min to reduce non-specific staining. Photo-oxidation was performed on a Zeiss Axiovert equipped with a 75 W xenon arc lamp light source. Specimens were viewed with a 40 × oil objective, n.a. 1.3. Hippocampal area CA1 was chosen for electron microscopic analysis. The appropriate areas were located with transmitted light and the pattern of fluorescent labeling was recorded by using the confocal attachment at a low laser power setting. The samples were immersed for 10 min in 0.1 m sodium cacodylate +ed with 2.8 mm diaminobenzidine (DAB; final pH 7.4) at 4 °C bubbled with pure O2 and then irradiated under conventional epifluorescence by using the xenon lamp. The DAB solution was changed every few minutes during the reaction process. Continuous observations were made during the photo-oxidation procedure by using transmitted light. After 6–8 min, a brownish reaction product began to appear in place of the fluorescence. The process was terminated via halting of the excitation. After photo-oxidation, tissue sections were rinsed in 0.1 m sodium cacodylate several times and incubated for 30 min with 1% osmium tetroxide in 0.1 m sodium cacodylate, pH 7.4. Some sections were fixed for 1 h in 2.25% glutaraldehyde with 0.2% tannic acid added cacodylate buffer. Osmication was conducted with 0.75% OsO4 in cacodylate buffer, pH 6, for 1 h on ice. After several washes with ddH2O, slices were dehydrated in an ascending ethanol series, infiltrated with Durcopan ACM resin, and polymerized for 24 h at 60 °C. Thin sections (80–100 nm) were cut with Reichert Ultracut E by using glass knives. Thin sections were examined by using an electron microscope at 80–100 keV.
Cultures of hippocampal neurons
E18 hippocampal neuron cultures were prepared as previously described (Apperson et al., 1996). Cultures were plated in Petri dishes with no. 0 glass bottoms (MatTek) or 24-well plates with cover slips facing up. We used a higher density culture (~ 500 cells mm2) for the study of GFP-CaMKIIα translocation, and a lower density culture (~ 200 cells/mm2) for dextran labeling. Fifty micromolar kynurenic acid was applied to reduce basal neuronal activity when cultures needed to be moved for experiments.
GFP-CaMKII transfection
The GFP-CaMKIIα construction, as previously described (Shen & Meyer, 1999), was kindly provided by Dr Tobias Meyer of Stanford University. The GFP-CaMKIIα construction was transfected using the standard lipofectamine method according to the protocol provided by the manufacturer (Life Technologies). Following transfection, the neuron cultures were incubated for 2 days without disturbance to allow GFP-CaMKIIα to be expressed. Typically, fewer than four healthy neurons from a culture dish (14–21 days after plating) could be observed with expression of a detectable amount of GFP protein. Extra caution was practiced when handling cultures as neurons can be activated easily and F-actin can be depolymerized in dendritic spines before application of designed manipulations. Kynurenic acid is useful for reducing the basal level activities. GFP-CaMKII translocation was induced using the protocol previously described by Shen & Meyer (1999).
Patch-clamping and dye labeling
Neurons (21 days after plating) with clear soma were selected for patch-clamping and dye labeling. After successful patching, neurons were subjected to a test protocol measuring the current–voltage (I–V) relation at test potentials in 15-mV increments over the voltage range from −150 mV to 60 mV. Currents were measured with a patch-clamp amplifier (Axopatch 200, Axon Instruments), filtered at 5 kHz, digitized at 10 kHz and recorded on a computer (pCLAMP 8, Axon Instruments). No significant difference was observed between neurons with or without jasplakinolide treatment. Cells were voltage-clamped at a holding potential of −80 mV. After 15 min of dye injection, the test protocol was repeated. Neurons were then fixed with 4% paraformaldehyde for 1 h and then immunolabeled for PSD95 to identify dendritic spines.
Results
Induction of LTP led to a transient decrease in F-actin labeling in hippocampus in vitro and in vivo
We first investigated whether the induction of LTP by tetanus led to a change in F-actin. To address this question, we evaluated changes in labeling of F-actin in rat acute hippocampal slices upon induction of LTP by the standard two-pathway paradigm as previously described (Ouyang et al., 1997, 1999). Electrophysiological signals were recorded for 30 min after LTP was induced by tetanic stimulation (Fig. 1A), and then slices were fixed and labeled with rhodamine-conjugated Phalloidin, as described in Materials and methods. Phalloidin was chosen because it specifically binds to F-actin but not to G-actin (Faulstich et al., 1988). In this experiment, we found that the induction of LTP led to decreased phalloidin rhodamine labeling of F-actin in the stratum radiatum of CA1 in hippocampal slices (Fig. 1B). In contrast, an increase in MAP2 labeling was observed in the same area of the same slice (Fig. 1C). At a higher magnification, we observed that the labeling of F-actin with phalloidin rhodamine was concentrated at spine-like structures (Fig. 2) and significantly decreased in the region where tetanic stimulation was given. Concordantly, previous studies have demonstrated that actin is concentrated in the dendritic spines of neurons (Fifkova & Delay, 1982; Matus et al., 1982). Furthermore, observations with electron microscopy indicate that phalloidin labeling is mainly concentrated in dendritic spines (Capani et al., 2001). Our results indicate that F-actin becomes depolymerized within 30 min after tetanic stimulation. We further conducted quantitative measurements using a previously described method (Ouyang et al., 1997, 1999), and observed a statistically significant decrease in F-actin labeling only in the slices where LTP was induced (Fig. 1D). No changes in F-actin labeling were observed in control slices, and the patterns of labeling were similar to CaMKII labeling as we reported previously (Ouyang et al., 1997). Furthermore, this LTP-induced decrease in F-actin labeling was blocked by APV, a N-methyl-d-aspartate (NMDA) receptor antagonist blocking the induction of LTP. These data indicate that the decrease in F-actin is LTP dependent and requires activation of NMDA receptors.
FIG. 1.
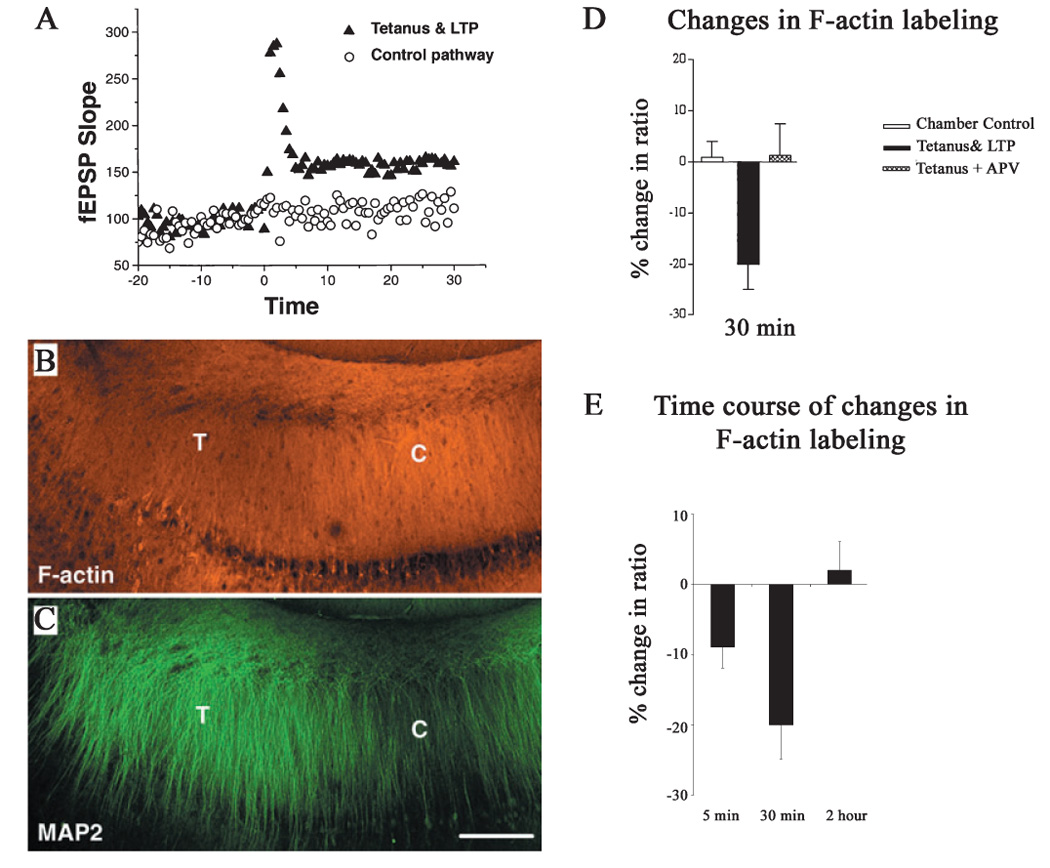
Induction of long-term potentiation (LTP) by tetanus decreased F-actin while increasing MAP2 labeling in hippocampal slices. (A) The field EPSP (fEPSP) increased with LTP induction after tetanic stimulation. (B) and (C) Double-labeling images from a whole CA1 region of a hippocampal section stained with phalloidin rhodamine for F-actin in red (B) and immunolabeled for MAP2 in green (C). Compared with the control side (marked with white C), decreased F-actin staining was observed at the tetanized side, while increased MAP2 labeling was noted (marked with white T). (D) Quantitative analysis of changes in the ratio (tetanized side/control side) of F-actin labeling associated with induction of LTP 30 min after tetanus, as previously described (Ouyang et al., 1997, 1999). The intensity of F-actin labeling was significantly lower in LTP slices than in their adjacent chamber control slices (P < 0.002, n = 7 animals). Application of APV, a blocker of NMDA receptors, abolished the changes in F-actin (n = 4 animals). (E) Time course of changes in labeling of F-actin in striatum radiatum of CA1 after tetanic stimulations. Decrease in F-actin labeling can be observed both 5 min after tetanus (n = 5 animals) and 30 min after tetanus. The labeling returns to control levels 2 h after tetanus (n = 4 animals). Scale bar: 200 µm.
FIG. 2.

Tetanic stimulation led to a decrease in F-actin labeling in dendritic spines. Confocal images were obtained from the same section as in Fig. 1, but with a higher magnification. Punctuate labeling of phalloidin rhodamine was noted in dendritic spines. Decreased spine-like labeling for F-actin in the tetanic stimulated side was noted (from white T side in Fig. 1) compared with the control side (from white C side in Fig. 1). Labeling of phalloidin rhodamine (in red) can be more easily identified in spines in the overlay images with MAP2 labeling (in green). MAP2 labeling was concentrated in dendritic shafts. Both images were taken with the same settings, and oversaturation was intentionally avoided so that in some areas weaker labeling, although present, is not easily visible in the figure without increased contrast. Scale bar: 15 µm.
We then examined the time course of the changes in F-actin in hippocampal slices after tetanic stimulation (Fig. 1E). In addition to the decrease in F-actin labeling 30 min after tetanic stimulation, we found that the decrease in F-actin occurs as soon as 5 min (percent of change was −8.87 ± 3 vs. −0.34 ± 2 for chamber controls, P < 0.01 for paired t-test, n = 5 for both), the earliest time point we could make a measurement after first tetanic stimulation was applied. It has been reported that actin is not only very dynamic, but also tends to continue to form filament (F-actin) and reach the steady state in 2 h in vitro (Korn et al., 1987). Interestingly, the labeling of F-actin returned to the control level 2 h after tetanic stimulations in hippocampal slices (percent of change was 2.01 ± 4 vs. −1.5 ± 4 for chamber controls, P = 0.65 for paired t-test, n = 4 for both).
We also examined the time course of F-actin changes following LTP induction in dentate gyrus of hippocampus in vivo. We used a similar procedure to induce LTP with tetanic stimulation (Fukazawa et al., 2003). Similar to the results in hippocampal slices, we observed an initial, transient decrease in F-actin 5 min after LTP induction, which returned to control levels by 30 min following tetanic stimulation (Fig. 3). Subsequently, there was a later increase in F-actin at 1 h after LTP induction, consistent with the previous results of Fukazawa et al. (2003).
FIG. 3.

Time course of changes in F-actin labeling in the middle molecular layer (MML) of dentate gyrus after LTP induction in vivo. (A) Pairs of superimposed traces represent the pre-tetanus baseline response (dotted) and the post-tetanus response (solid) for three different animals that were killed 5, 30 and 60 min (indicated under each pair of traces) after the tetanic stimuli were applied to induce LTP. In each case there was post-tetanic potentiation, and longer time points demonstrate LTP. Calibration 3 ms, 1 mV. (B) The micrographs show labeling of F-actin in dentate gyrus 5, 30 and 60 min after tetanic stimulation (Tetanus) next to corresponding micrographs from the contralateral dentate gyrus of the same section (Control). The inset graph shows the time course of changes in F-actin labeling following LTP induction. The percentage change in F-actin in the MML in tetanic stimulated dentate gyrus (left side of the brain) was normalized with the labeling in CA1 in the same image, which is not directly dependent on the stimulation of dentate gyrus and then the right side of dentate gyrus in the same section [ratio = (brightness of left MML/brightness of left CA1)/(brightness of right MML/brightness of right CA1)]. Data obtained from three sections (about 200 µm around recording site) were used from each animal. Decrease in F-actin was observed in MML 5 min after tetanic stimulation (n = 4 animals). There was no obvious difference 30 min after the tetanus (n = 4 animals). An increase in F-actin labeling was observed 1 h after tetanic stimulation (n = 3 animals). Scale bar: 100 µm.
Electron microscopy observations and quantitative analysis of changes in F-actin labeling at dendritic spines in hippocampal slices after induction of LTP
We examined the change in F-actin at dendritic spines in hippocampal slices in more detail using electron microscopy. As shown in Fig. 4, labeling of F-actin in dendritic spines was observed in many dendritic spines in a non-tetanized region of a hippocampal slice (left panel). In contrast, this F-actin labeling in dendritic spines was decreased after 30 min in the LTP area of the same slice. Statistical analysis indicated that after the induction of LTP, the number of dendritic spines with F-actin labeling was reduced by approximately 50%. This result further supported the contention that the induction of LTP is associated with an initial depolymerization of F-actin in dendritic spines.
FIG. 4.

Electron microscopy observation further confirmed a decrease in F-actin in dendritic spines after induction of LTP by tetanus. Slices were prepared as described in Fig. 1 and in the Materials and methods. Dendritic spines can be identified by the typical structure of the postsynaptic density and their connection to presynaptic terminals, which contains many synaptic vesicles. Numbers of spines with strong labeling of F-actin were counted and statistically analysed. Compared with the control, a 50% decrease in F-actin-labeled dendritic spines was noted upon tetanus stimulation [*P < 0.01, n = 879 control spines and 1156 long-term potentiation (LTP) spines from three animals].
Activation of hippocampal slices resulted in decreased p-cofilin and F-actin
We asked whether increased neuronal activity could decrease the phosphorylation of cofilin, which should increase cofilin activation and trigger the depolymerization of F-actin in hippocampal slices. KCl (50 mm) was used to treat the hippocampal neurons and to induce neuronal activity. Hippocampal slices were prepared and rested as previously described (Ouyang et al., 1997, 1999). After incubation in oxygenized ACSF with 50 mm KCl for 5 min, slices were homogenized with ice-cold buffer for Western blot analysis. In each experiment, we chose adjacent slices as immunolabeling controls. In addition, a slice from each condition was monitored for EPSPs to ensure that the slices were in good condition.
Labeling of F-actin was decreased in slices exposed to 50 mm KCl (Fig. 5), suggesting that F-actin may be depolymerized in response to synaptic potentiation. Western blot data showed that the amount of phosphorylated cofilin was significantly (P < 0.04 in paired t-test, n = 3) decreased (about 80% of the control level) when the slides were exposed to 50 mm KCl, while no changes in the total amount of actin and cofilin were noted. Our results demonstrated that activating hippocampal slices resulted in decreased p-cofilin and F-actin.
FIG. 5.

Detection of decreases in F-actin and p-cofilin in hippocampal slices exposed to 50 mm KCl for 5 min. Slices were prepared in the same way for LTP experiments. After treatment, slices were fixed for F-actin labeling or homogenized for Western blots. The confocal images show F-actin labeling with phalloidin rhodamine in hippocampal CA1 region, same as in Fig 1 and Fig 2. Compared with the control, the brightness of labeling for F-actin in the dendritic spine area is much lower in the slice exposed to 50 mm KCl for 5 min. However, labeling in blood vesicles remains high in the same area, suggesting that the reduction of F-actin labeling was due to high potassium-induced neuronal activities. Change in phosphorylation of cofilin was detected by Western blot. While p-cofilin was easily detected in controls (C) by Western blot, p-cofilin was barely observed in samples exposed to 50 mm KCl (K). Quantitative analysis revealed that the reduction of p-cofilin was about 80% (P < 0.04 in paired t-test, n = 4 experiments with four animals). In contrast, there were little differences between the two groups in total cofilin and actin. The two groups of slices were prepared by adjacent pairs in order to reduce variation of slice size.
Stabilization of F-actin blocks the translocation of GFP-CaMKII into dendritic spines
We then investigated whether the stabilization of F-actin could alter protein translocation into dendritic spines. The experiment was conducted using cultured hippocampal neurons transfected with GFP-CaMKII. The protocol to triggering GFP-CaMKII translocation has been previously described (Shen & Meyer, 1999). Jasplakinolide (2 µm for 3 h) was used to increase the rate of actin polymerization and to stabilize F-actin (Bubb et al., 2000). Morphology of the neurons in culture was examined by double-labeling neurons for F-actin in dendritic spines (in red) and MAP2 in dendritic shafts (in green) in Fig. 6E. A basal level of translocation of GFP-CaMKII was observed in the cell body, dendritic shafts and a few spines, and the number of labeled spines per 100 µm of dendrite was 9.0 ± 2.0 (Fig. 6A, n = 10 neurons). It was possible that GFP-CaMKII was translocated into these spines due to the spontaneous activity of the cultured neurons during 2 days of incubation. In the neurons treated with jasplakinolide, the number of labeled spines per 100 µm dendrites was 7.2 ± 4.9 (Fig. 6B, n = 5). When activating GFP-CaMKII-transfected neurons with glutamate and glycine, this led to significantly increased numbers of spines filled with GFP-CaMKII (Fig. 6C, 42.8 ± 3.7, n = 22). Furthermore, this glutamate- and glycine-induced translocation of GFP-CaMKII was totally blocked by treating the neurons with jasplakinolide (Fig. 6D, 7.2 ± 3.5, n = 10, P < 0.0002), and the numbers of spines filled with GFP-CaMKII was similar to jasplakinolide-treated control neurons (7.2 ± 3.5 vs. 7.2 ± 4.9). Thus, these results suggest that stabilization of F-actin can significantly block neurotransmitter-induced GFP-CaMKII translocation into dendritic spines.
FIG. 6.

Stabilization of F-actin by jasplakinolide blocked the translocation of GFP-CaMKIIα triggered with glutamate and glycine. Neurons were transfected with GFP-CaMKIIα and expressed for 2 days without disruption. (A) A normal control neuron. (B) A neuron incubated with 2 µm jasplakinolide (J). (C) A neuron treated with 100 µm glutamate and 10 µm glycine (G). (D) A neuron first treated with 2 µm jasplakinolide and then with 100 µm glutamate and 10 µm glycine (J + G). (E) A neuron from the sister culture that immunolabeled for MAP2 in green and F-actin in red. Many dendritic spines can be observed in neurons under the condition of culture. (F) Statistical analysis indicates that stabilization of F-actin by jasplakinolide can significantly block CaMKIIα translocation triggered by glutamate and glycine (P < 0.0002, n = 22 neurons without jasplakinolide and 10 neurons with jasplakinolide). Scale bar: 10 µm.
Stabilization of F-actin inhibits the diffusion of 3-kDa dextran into dendritic spines
We then sought to determine whether the blockage of translocation by the stabilization of F-actin applied to any macromolecules, and was not limited to functional proteins such as CaMKII. To that end, we used 3-kDa fluorescein-conjugated dextran to test if the diffusion of molecules of this size into dendritic spines would be inhibited by the stabilization of F-actin as well. Because 3-kDa dextran is much smaller than GFP-CaMKII and other functional proteins, we can assume that the stabilization of F-actin will generally block protein translocation if inhibited diffusion of 3-kDa dextran is observed. Neurons were patched and the activity was tested as described in the Materials and methods. The patching with fluorescein-conjugated dextran in the pipette lasted for 15 min to allow the dextran to diffuse into distal dendrites. After fixation with 4% paraformaldehyde, neurons were immunolabeled with PSD95 to identify dendritic spines. As shown in Fig. 7, 3-kDa dextran was capable of diffusing into some of the spines on the distal dendrites under control conditions. This diffusion was blocked after treating the neurons with jasplakinolide, even though the spines were very close to the soma of neurons where the dextran had originally diffused in from the electrode. We further conducted a blind analysis to calculate the percentage of spines labeled with dextran 3 kDa vs. the percentage labeled with PSD95 (for identifying dendritic spines). The overlap of labeling indicated that the 3-kDa dextran had diffused into spines. We found that a statistically significant higher number of spines (35.3 ± 5.0%, n = 7) was labeled with the 3-kDa dextran in the control than those in the neurons treated with jasplakinolide (21.7 ± 5.3%, n = 7, P < 0.04). This result suggested that the stabilization of F-actin at steady state increases its capability of blocking macromolecules from getting into dendritic spines. Such a barrier appears to be present in some of the spines even under control conditions, because not every spine labeled with PSD95 was labeled with the 3-kDa dextran as well. On the other hand, the 3-kDa dextran entered about 20% of spines even in the jasplakinolide-treated condition. However, because the molecular weight of proteins is much higher than 3 kDa, it is logical to expect that F-actin should be more effective in blocking non-specific protein translocation. Therefore, F-actin at the steady state could serve as an underlying mechanism to regulate synaptic specificity by blocking translocation of proteins into unactivated spines.
FIG. 7.
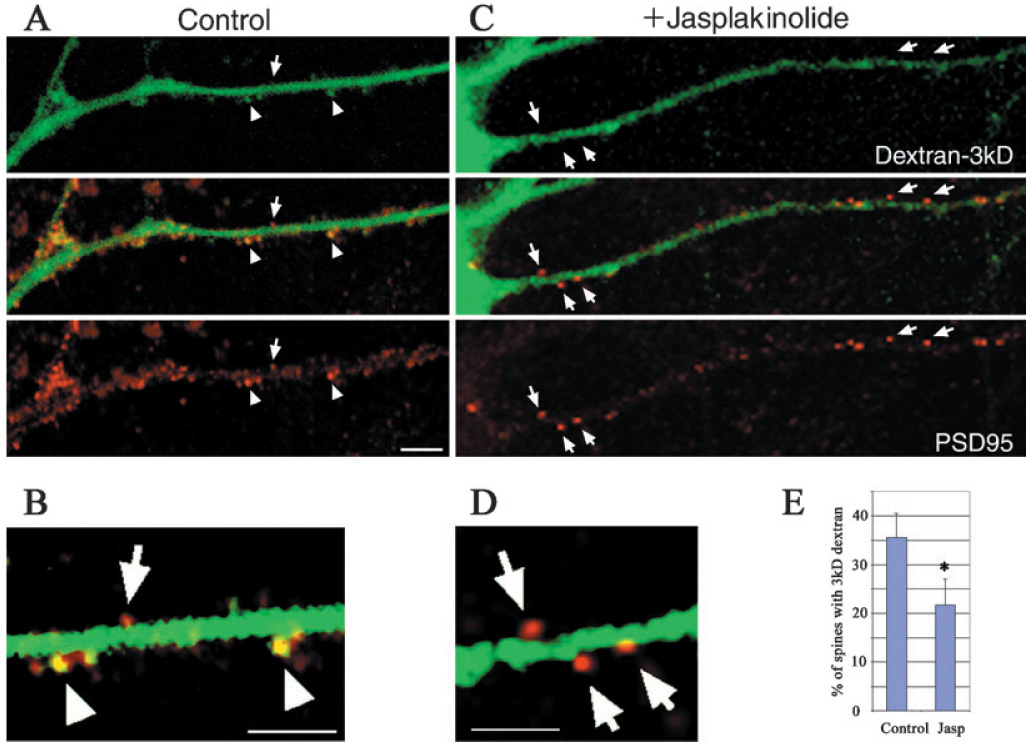
Stabilization of F-actin by jasplakinolide inhibited the diffusion of a 3-kDa dextran into dendritic spines. Fluorescent conjugated dextran was allowed to diffuse into neurons for 15 min by patching. Dextran labeling (green) is shown at the top panel and PSD95 immunolabeling (red) shown at the bottom in A and C. The middle row shows the overlays. (A) The control condition in which some of the spines at a distal dendrite were labeled with both the fluorescent and PSD95, as indicated by arrowheads. Some spines were labeled only with PSD95, as indicated with arrows. (B) is enlarged from A, and the contrast of image was adjusted to show double-labeling of 3-kDa dextran and PSD95 some dendritic spines in yellow (arrowhead). (C) A neuron treated with jasplakinolide before being patched for dextran labeling. Spines in a dendrite close to soma were labeled with PSD95 but not 3-kDa dextran, as indicated with arrows. (D) is enlarged from (C) with the same contrast adjustment as in B. (E) Statistical result that stabilization of F-actin by jasplakinolide can significantly inhibit the diffusion of a 3-kDa dextran into dendritic spines (*P < 0.04, n = 7 neurons in each condition). Scale bar, 10 µm.
Activating cultured neurons resulted in decreased phosphorylation of cofilin
Although we showed that the stabilization of F-actin blocks protein translocation into dendritic spines, it remained unclear whether this phenomenon was mediated through the decreased phosphorylation of cofilin (p-cofilin). To address this question, we used Western blot to measure changes in the phosphorylation of cofilin. The cultured neurons were activated by exposing them to 100 µm glutamate and 10 µm glycine or 50 mm KCl in culture medium (30 s at 37 °C) prior to harvesting. Samples from cultured neurons were collected with an ice-cold buffer. While no changes in the total levels of cofilin and actin were observed, we found that the level of cofilin phosphorylation was decreased upon neuron activation. As shown in Fig. 8, a 27% decrease in the phosphorylation of cofilin (P < 0.008 in paired t-test, n = 5) was detected in neurons treated with glutamate and glycine. Exposing neurons to 50 mm KCl resulted in a more profound decrease in the phosphorylation of cofilin (81%, P < 0.0001 in paired t-test, n = 5). Taken together, activating neurons with glutamate or a high concentration of potassium showed a decrease in the phosphorylation of cofilin. These results were consistent with the findings in acute hippocampal slices.
FIG. 8.

Changes in p-cofilin, but not in total cofilin and total actin were measured using Western blots for samples prepared from hippocampal neuronal cultures exposed to 100 µm glutamate and 10 µm glycine (G) and 50 mm KCl (K) for 30 s at 37 °C. A 27% decrease in the phosphorylation of cofilin (P < 0.008 in paired t-test, n = 5 experiments) was detected in neurons treated with glutamate and glycine. Exposing neurons to 50 mm KCl resulted in a more profound decrease in the phosphorylation of cofilin (81%, P < 0.0001 in paired t-test, n = 5 experiments). Neuronal cultures were prepared as described in Fig. 5.
Discussion
Despite the identification of dendritic spines over 100 years ago (Cajal, 1888), the functional significance of these structures remains mysterious. In particular, as actin is highly concentrated specifically in spines, the potential role of actin in synaptic plasticity is a subject of great interest and importance. To explore the mechanisms underlying actin dynamics in protein translocation into dendritic spines, we used multidisciplinary approaches in this study. In addition to standard LTP experiment with acute hippocampal slices, we applied immunolabeling and imaging with both confocal and electron microscopy. We also used cultured neurons in combination with gene transfection and dextran tracing. Our data indicate that dendritic spines are capable of gating the diffusion of large molecules into them. We have found that stabilized F-actin in spines blocks the translocation of CaMKII, a functional protein, and inhibits diffusion of 3-kDa dextran, suggesting highly concentrated F-actin may contribute to the regulation of synaptic specificity of protein translocation through blocking nonspecific translocation of protein into unpotentiated spines. Our contention is further supported by a study by Shi (2001), which demonstrated that GFP-GluR1 was present in less than 1% of dendritic spines in the dendritic trees of CA1 pyramidal neurons transfected with GFP-GluR1. Similarly, De Simoni et al. (2004) reported that Alexa dye could not be equally distributed in every dendritic spine. Thus, we hypothesise that synaptic potentiation could selectively control the gating by regulating actin dynamics via cofilin. After the gate is opened by actin depolymerization, protein may be delivered into the potentiated dendritic spines.
In summary, our results suggest that F-actin may play a role in selective protein translocation to potentiated spines. Our data suggest the following model (Fig. 9) in a hippocampal pyramidal neuron: in the absence of appropriate activation, actin located in spines forms filaments (F-actin) and eventually reaches a low-energy steady state. The spines are therefore in a ‘writing protection’ mode so that no new proteins can be translocated into the structures. After the activation of NMDA receptors, F-actin is rapidly depolymerized to unlock the spines, thereby allowing translocation of new proteins or polyribosomes to their target sites to perform the necessary remodeling in response to LTP or memory input (Steward & Schuman, 2001; Malinow & Malenka, 2002; Ostroff et al., 2002). After this transient opening of the spine gate, highly concentrated actin polymerizes again to reach the steady state, resulting in stable filamentous networks that help to secure the change in synaptic strength and long-term information storage.
FIG. 9.

A proposed model of protein translocation mediated by actin dynamics. (A) F-actin indicated in red is concentrated in dendritic spines and serves as a core for a complex that could block non-specific translocation of protein into the spines. (B) After synaptic potentiation induced by tetanus, F-actin in the spine will be dramatically depolymerized. (C) Proteins can be synthesized locally at dendrite after induction of long-term potentiation (LTP) by tetanus. The potentiated spine opens allowing new proteins to be translocated into it to increase the synaptic strength. Formation of new F-actin may occur. Dynamic exchange of actin subunits between the monomeric pool and F-actin may be necessary for this strictly targeted translocation of protein. (D) After proteins are translocated into the potentiated spine, actin dynamics may eventually reach the steady state and the spine returns to the resting state with a secured increase in synaptic strength. The size of the spine may be enlarged with more actin filaments. LTP may then be induced in another spine without interfering with the previously potentiated spine.
A transient depolymerization of F-actin may contribute to the regulation of synaptic specificity of protein translocation
F-actin is believed to play roles in many cellular functions. In addition to maintaining morphological structure, F-actin also mediates protein trafficking (Ziff, 1997; Halpain, 2000; Matus, 2000). Formation of new F-actin can play a role in selective protein transportation as well as in targeting. As reported by Allison et al. (2000), blocking the formation of new F-actin by latrunculin A leads to a lower level of CaMKII immunolabeling in distal dendrites. Application of latrunculin A is also reported to decrease LTP in the CA1 of hippocampal slices (Krucker et al., 2000). Recent studies report an increase of F-actin after stimulation of neurons (Fukazawa et al., 2003; Okamoto et al., 2004). Fukazawa et al. (2003) found that F-actin was increased after induction of LTP in vivo and was necessary for a late phase of LTP. Collectively, most of these previous studies support the importance of F-actin polymerization for LTP formation and protein translocation into spines. In contrast, the possibility of a shorter-term decrease in F-actin has not been previous explored. Our data demonstrate a biphasic response of F-actin with a transient decrease, followed by a subsequent increase, in F-actin after LTP in vivo. It took about 30 min for the labeling of F-actin to return to control levels in dentate gyrus in vivo and 2 h in striatum radiatum of CA1 in hippocampal slices. Compared with our results, the initial transient phase of depolymerization may not have been detected in previous studies because only later time points were assayed or a steady-state of F-actin may not have been established at the outset of the experiments. This transient phase of F-actin depolymerization may represent a critical window of opportunity allowing protein translocation to occur prior to longer-term stabilization of spines.
F-actin depolymerization may be regulated by synaptic potentiation through phosphorylation of cofilin
Previous studies indicate that the depolymerization of F-actin can be regulated by controlling the phosphorylation of cofilin. Reduction of phosphorylation at Ser3 facilitates cofilin to bind to F-actin, and is subsequently effective in depolymerizing F-actin (Agnew et al., 1995; Moriyama et al., 1996). Our results indicated that F-actin depolymerization may be regulated by synaptic potentiation through the phosphorylation of cofilin. This contention was supported by our findings that synaptic potentiation initiated by chemicals such as KCl or tetanic stimulation led to decreased phosphorylation of cofilin and F-actin in dendritic spines in both cultured primary neurons and hippocampal slices.
Interestingly, it appears that a partial decrease in phosphorylation of cofilin is sufficient to allow protein translocation into dendritic spines. Only a 27% decrease in p-cofilin was detected after neurons were exposed to 100 µm glutamate and 10 µm glycine in our study. This activation with glutamate and glycine is sufficient to trigger translocation of GFP-CaMKII into dendritic spines (Fig. 5, and Shen & Meyer, 1999). In contrast, no significant change in p-cofilin was detected in the hippocampal slices that were exposed to 100 µm glutamate and 10 µm glycine for 5 min (data not shown). This discrepancy is likely due to the fact that the glutamate could not easily penetrate synaptic clefts in the tissue. Nevertheless, 50 mm KCl induced a stronger decrease in p-cofilin (approximately 80%) in both cultured neurons and hippocampal slices.
In summary, we found that synaptic potentiation can lead to a decrease in F-actin in dendritic spines probably through cofilin dephosphorylation. Further investigations are warranted to understand the signaling pathways that could underlie the potentiation-mediated F-actin depolymerization.
F-actin may be depolymerized before reaching the steady state
Dendritic spines are very stable in vivo (Grutzendler et al., 2002) and in acute brain slices, but are very motile in organic cultured slices (Fischer et al., 1998). The motility of dendritic spines is primarily driven by the dynamics of actin (Fischer et al., 1998; Okamoto et al., 2004). De Simoni et al. (2003) reported that compared with acute slices, organotypic slices showed a four–fivefold increase in the absolute frequency of glutamatergic miniature postsynaptic currents. Therefore, actin may be depolymerized again before reaching a steady state in dendritic spines in cultured slices. While Fig. 9 may represent the dynamic cycle of regulated actin in most physiological conditions, actin in organic cultured slice may revert to a depolymerized state (as shown in Fig. 9B) before reaching the steady state (as indicated in Fig. 9D). Thus, without fully appreciating the whole cycle of actin dynamics and carefully defining the controls in experiments, the use of different preparations may lead to seemingly contradictory observations and implicate different mechanisms related to the role of actin in mediating protein trafficking and synaptic plasticity.
Future directions
The functional significance of this gating mechanism of dendritic spines in vivo remains to be investigated and recognized. One application of this mechanism is that it may provide a possible explanation at the cellular level for complicated observations such as why there is an increase in LTP but a disruption in learning tests in the LIMK1 knockout mice (Meng et al., 2002). If F-actin is not able to reach steady state, as likely occurs in LIMK1 knockout mice, then several abnormalities will result. On one hand, information storage in spines (synapses) may be disrupted so that LIMK1 knockout mice exhibit impaired learning (Meng et al., 2002), and patients with Williams’ syndrome have defects in visuo-spatial cognition (Frangiskakis et al., 1996). On the other hand, LTP could be increased in hippocampal slices from LIMK1 mice, as new proteins may enter more spines without the gating and lead to more potentiated synapses. Therefore, the gating mechanism of dendritic spines could be critical.
It remains to be investigated how specific signaling pathways are regulated during synaptic plasticity. Activation of NMDA receptors leads to subsequent activation of Rho (Li et al., 2000; Wong et al., 2000), which involves the regulation of actin dynamics (Maekawa et al., 1999; Nakayama et al., 2000). In addition to depolymerizing F-actin, the induction of LTP is associated with many other changes such as an increase in CaMKII autophosphorylation and synthesis (Ouyang et al., 1997, 1999). Clearly, more studies are needed to understand the complicated function of actin dynamics on protein translocation and underlying mechanisms. Nevertheless, our results indicate that depolymerization of F-actin from the steady state is necessary to allow specific protein translocation into spines in response to potentiation. This may serve as one of the basic mechanisms underlying the synaptic specificity of learning and memory.
Acknowledgements
This work was supported by Department of Neurology of Washington University School of Medicine for Yannan Ouyang and NIH grants for Drs Mark H. Ellisman (RR04050, NS046068, NS014718), Kelvin Yamada (NS 042744), Jane Y. Wu (R01AG1751804), Mary B. Kennedy (NS017660), Henry A. Lester (DA009121), Steven M. Rothman (R01NS42936 and R21NS045652), Michael Wong (1K02NS045583) and Yannan Ouyang (NS10660). We thank Dr Aili Cai and Mr Alan Rosenstein for their technical assistance. We thank many colleagues and friends for their help.
Abbreviations
- ACSF
artificial cerebrospinal fluid
- DAB
diaminobenzidine
- LTD
long-term depression
- LTP
long-term potentiation
- NMDA
N-methyl-d-aspartate
- SDS–PAGE
sodium dodecyl sulphate–polyacrylamide gel electrophoresis
References
- Agnew BJ, Minamide LS, Bamburg JR. Reactivation of phosphorylated actin depolymerizing factor and identification of the regulatory site. J. Biol. Chem. 1995;270:17582–17587. doi: 10.1074/jbc.270.29.17582. [DOI] [PubMed] [Google Scholar]
- Allison DW, Chervin AS, Gelfand VI, Craig AM. Postsynaptic scaffolds of excitatory and inhibitory synapses in hippocampal neurons: maintenance of core components independent of actin filaments and microtubules. J. Neurosci. 2000;20:4545–4554. doi: 10.1523/JNEUROSCI.20-12-04545.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apperson ML, Moon IS, Kennedy MB. Characterization of densin-180, a new brain-specific synaptic protein of the O-sialoglycoprotein family. J. Neurosci. 1996;16:6839–6852. doi: 10.1523/JNEUROSCI.16-21-06839.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bamburg JR, McGough A, Ono S. Putting a new twist on actin: ADF/cofilins modulate actin dynamics. Trends Cell Biol. 1999;9:364–370. doi: 10.1016/s0962-8924(99)01619-0. [DOI] [PubMed] [Google Scholar]
- Bubb MR, Spector I, Beyer BB, Fosen KM. Effects of jasplakinolide on the kinetics of actin polymerization. An explanation for certain in vivo observations. J. Biol. Chem. 2000;275:5163–5170. doi: 10.1074/jbc.275.7.5163. [DOI] [PubMed] [Google Scholar]
- Cajal SR. Estructural de los centros nerviosos de la saves. Rev. Trim. Histol. Norm. Pat. 1888;1:1–10. [Google Scholar]
- Capani F, Martone ME, Deerinck TJ, Ellisman MH. Selective localization of high concentrations of F-actin in subpopulations of dendritic spines in rat central nervous system: a three-dimensional electron microscopic study. J. Comp. Neurol. 2001;435:156–170. doi: 10.1002/cne.1199. [DOI] [PubMed] [Google Scholar]
- Chen H, Bernstein BW, Bamburg JR. Regulating actin-filament dynamics in vivo. Trends Biochem. Sci. 2000;25:19–23. doi: 10.1016/s0968-0004(99)01511-x. [DOI] [PubMed] [Google Scholar]
- Condeelis J. How is actin polymerization nucleated in vivo? Trends Cell Biol. 2001;11:288–293. doi: 10.1016/s0962-8924(01)02008-6. [DOI] [PubMed] [Google Scholar]
- Davis HP, Squire LR. Protein synthesis and memory: a review. Psychol. Bull. 1984;96:518–559. [PubMed] [Google Scholar]
- De Simoni A, Fernandes F, Edwards FA. Spines and dendrites cannot be assumed to distribute dye evenly. Trends Neurosci. 2004;27:15–16. doi: 10.1016/j.tins.2003.10.006. [DOI] [PubMed] [Google Scholar]
- De Simoni A, Griesinger CB, Edwards FA. Development of rat CA1 neurones in acute versus organotypic slices: role of experience in synaptic morphology and activity. J. Physiol. 2003;550:135–147. doi: 10.1113/jphysiol.2003.039099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deller T, Merten T, Roth SU, Mundel P, Frotscher M. Actin-associated protein synaptopodin in the rat hippocampal formation: localization in the spine neck and close association with the spine apparatus of principal neurons. J. Comp. Neurol. 2000;418:164–181. doi: 10.1002/(sici)1096-9861(20000306)418:2<164::aid-cne4>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- Faulstich H, Zobeley S, Rinnerthaler G, Small JV. Fluorescent phallotoxins as probes for filamentous actin. J. Muscle Res. Cell Motil. 1988;9:370–383. doi: 10.1007/BF01774064. [DOI] [PubMed] [Google Scholar]
- Fifkova E, Delay RJ. Cytoplasmic actin in neuronal processes as a possible mediator of synaptic plasticity. J. Cell Biol. 1982;95:345–350. doi: 10.1083/jcb.95.1.345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer M, Kaech S, Knutti D, Matus A. Rapid actin-based plasticity in dendritic spines. Neuron. 1998;20:847–854. doi: 10.1016/s0896-6273(00)80467-5. [DOI] [PubMed] [Google Scholar]
- Frangiskakis JM, Ewart AK, Morris CA, Mervis CB, Bertrand J, Robinson BF, Klein BP, Ensing GJ, Everett LA, Green ED, Proschel C, Gutowski NJ, Noble M, Atkinson DL, Odelberg SJ, Keating MT. LIM-kinase1 hemizygosity implicated in impaired visuospatial constructive cognition. Cell. 1996;86:59–69. doi: 10.1016/s0092-8674(00)80077-x. [DOI] [PubMed] [Google Scholar]
- Frey U, Morris RG. Synaptic tagging and long-term potentiation. Nature. 1997;385:533–536. doi: 10.1038/385533a0. [DOI] [PubMed] [Google Scholar]
- Fukazawa Y, Saitoh Y, Ozawa F, Ohta Y, Mizuno K, Inokuchi K. Hippocampal LTP is accompanied by enhanced F-actin content within the dendritic spine that is essential for late LTP maintenance in vivo. Neuron. 2003;38:447–460. doi: 10.1016/s0896-6273(03)00206-x. [DOI] [PubMed] [Google Scholar]
- Grutzendler J, Kasthuri N, Gan WB. Long-term dendritic spine stability in the adult cortex. Nature. 2002;420:812–816. doi: 10.1038/nature01276. [DOI] [PubMed] [Google Scholar]
- Halpain S. Actin and the agile spine: how and why do dendritic spines dance? Trends Neurosci. 2000;23:141–146. doi: 10.1016/s0166-2236(00)01576-9. [DOI] [PubMed] [Google Scholar]
- Hartwin . An ultrastructural approach to understanding the cytoskeleton. In: Carraway KL, Carraway CAC, editors. The Cytoskeleton – a Practical Approach. Oxford: Oxford University Press; 1992. pp. 23–45. [Google Scholar]
- Holt MR, Koffer A. Cell motility: proline-rich proteins promote protrusions. Trends Cell Biol. 2001;11:38–46. doi: 10.1016/s0962-8924(00)01876-6. [DOI] [PubMed] [Google Scholar]
- Kim CH, Lisman JE. A role of actin filament in synaptic transmission and long-term potentiation. J. Neurosci. 1999;19:4314–4324. doi: 10.1523/JNEUROSCI.19-11-04314.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korn ED, Carlier MF, Pantaloni D. Actin polymerization and ATP hydrolysis. Science. 1987;238:638–644. doi: 10.1126/science.3672117. [DOI] [PubMed] [Google Scholar]
- Krucker T, Siggins GR, Halpain S. Dynamic actin filaments are required for stable long-term potentiation (LTP) in area CA1 of the hippocampus. Proc. Natl. Acad. Sci. USA. 2000;97:6856–6861. doi: 10.1073/pnas.100139797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landis DM, Reese TS. Cytoplasmic organization in cerebellar dendritic spines. J. Cell Biol. 1983;97:1169–1178. doi: 10.1083/jcb.97.4.1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Van Aelst L, Cline HT. Rho GTPases regulate distinct aspects of dendritic arbor growth in Xenopus central neurons in vivo. Nat. Neurosci. 2000;3:217–225. doi: 10.1038/72920. [DOI] [PubMed] [Google Scholar]
- Linden DJ. A protein synthesis-dependent late phase of cerebellar long-term depression. Neuron. 1996;17:483–490. doi: 10.1016/s0896-6273(00)80180-4. [DOI] [PubMed] [Google Scholar]
- Maekawa M, Ishizaki T, Boku S, Watanabe N, Fujita A, Iwamatsu A, Obinata T, Ohashi K, Mizuno K, Narumiya S. Signaling from Rho to the actin cytoskeleton through protein kinases ROCK and LIM-kinase. Science. 1999;285:895–898. doi: 10.1126/science.285.5429.895. [DOI] [PubMed] [Google Scholar]
- Malinow R, Malenka RC. AMPA receptor trafficking and synaptic plasticity. Annu. Rev. Neurosci. 2002;25:103–126. doi: 10.1146/annurev.neuro.25.112701.142758. [DOI] [PubMed] [Google Scholar]
- Matus A. Actin-based plasticity in dendritic spines. Science. 2000;290:754–758. doi: 10.1126/science.290.5492.754. [DOI] [PubMed] [Google Scholar]
- Matus A, Ackermann M, Pehling G, Byers HR, Fujiwara K. High actin concentrations in brain dendritic spines and postsynaptic densities. Proc. Natl. Acad. Sci. USA. 1982;79:7590–7594. doi: 10.1073/pnas.79.23.7590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng Y, Zhang Y, Tregoubov V, Janus C, Cruz L, Jackson M, Lu WY, MacDonald JF, Wang JY, Falls DL, Jia Z. Abnormal spine morphology and enhanced LTP in LIMK-1 knockout mice. Neuron. 2002;35:121–133. doi: 10.1016/s0896-6273(02)00758-4. [DOI] [PubMed] [Google Scholar]
- Moriyama K, Iida K, Yahara I. Phosphorylation of Ser-3 of cofilin regulates its essential function on actin. Genes Cells. 1996;1:73–86. doi: 10.1046/j.1365-2443.1996.05005.x. [DOI] [PubMed] [Google Scholar]
- Nakayama AY, Harms MB, Luo L. Small GTPases Rac and Rho in the maintenance of dendritic spines and branches in hippocampal pyramidal neurons. J. Neurosci. 2000;20:5329–5338. doi: 10.1523/JNEUROSCI.20-14-05329.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto K, Nagai T, Miyawaki A, Hayashi Y. Rapid and persistent modulation of actin dynamics regulates postsynaptic reorganization underlying bidirectional plasticity. Nat. Neurosci. 2004;7:1104–1112. doi: 10.1038/nn1311. [DOI] [PubMed] [Google Scholar]
- Ostroff LE, Fiala JC, Allwardt B, Harris KM. Polyribosomes redistribute from dendritic shafts into spines with enlarged synapses during LTP in developing rat hippocampal slices. Neuron. 2002;35:535–545. doi: 10.1016/s0896-6273(02)00785-7. [DOI] [PubMed] [Google Scholar]
- Ouyang Y, Kantor D, Harris KM, Schuman EM, Kennedy MB. Visualization of the distribution of autophosphorylated calcium/calmodulin-dependent protein kinase II after tetanic stimulation in the CA1 area of the hippocampus. J. Neurosci. 1997;17:5416–5427. doi: 10.1523/JNEUROSCI.17-14-05416.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouyang Y, Rosenstein A, Kreiman G, Schuman EM, Kennedy MB. Tetanic stimulation leads to increased accumulation of Ca (2+)/calmodulin-dependent protein kinase II via dendritic protein synthesis in hippocampal neurons. J. Neurosci. 1999;19:7823–7833. doi: 10.1523/JNEUROSCI.19-18-07823.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollard TD, Blanchoin L, Mullins RD. Molecular mechanisms controlling actin filament dynamics in nonmuscle cells. Annu. Rev. Biophys. Biomol. Struct. 2000;29:545–576. doi: 10.1146/annurev.biophys.29.1.545. [DOI] [PubMed] [Google Scholar]
- Schuman EM. Synapse specificity and long-term information storage. Neuron. 1997;18:339–342. doi: 10.1016/s0896-6273(00)81234-9. [DOI] [PubMed] [Google Scholar]
- Shen K, Meyer T. Dynamic control of CaMKII translocation and localization in hippocampal neurons by NMDA receptor stimulation. Science. 1999;284:162–166. doi: 10.1126/science.284.5411.162. [DOI] [PubMed] [Google Scholar]
- Shi SH. Amersham Biosciences & Science Prize. AMPA receptor dynamics and synaptic plasticity. Science. 2001;294:1851–1852. doi: 10.1126/science.1067844. [DOI] [PubMed] [Google Scholar]
- Shi SH, Hayashi Y, Petralia RS, Zaman SH, Wenthold RJ, Svoboda K, Malinow R. Rapid spine delivery and redistribution of AMPA receptors after synaptic NMDA receptor activation. Science. 1999;284:1811–1816. doi: 10.1126/science.284.5421.1811. [DOI] [PubMed] [Google Scholar]
- Steward O, Schuman EM. Protein synthesis at synaptic sites on dendrites. Annu. Rev. Neurosci. 2001;24:299–325. doi: 10.1146/annurev.neuro.24.1.299. [DOI] [PubMed] [Google Scholar]
- Wear MA, Schafer DA, Cooper JA. Actin dynamics: assembly and disassembly of actin networks. Curr. Biol. 2000;10:R891–R895. doi: 10.1016/s0960-9822(00)00845-9. [DOI] [PubMed] [Google Scholar]
- Wong WT, Faulkner-Jones BE, Sanes JR, Wong RO. Rapid dendritic remodeling in the developing retina: dependence on neurotransmission and reciprocal regulation by Rac and Rho. J. Neurosci. 2000;20:5024–5036. doi: 10.1523/JNEUROSCI.20-13-05024.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyszynski M, Lin J, Rao A, Nigh E, Beggs AH, Craig AM, Sheng M. Competitive binding of alpha-actinin and calmodulin to the NMDA receptor. Nature. 1997;385:439–442. doi: 10.1038/385439a0. [DOI] [PubMed] [Google Scholar]
- Ziff EB. Enlightening the postsynaptic density. Neuron. 1997;19:1163–1174. doi: 10.1016/s0896-6273(00)80409-2. [DOI] [PubMed] [Google Scholar]