

Abstract
Central sleep apnea (CSA) is characterized by a lack of drive to breathe during sleep, resulting in repetitive periods of insufficient ventilation and compromised gas exchange. These nighttime breathing disturbances can lead to important comorbidity and increased risk of adverse cardiovascular outcomes. There are several manifestations of CSA, including high altitude-induced periodic breathing, idiopathic CSA, narcotic-induced central apnea, obesity hypoventilation syndrome, and Cheyne-Stokes breathing. While unstable ventilatory control during sleep is the hallmark of CSA, the pathophysiology and the prevalence of the various forms of CSA vary greatly. This brief review summarizes the underlying physiology and modulating components influencing ventilatory control in CSA, describes the etiology of each of the various forms of CSA, and examines the key factors that may exacerbate apnea severity. The clinical implications of improved CSA pathophysiology knowledge and the potential for novel therapeutic treatment approaches are also discussed.
Keywords: apnea threshold, arousal, chemoresponsiveness, control of breathing, hypercapnia, hypoxia, sleep-disordered breathing
Central sleep apnea (CSA) is characterized by a lack of drive to breathe during sleep, resulting in insufficient or absent ventilation and compromised gas exchange. In contrast to obstructive sleep apnea (OSA), in which ongoing respiratory efforts are observed, central apnea is defined by a lack of respiratory effort during cessations of airflow. However, as will be discussed, considerable overlap exists in the pathogenesis and pathophysiology of obstructive and central apnea, making this distinction somewhat difficult at times. CSA, like OSA, is associated with important complications, including frequent nighttime awakenings, excessive daytime sleepiness, and increased risk of adverse cardiovascular outcomes.1,2 There are several manifestations of CSA. These include high altitude-induced periodic breathing, idiopathic CSA (ICSA), narcotic-induced central apnea, obesity hypoventilation syndrome (OHS), and Cheyne-Stokes breathing (CSB). While the precise precipitating mechanisms involved in the various types of CSA may vary considerably, unstable ventilatory drive during sleep is a principal underlying feature.
The prevalence of CSA varies greatly between the various forms of CSA. Most healthy individuals will have periodic breathing on high-altitude ascent, provided the magnitude of the ascent is sufficient to cause substantial alveolar hypoxia.3 Given the global increase in obesity, the prevalence of OHS is likely on the rise.4 ICSA is relatively uncommon and may constitute < 5% of patients referred to a sleep clinic.5 Conversely, within certain clinical populations the presence of CSA may be extremely high. For example, a recent, prospective prevalence study6 of patients with heart failure and left ventricular ejection fraction < 45% revealed that 37% of patients had CSA. Interestingly, OSA is not uncommon in this population at 12%.6 Indeed, instances whereby central respiratory events lead to obstructive respiratory events in patients with vulnerable pharyngeal anatomy, and vice versa, are observed in the majority of sleep apnea patients.7 The overlap between CSA and OSA suggests that common mechanistic traits are likely involved. Typically, CSA is considered to be the primary diagnosis when ≥ 50% of apneas are scored as central in origin (ie, > 10 s cessation of breathing in the absence of respiratory effort); however, such thresholds are clearly arbitrary.
Chemical Control of Breathing
Chemoreceptor inputs (medullary neurons responding to CO2 via shifts in H+ concentration and peripherally at the carotid body via Pao2 and Paco2) play a key role in modulating ventilation. The ventilatory output to a given change in Pao2 or Paco2 (“chemosensitivity”) can vary greatly between individuals and with disease status. Highly sensitive chemoresponses can place an individual at risk for unstable breathing patterns because these individuals “overrespond” to small changes in chemical stimuli. The inherent delays in the negative feedback loop controlling ventilation also contribute to the risk for developing instability. For example, for a given increase in Paco2, an individual with high chemosensitivity will respond by increasing ventilation to a greater extent than someone with low chemosensitivity. This increased ventilation will continue until the resultant reduction in Paco2 (caused by the response) is detected at the chemoreceptors. Thus, individuals with high chemoresponsiveness will hyperventilate markedly to a perturbation potentially, lowering Paco2 below the eupnic level and leading to hypoventilation and potential apnea. Similarly, an individual with a long delay in the loop, such as individuals with reduced cardiac output, may have more prolonged hyperventilation, also leading to greater hyperventilation and a subsequent unstable breathing. The details of the control of the respiratory system feedback loop, otherwise known as loop gain, are described in detail elsewhere.8–10 Just as high chemosensitivity can be destabilizing to the respiratory system, severely blunted chemosensitivity can also be deleterious to cardiorespiratory homeostasis because extremely severe blood gas disturbances occur before a response is mounted.
Other Important Components that Regulate Breathing
In addition to chemical control, there are several other important homeostatic feedback mechanisms that regulate ventilation to maintain gas exchange within tightly controlled limits. Afferent information from Golgi tendon organs and muscle spindles from the chest wall and respiratory muscles also play an important role in regulating the rate and depth of breathing. During wakefulness nonrespiratory behavioral influences are also capable of modulating ventilatory activity. Examples include strong emotional expressions involving limbic forebrain structures and performing secondary tasks such as speech and ingestion of food. An independent background augmentation in respiratory drive known as the wakefulness drive to breathe is also present.11
State-Related Changes in the Control of Breathing
Transition to Sleep
The transition from wakefulness to sleep is an inherently unstable period in terms of cardiorespiratory control.12,13 With sleep onset, there is a loss of the wakefulness stimulus and behavioral influences.14 In addition, several respiratory control mechanisms are down regulated at sleep onset. Upper airway (UA) dilator and respiratory pump muscle tone is reduced, and there is an accompanying increase in UA resistance leading to a reduction in ventilation for a given level of drive.15,16 Chemosensitivity is also likely reduced at sleep onset.17 Al-though of variable magnitude and rate, these normal physiologic responses occur in all individuals. Should the withdrawal of the wakefulness drive be rapid at sleep onset, this in itself may be sufficient to promote hypopnea/apnea due to the delay required to elicit an appropriate compensatory response from the chemoreceptors.18 Thus, the dysrhythmic breathing characteristics observed even in healthy individuals at sleep onset likely relates to a combination of state instability and the associated changes in chemoreceptor sensitivity.19
Apnea Threshold
While behavioral influences and neurocompensatory responses strongly appose apnea even in the presence of marked decreases in Paco2 during wake-fulness, this is not the case during sleep. Indeed, during sleep all individuals are susceptible to breathing cessation should the Paco2 fall below a critical threshold known as the apnea threshold. The apnea threshold is usually 2 to 6 mm Hg below the eucapnic sleeping Paco2 level. This typically equates to the wakefulness eucapnic Paco2 level or marginally lower20,21 (see Dempsey22 for details).
Stable Sleep Changes
In addition to the changes that occur at sleep onset, ventilatory responses to hypoxia and hyper-capnia and respiratory load compensation are reduced across sleep stages, particularly during rapid eye movement (REM) sleep.23–25 The resultant reduction in ventilation with progressive sleep is coupled with a gradual rise in Paco2 on the order of approximately 3 to 8 mm Hg,26 depending on the prevailing metabolic conditions. Provided stable sleep is achieved, a new sleep-specific CO2 set point is established. Thus, during sleep, chemoreceptor and respiratory reflex feedback become critical components that regulate ventilation, albeit at a reduced homeostatic level compared to wakefulness.
Transition to Wake
Arousal from sleep is an integrated physiologic process that can serve as an important protective response. For example, during periods of compromised ventilation, arousal may be an important mechanism for restoring gas exchange when other compensatory mechanisms fail. However, arousal from sleep can also be deleterious to respiratory control stability. The propensity to develop central apnea is likely influenced by two important components of arousal sensitivity: arousal threshold and the ventilatory response to arousal.
Arousal Threshold
Regardless of the underlying cause of arousal from sleep (ie, spontaneous arousal, periodic leg movements, respiratory load induced arousal), an individual with a low arousal threshold (ie, susceptible to waking up easily) will be vulnerable to sleep state instability. That is, the combination of a predisposition to sleep transition apnea and a low arousal threshold may be sufficient to facilitate a repetitive CSA cycle as the individual oscillates between wake-fulness and sleep. The arousal threshold does, however, increase with progressively deeper sleep stage,27 as does breathing stability provided slow wave sleep can be achieved. However, it remains controversial whether slow wave sleep is intrinsically more stable from a respiratory standpoint, or if stable breathing allows sleep to deepen.
Ventilatory Response to Arousal
The rapid switch from sleep to wakefulness that occurs with arousal causes a sudden shift in the underlying homeostatic control of the cardiorespiratory system. The eucapnic set point rapidly shifts from the sleep set point (approximately 45 mm Hg) to the wakefulness level (approximately 40 mm Hg) creating a state of relative hypercapnia. In addition, sleep-induced UA resistance is removed and the wakefulness drive is reintroduced. Accordingly, a ventilatory response is evoked, the magnitude of which is determined by the extent of the shift between the various state-related physiologic changes. In addition, there is evidence to suggest there may be an additional waking reflex that further augments this response.28,29 The brisk ventilatory response causes a rapid reduction in Paco2, such that central apnea may ensue during subsequent sleep if the hypocapnia is sufficient to cross the apnea threshold30 (Fig 1).
Figure 1.
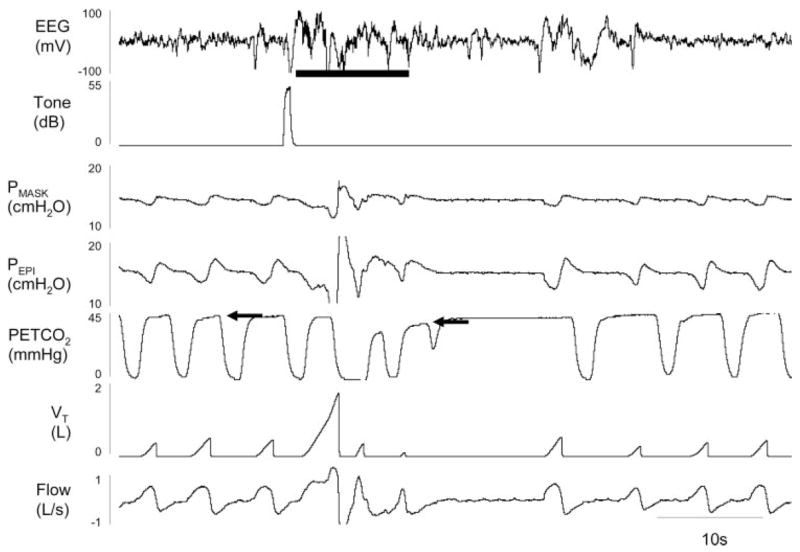
An example of experimentally induced arousal leading to central apnea. During stable stage 2 sleep, a 55-decibel (db) tone was played to induce an arousal from sleep (shown by solid line under EEG) in a 33-year-old woman (follicular menstrual phase) with severe OSA who was receiving CPAP (14 cm H2O). A brisk ventilatory response ensues driving end-tidal Pco2 (PETCO2) from 44 mm Hg during stable sleep (first arrow, note there is an approximate 3-s sampling delay between ventilation and end-tidal Pco2) to 38 mm Hg by the return to sleep (second arrow) and was accompanied by an approximate 10-s central apnea as documented by no change in epiglottic pressure (Pepi). VT = tidal volume. PMASK = pressure at the mask.
Manifestations of CSA
CSA syndromes can be broadly classified into two groups according to the wakefulness CO2 levels (hypercapnic vs nonhypercapnic), although the prevailing abnormalities in these two groups can be quite disparate.31 These underlying physiologic differences contribute to the varying CSA etiologies.
Hypercapnic CSA
By definition, patients with impaired ventilatory output during wakefulness will have some degree of daytime hypercapnia. Undoubtedly with the removal of the wakefulness drive and other behavioral influences, hypercapnia will worsen during sleep. Hence, the term sleep hypoventilation is often used to highlight an underlying condition of hypercapnia that worsens with sleep. From a physiologic perspective, patients with hypercapnia can be broadly classified into abnormal central pattern generator output (“won’t breathe”) or impairment of respiratory motor output caudal to the respiratory pattern generator (“can’t breathe”).
Impaired Central Drive (“Won’t Breathe”)
Tumors or trauma-induced lesions to brainstem structures may directly diminish ventilatory output, which on removal of wakefulness/behavioral drive is subject to further decline during sleep resulting in CSA. One form of hypercapnic CSA is congenital central hypoventilation syndrome (CCHS, formerly known as the Ondine curse), which is likely genetic in etiology without clear anatomic pathology. This rare condition is characterized by marked alveolar hypoventilation during sleep often resulting in severe hyper-capnia and hypoxemia.32 Further complications associated with this condition may include secondary polycythemia, pulmonary hypertension, and cor pulmonale. The breathing pattern during sleep is characterized by near-normal respiratory rate with diminished tidal volume.33 Unlike OSA, ventilation in CCHS tends to be more stable during REM compared to non-REM sleep,34 presumably due to the presence of additional respiratory stimulation during REM. Ventilatory responses and sensations of dyspnea to hypercapnia and hypoxia are often absent or greatly diminished in children with CCHS.32 While most cases of CCHS present in the newborn period, a recent report35 has revealed mild cases can present in adulthood.
The respiratory depressant effects of acute use of opioid-based medications are well known36,37 but have long been believed to subside with longer-term usage.38 However, evidence39,40 suggests that long-term use may lead to an increased propensity for CSA in up to 50% of patients. Because of the high prevalence of chronic pain and narcotic use,41,42 opioid-induced sleep-disordered breathing (SDB) is likely a major issue although only beginning to be recognized. Reported features of opioid-induced CSA may include prolonged periods of hypoventilation with marked hypoxemia and repetitive central apneas (Fig 2, top, A). However, again SDB tends to improve during REM sleep.39 While the precise underlying mechanisms are not clear, opioid-induced impairment of the hypercapnic and hypoxic ventilatory responses likely contribute,43 effects that are thought to be dose dependent (Fig 2, bottom, B). However, the consequences of more long-term opioid medication use on the hypoxic ventilatory response and the development of SDB are less clear.44 There is also an emerging literature that disruption of sleep can worsen physical pain, leading to the intriguing hypothesis that narcotic-induced central apnea may worsen narcotic requirements.
Figure 2.
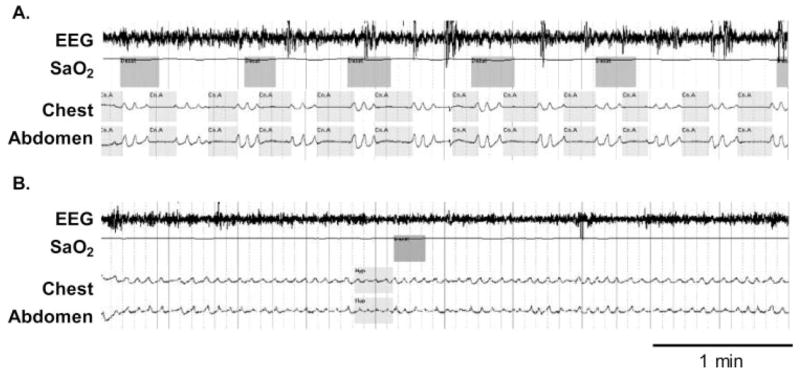
Top, A: An example of a patient receiving high-dose opioid medication for back pain experiencing repetitive central apneas as demonstrated by a lack of movement of respiratory effort bands (both abdominal and thoracic) with associated oxygen desaturations. Bottom, B: Marked improvement in SDB following gradual dose reduction of opioid medication. Sao2 = arterial oxygen saturation
Another form of hypercapnic CSA is OHS,45 the prevalence of which is likely on the rise.4 This disorder is typically defined as a combination of obesity (body mass index > 30 kg/m2) and arterial hypercapnia (Paco2 >45 mm Hg) during wakefulness not explained by other known causes of hypoventilation.46 Hypoventilation worsens during non-REM sleep and further during REM sleep, resulting in marked hypercapnia with accompanying hypoxemia. Typical symptoms may be similar to patients with OSA, including morning headaches and daytime hypersomnolence.47 Indeed, some patients with OHS also have OSA, suggesting there is mechanistic overlap between these obesity-related forms of SDB. The underlying mechanisms and the reasons why some obese patients have OHS but not others remains a major unresolved issue within the field. The inability of some patients to compensate for their obesity-related impairment in respiratory mechanics may be related to differences in the anatomic distribution of fat combined with ventilatory control deficits such as blunted chemosensitivity.45,48,49 Studies50–52 raise the possibility that the hormone leptin, secreted by adipocytes, may also be important in obesity-related hypoventilation in some patients.
Impaired Respiratory Motor Control (“Can’t Breathe”)
Hypercapnic patients with primarily intact central respiratory output from pattern generator neurons who have CSA may have abnormalities from upper motor neurons right down the neuromotor axis to the respiratory muscles. This encompasses a wide range of neuromuscular disorders, including myasthenia gravis (neuromuscular junction), amyotrophic lateral sclerosis (motor neuron disease), post-polio syndrome, and myopathies (eg, acid maltase deficiency). Chest wall syndromes such as kyphoscoliosis can also be associated with hypoventilation and CSA. The etiology and severity of CSA in these types of patient populations varies according to the extent and nature of the underlying abnormality.5
Nonhypercapnic CSA
The factors underlying central apnea in patients who are eucapnic or hypocapnic can be quite different from patients with hypercapnic CSA.
CSB
CSB is characterized by a waxing and waning pattern of ventilation (Fig 3). This disorder is most commonly observed in patients with congestive heart failure (CHF) and left ventricular systolic dysfunction. Apneas or hypopneas occur at the nadir of the characteristic crescendo/decrescendo ventilatory pattern and are most common during lighter sleep (stages 1 and 2). The cycle time of this pattern of unstable ventilation (typically 60 to 90 s) is much longer than other forms of CSA, due to prolonged circulation time in patients with CHF. Arousal typically occurs mid-cycle at the peak of ventilatory effort rather than at the cessation of apnea.53 Recent data54 suggest that SDB is more severe in the supine vs lateral position in patients with CSB, independent of postural effects on the UA. Characteristic symptoms may include fragmented sleep, paroxysmal nocturnal dyspnea, orthopnea, and daytime fatigue.55 Multiple features likely contribute to the development of the distinctive CSB pattern. These include factors that promote unstable breathing such as high ventilatory drive,55,56 minimal difference between the apnea threshold and sleeping eucapnic Paco2,57 long circulation time resulting in a mismatch between arterial blood gas concentration with the respiratory controllers,18,58 and impaired cere- brovascular reactivity to CO2.59 Animal data60 suggest that pulmonary congestion activates afferent C fibers (J receptors) causing sensory information to relay to the respiratory control centers to elicit a strong inhibitory reflex resulting in apnea followed by a period of hyperventilation that is likely to further destabilize breathing. A strong relationship between pulmonary capillary wedge pressure, hypocapnia, and CSA severity exists in patients with CHF,61 suggesting that similar reflex mechanisms may exist in humans.
Figure 3.
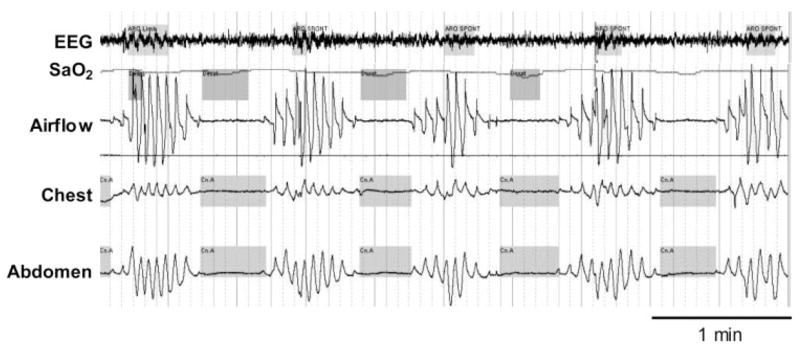
An example of a patient with CSB. Note the characteristic crescendo/decrescendo pattern of breathing, long circulation time (each oxygen desaturation corresponds to the previous apnea), and arousal occurring at the peak of respiratory effort. See Figure 2 legend for expansion of abbreviation.
ICSA
While there is clearly mechanistic overlap between ICSA and CSB, patients who have central apneas during sleep that do not display the typical CSB pattern or sleep transition apnea with normocapnia or hypocapnia during wakefulness fall into the category of ICSA. Central apneas in ICSA may occur as distinct features or in a repetitive cyclical manner (Fig 4). The duration of the cycle time (typically 20 to 40 s) is much less than CSB, and desaturations associated with events tend to be less severe. Similar to CSB, apneas are most commonly observed during stages 1 and 2 in ICSA. However, arousals typically occur at the termination of central apnea. Insomnia or hypersomnolence are common presenting symptoms. Typically, these patients are thinner and snore less than patents with OSA, although male predominance is likely a common trait.62 As the name implies, the underlying mechanisms for this disorder are not fully understood. Elevated hypercapnic ventilatory responses56,63,64 leading to hypocapnia and respiratory control instability are believed to be particularly important. Arousal and the accompanying hyperventilation, which due to the brief reintroduction of the waking stimulus and altered chemo-sensitivity, likely play an important role in triggering hypocapnia in patients with ICSA, resulting in further destabilization of breathing.30 These destabilizing factors render the patient vulnerable to crossing the apnea threshold, which may be very close to thesleeping eucapnic Paco2, particularly in patients with daytime hypocapnia. An inherently long transition duration between wakefulness and stable sleep, leading to greater exposure for state related breathing instability and high efficiency of CO2 excretion, may also be a causative factor.
Figure 4.
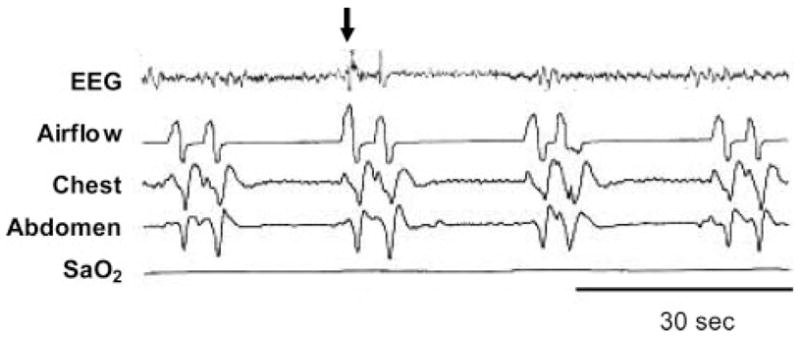
An example of ICSA. Note the shortened cycle time (approximately 25 s in this example) compared to CSB and that arousal (arrow) occurs at the cessation of apnea. See Figure 2 legend for expansion of abbreviation. Adapted from Malhotra et al5 with permission.
According to the Chicago criteria,65 the term periodic breathing is generally reserved for altitude-induced breathing instability. Nonetheless, this form of unstable breathing likely shares common mechanistic elements with other forms of nonhypercapnic CSA, such as vulnerability to crossing the apnea threshold due to a relative state of hypocapnia and the propensity for arousal to lead to further ventilatory control instability.66
Physiologic Factors Likely to Influence CSA Severity
Hypoxia
While deviations in chemosensitivity from normality may contribute to the pathophysiology of the various forms of CSA, there is evidence to suggest that the depressive effects of hypoxia may further increase disease severity. The different forms of CSA result in varied magnitude and duration of hypoxia, which are likely important determinants for the possible development of hypoxia-induced depressive effects. ICSA and CSB are characterized by intermittent hypoxia during sleep, while OHS is typically characterized by prolonged periods of sustained hypoxia during sleep. Hypoxia-induced central depression has been proposed to be an important contributing mechanism to SDB that occurs at altitude.67 Data68,69 suggest that acute sustained hypoxia impairs respiratory sensory processing and arousal responses to respiratory stimuli during sleep. These findings raise the possibility that hypoxia may impair respiratory sensory feedback mechanisms and increase disease severity in conditions characterized by sustained hypoxia such as sleep hypoventilation syndrome. Animal data70 demonstrate that the combination of acute hypoxia and pulmonary congestion, which stimulates afferent C fibers (J receptors) to evoke an inhibitory respiratory reflex, may lead to prolonged apnea, which may further perpetuate cyclical breathing in patients with CSB.
UA Anatomy
UA dilator muscles such as the genioglossus muscle receive neural input from central pattern generator neurons. An individual with an anatomically narrow UA is extremely reliant on neural drive to UA muscles for maintaining an open UA, whereas an anatomically larger UA is mechanically less reliant on neural drive. Thus, it is not surprising that in the absence of neural drive (central apnea), depending on the properties of the UA, varying degrees of UA collapse can ensue.7 Correcting an anatomically narrow UA with continuous positive airway pressure (CPAP) in a patient with primarily OSA can also lead to apparent treatment emergent central apnea. Although this phenomenon has been minimally studied,71 CPAP reduces UA resistance thereby improving the efficiency of CO2 excretion, rendering the hypocapnic patient vulnerable to crossing the apnea threshold. Activation of stretch reflexes that may inhibit ventilation secondary to increased lung volume effects of CPAP (especially if overtitrated) may also contribute.5 While recent data71 have revealed a greater male predominance of treatment-emergent CSA than OSA or other forms of CSA, the clinical presentation of patients with treatment-emergent CSA appears similar. Although there are currently no long-term data available, clinical experience suggests that these treatment emergent central apneas resolve with ongoing treatment, since CSA is relatively uncommon among patients receiving stable CPAP.5
Therapeutic Interventions
Given the range of pathophysiologic factors contributing to the varied forms of CSA (summarized schematically in Fig 5), treatment approaches also vary considerably. Gradual dose reduction of opioid medication may improve high-dose narcotic-induced CSA (Fig 2, bottom, B). Weight loss is likely to lead to improvement of SDB in patients with OHS.46 In practice, both these goals may be difficult to achieve. However, surgical weight loss may be an effective alternative option for morbidly obese patients with OHS.72 Varied strategies to manipulate chemosensitivity and respiratory drive offer promise for stabilizing unstable breathing patterns during sleep for many forms of CSA. However, concerns regarding potential adverse effects given the current lack of long-term randomized controlled trials warrant caution when considering such approaches as therapeutic options. Interventions that improve cardiac status for patients with an underlying heart condition may also attenuate SDB. The current level of evidence, mechanistic action and potential adverse effects for the main treatment options for CSA are discussed in the following section and summarized in Tables 1 and 2. For a comprehensive approach to the management of CCHS, the reader is referred to the American Thoracic Society guidelines32 and a recent report35 incorporating investigation for the PHOX2B gene mutation.
Figure 5.
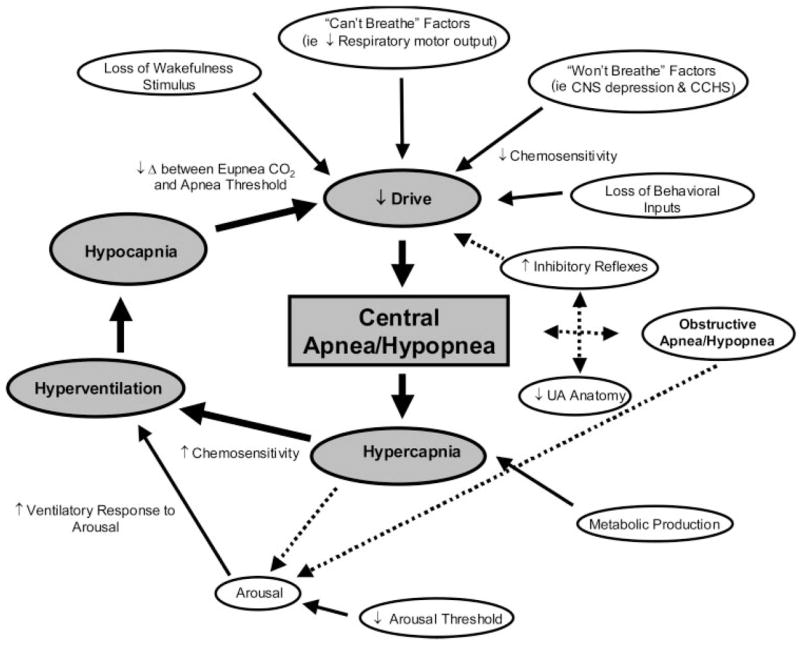
Schematic of the many potential mechanisms contributing to CSA/hypopnea. The gray boxes and largest solid arrows represent the key components contributing to unstable breathing and central apnea/hypopnea during sleep. The smaller solid arrows denote the main factors that lead to or modulate unstable breathing during sleep. Dashed arrows highlight the potential interactive links between obstructive and central apnea/hypopnea and for hypercapnia to cause arousal. Some arrows have been omitted to simplify the Figure. Refer to the text for further detail.
Table 1.
Summary of Potential Treatment Strategies and Their Effectiveness for Hypercapnic CSA*
| Variables | Intervention | Benefits | Limitations | Level of Evidence |
|---|---|---|---|---|
| OHS (won’t breathe) | Weight loss | Likely ↓ in SDB and other health-related benefits | Difficult to achieve | No published data in this population |
| CPAP | Variable ↓ in SDB; ↑ QOL | Not effective for all patients | Several small, short-term non- RCTs73–75 | |
| Bilevel PAP/bilevel PAP plus backup mode | Normalizes AHI; ↑QOL; ↑ Pao2; ↓Paco2 | Long-term effectiveness unknown | Several, small, short-term non- RCTs73,76 and one small RCT77 | |
| O2 | May ↓ hypoventilation in certain patients | Very limited data available | Case report78 | |
| Progesterone | May improve daytime gas exchange; ↑ hypercapnic chemoresponsiveness | Effects on AHI unknown in this population and no long- term safety data | One small, moderate-term non- RCT79 | |
| Narcotic-induced CSA (can’t breathe) | Dose reduction | Likely ↓ in SDB | Difficult to achieve | Case report (Fig 2) |
| Impaired respiratory motor control | Bilevel PAP | Likely ↓ in SDB | Limited data available, and patient tolerance may be poor | Several non-RCTs (refer to Malhotra et al5 and Schneerson et al76 for detail) |
PAP = positive airway pressure; QOL = quality of life; RCT = randomized control trial; ↑ = increase; ↓ = decrease.
Table 2.
Summary of Potential Treatment Strategies and Their Effectiveness for Nonhypercapnic CSA*
| Variables | Intervention | Benefits | Limitations | Level of Evidence |
|---|---|---|---|---|
| CSB/CSA in heart failure | Optimization of therapy | Likely ↓ in SDB | Several, small, non-RCTs61,80–82 | |
| CPAP | Approximately 50% ↓ in AHI | No evidence for ↓ mortality | Several, including one large study83 | |
| Adaptive NIV | Approximate 85% ↓ in AHI; ↑ in LVEF and QOL | Further studies on long-term hemodynamic effects required | Several, small, short/moderate-term RCTs84–86 and one non-RCT87 | |
| CO2; ↑ dead space | Normalizes AHI | No long-term safety data; may lead to anxiety/panic/insomnia | Several, small, short-term non- RCTs88,89 | |
| O2 | Approximate 50% ↓ in AHI | Hemodynamic effects not consistent | Several, small, short-term, non- RCTs70,91 and RCTs92,93 | |
| CRT | Normalizes AHI in some patients; ↑in LVEF | Not effective for all patients | Two small, short/moderate-term non-RCTs94,95 | |
| Theophylline | Approximate 50% ↓ in AHI | No change in LVEF; ↑ risk of cardiac events | One small, short-term RCT96 | |
| Acetazolamide | Approximate 40% ↓ in AHI; ↓ DTS and fatigue | No long-term safety data | One small, short-term RCT97 | |
| ICSA | CPAP | May normalize AHI in some patients | Very limited data available | Case report98 and one small, short- term non-RCT99 |
| CO2; ↑ dead space | Normalizes AHI | No long-term safety data; may lead to anxiety/panic/ insomnia | One small, short-term non-RCT100 | |
| O2 | Normalizes AHI | Very limited data available | One small (two ICSA patients) non-RCT90 | |
| Atrial overdrive pacing | Approximate 60% ↓ in AHI in one study (OSA plus CSA) | Several subsequent negative studies | Several, small short-term RCTs101–107 | |
| Acetazolamide | Approximate 70% ↓ in AHI; ↓ DTS | No long-term safety data | One small, short-term non-RCT108 |
CRT = cardiac resynchronization therapy; DTS = daytime sleepiness; LVEF = left ventricular ejection fraction; NIV = noninvasive ventilation. See Table 1 for expansion of abbreviation.
O2
Nonhypercapnic CSA patients with heightened chemosensitivity may benefit from the stabilizing respiratory control effects associated with O2 therapy. Indeed, several short-term trials have demonstrated that SDB improves with O2 administration in patients with ICSA90 and CSB,90–93 and potentially in certain patients with hypoventilation syndrome.78 Although minimally studied, there is some evidence to suggest that sleep efficiency parameters may be more favorable on O2 therapy than CPAP.93 To date, no large-scale long-term trials have been performed to determine which patients will likely benefit from O2 therapy and its long-term efficacy. There are some concerns that O2 therapy may have cardiodepressant effects mediated via O2 radicals.109 While larger trials are required before O2 therapy can be recommended for the treatment of CSA in patients with heart failure, evidence suggesting favorable cardiovascular function is beginning to emerge.110,111
CO2
Several studies have demonstrated that mild increases in inspired CO2, delivered directly or via the application of increased dead space, can be highly effective in treating CSA. Overnight trials involving small numbers of subjects have reported marked decreases in the apnea-hypopnea index (AHI) in patients with ICSA100 and CSB,88 without apparent acute cardiovascular adverse effects.89 Improvement is likely the result of a widening in the difference between eucapnic sleeping Pco2 and the apnea threshold. However, other reports suggest that despite marked improvement in AHI, increased CO2 does not improve sleep quality112 or reduce the arousal index113 and may lead to marked sympatho-excitation.114 Clearly, larger trials are required to determine the long-term efficacy and safety of increased CO2 for the treatment of CSA.
Noninvasive Ventilation
While the need to mechanically ventilate patients with severe hypercapnic CSA may be clear (ie, infants with severe CCHS and end-stage patients), the role of noninvasive ventilation in less severe forms of CSA is somewhat less clear. Nasal CPAP has been shown to be effective in some patients with ICSA.98,99 The mechanism for improvement in these patients is not clear but may relate to prevention of inhibitory reflex mechanisms that arise during airway closure and potentially CPAP-induced increases in lung volume/O2 stores.115 CPAP treatment improves hemodynamics and SDB in heart failure patients.116 However, the largest trial83 investigating CPAP in CSA did not reveal improvement in mortality and only partially improved the AHI. The combination of CPAP and increased CO2 may be highly effective in treating ICSA117 and mixed central and obstructive SDB.118 However, similar to the effect of increased inhaled CO2 alone, the lack of long-term trials and the potential for sympathoexcitation prevent the use of this approach in routine clinical practice at this time. Bilevel positive airway pressure may be deleterious to certain CSA patients by inducing hypocapnia119 but effective in others.73,76,120 Indeed, when used with a backup rate, bilevel positive airway pressure may lead to significant improvements in ventilation during sleep and a marked reduction in Paco2 in patients with OHS.77 Other new-generation, adaptive machines may also be effective in treating central apnea.84,87 Evidence85,86 suggests that such devices may improve patient adherence that would be predicted to improve symptoms and cardiac function, although data are currently equivocal.
Strategies To Improve Cardiac Function to Treat SDB
Optimization of medications for patients with an underlying heart condition can lead to significant improvement in SDB for patients with CSB.61,80–82 Restoration of cardiac function in patients with more severe disease via heart transplantation may also improve CSA, although some patients subsequently acquire OSA.121–123 A novel strategy of atrial overdrive pacing to increase heart rate by 15 beats/min significantly improved SDB in patients with symptomatic sinus bradycardia with normal or mildly depressed left ventricular function.101 This beneficial effect was attributed to increased cardiac output and decreased pulmonary congestion (decreased loop gain), although the findings remain controversial. The majority of patients with SDB have observed no benefit of overdrive pacing.102–107,124 Resynchronization pacemaker therapy has been shown recently to improve CSA and cardiac function in patients with heart failure in two small studies.94,95
Respiratory Stimulants
The respiratory stimulants acetazolamide and theophylline have been shown to improve CSA in patients with heart failure.96,97 Acetazolamide and has also been shown to improve SDB in ICSA.108 The carbonic anhydrase inhibitor acetazolamide leads to metabolic acidosis that likely shifts the hypercapnic ventilatory response and lowers the Paco2 apnea threshold.108,125 Theophylline likely improves SDB via increasing central respiratory drive and cardiac contractility. Progesterone increases chemoresponsiveness and may lead to improvement in daytime gas exchange in patients with OHS.79,126 However, respiratory stimulants cannot be recommended for routine CSA treatment at this time. Theophylline may increase the risk of cardiac arrhythmias and sudden death in these patients,127,128 and long-term trials exploring the efficacy and safety of acetazolamide and progesterone are not yet available.
Summary
In summary, CSA encompasses a wide range of distinct yet interrelated forms of unstable breathing that can lead to substantial comorbidity and increased risk of adverse cardiovascular outcomes. The underlying pathophysiology and the prevalence of the various forms of CSA varies greatly. Given the range of pathophysiologic factors contributing to the varied forms of CSA, treatment approaches also vary considerably. NIV remains a major treatment approach for many patients. While short-term studies have highlighted the potential for alternate treatment options, there is currently a lack of long-term randomized trials, an area of investigation that clearly needs to be pursued.
Acknowledgments
The authors are grateful to Dr. Nick Antic and Dr. Rajeev Ratnavadivel of the Adelaide Institute for Sleep Health for providing polysomnography examples of narcotic-induced CSA and CSB, respectively.
Abbreviations
- AHI
apnea-hypopnea index
- CCHS
congenital central hypoventilation syndrome
- CHF
congestive heart failure
- CPAP
constant positive airway pressure
- CSA
central sleep apnea
- CSB
Cheyne-Stokes breathing
- ICSA
idiopathic central sleep apnea
- OHS
obesity hypoventilation syndrome
- OSA
obstructive sleep apnea
- REM
rapid eye movement
- SDB
sleep-disordered breathing
- UA
upper airway
Dr. Eckert is a recipient of the Thoracic Society of Australia and New Zealand/Allen and Hanbury’s respiratory research fellowship. Dr. Jordan is supported by a grant from the American Heart Association. Dr. Malhotra is funded by National Institute of Aging, Beeson Award (from 2004 to 2008), “Aging Influence on the Development of Sleep Apnea” (AG024837–01), National Institutes of Health (from 2004 to 2008), “Sleep Apnea and Obesity: Cardiovascular Risk Assessment” (RO1-HL73146–01), and National Institutes of Health, Specialized Center of Research Project 1.
Drs. Eckert, Jordan, and Merchia have no conflict of interest to declare in relation to the subject matter contained within this review article. Dr. Malhotra is a consultant for Respironics, Restore Medical, and Inspiration Medication), receiving < $20,000 per year from each of these companies. He has received an unrestricted research grant from Respironics for $100,000 to study the cardiovascular complications of sleep apnea. He has received an industry grant from Restore Medical for $100,000 to develop a computational model of the upper airway.
Reproduction of this article is prohibited without written permission from the American College of Chest Physicians (www.chestjournal.org/misc/reprints.shtml).
References
- 1.Lanfranchi PA, Somers VK, Braghiroli A, et al. Central sleep apnea in left ventricular dysfunction: prevalence and implications for arrhythmic risk. Circulation. 2003;107:727–732. doi: 10.1161/01.cir.0000049641.11675.ee. [DOI] [PubMed] [Google Scholar]
- 2.Javaheri S, Parker TJ, Liming JD, et al. Sleep apnea in 81 ambulatory male patients with stable heart failure: types and their prevalences, consequences, and presentations. Circulation. 1998;97:2154–2159. doi: 10.1161/01.cir.97.21.2154. [DOI] [PubMed] [Google Scholar]
- 3.White DP, Gleeson K, Pickett CK, et al. Altitude acclimatization: influence on periodic breathing and chemoresponsiveness during sleep. J Appl Physiol. 1987;63:401–412. doi: 10.1152/jappl.1987.63.1.401. [DOI] [PubMed] [Google Scholar]
- 4.Poulain M, Doucet M, Major GC, et al. The effect of obesity on chronic respiratory diseases: pathophysiology and therapeutic strategies. Can Med Assoc J. 2006;174:1293–1299. doi: 10.1503/cmaj.051299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Malhotra A, Berry RB, White DP. Central sleep apnea. In: Carney PR, Berry RB, Geyer JD, editors. Clinical sleep disorders. Philadelphia, PA: Lippincott Williams and Wilkins; 2004. pp. 331–346. [Google Scholar]
- 6.Javaheri S. Sleep disorders in systolic heart failure: a prospective study of 100 male patients: the final report. Int J Cardiol. 2006;106:21–28. doi: 10.1016/j.ijcard.2004.12.068. [DOI] [PubMed] [Google Scholar]
- 7.Badr MS, Toiber F, Skatrud JB, et al. Pharyngeal narrowing/ occlusion during central sleep apnea. J Appl Physiol. 1995;78:1806–1815. doi: 10.1152/jappl.1995.78.5.1806. [DOI] [PubMed] [Google Scholar]
- 8.Khoo MC, Kronauer RE, Strohl KP, et al. Factors inducing periodic breathing in humans: a general model. J Appl Physiol. 1982;53:644–659. doi: 10.1152/jappl.1982.53.3.644. [DOI] [PubMed] [Google Scholar]
- 9.White DP. Pathogenesis of obstructive and central sleep apnea. Am J Respir Crit Care Med. 2005;172:1363–1370. doi: 10.1164/rccm.200412-1631SO. [DOI] [PubMed] [Google Scholar]
- 10.Wellman A, Malhotra A, Fogel RB, et al. Respiratory system loop gain in normal men and women measured with proportional-assist ventilation. J Appl Physiol. 2003;94:205–212. doi: 10.1152/japplphysiol.00585.2002. [DOI] [PubMed] [Google Scholar]
- 11.Orem J. The nature of the wakefulness stimulus for breathing. Prog Clin Biol Res. 1990;345:23–30. discussion 31. [PubMed] [Google Scholar]
- 12.Burgess HJ, Kleiman J, Trinder J. Cardiac activity during sleep onset. Psychophysiology. 1999;36:298–306. doi: 10.1017/s0048577299980198. [DOI] [PubMed] [Google Scholar]
- 13.Trinder J, Whitworth F, Kay A, et al. Respiratory instability during sleep onset. J Appl Physiol. 1992;73:2462–2469. doi: 10.1152/jappl.1992.73.6.2462. [DOI] [PubMed] [Google Scholar]
- 14.Skatrud JB, Berssenbrugge AD. Effect of sleep state and chemical stimuli on breathing. Prog Clin Biol Res. 1983;136:87–95. [PubMed] [Google Scholar]
- 15.Kay A, Trinder J, Bowes G, et al. Changes in airway resistance during sleep onset. J Appl Physiol. 1994;76:1600–1607. doi: 10.1152/jappl.1994.76.4.1600. [DOI] [PubMed] [Google Scholar]
- 16.Worsnop C, Kay A, Pierce R, et al. Activity of respiratory pump and upper airway muscles during sleep onset. J Appl Physiol. 1998;85:908–920. doi: 10.1152/jappl.1998.85.3.908. [DOI] [PubMed] [Google Scholar]
- 17.Dunai J, Wilkinson M, Trinder J. Interaction of chemical and state effects on ventilation during sleep onset. J Appl Physiol. 1996;81:2235–2243. doi: 10.1152/jappl.1996.81.5.2235. [DOI] [PubMed] [Google Scholar]
- 18.Khoo MC, Gottschalk A, Pack AI. Sleep-induced periodic breathing and apnea: a theoretical study. J Appl Physiol. 1991;70:2014–2024. doi: 10.1152/jappl.1991.70.5.2014. [DOI] [PubMed] [Google Scholar]
- 19.Dunai J, Kleiman J, Trinder J. Ventilatory instability during sleep onset in individuals with high peripheral chemosensitivity. J Appl Physiol. 1999;87:661–672. doi: 10.1152/jappl.1999.87.2.661. [DOI] [PubMed] [Google Scholar]
- 20.Meza S, Mendez M, Ostrowski M, et al. Susceptibility to periodic breathing with assisted ventilation during sleep in normal subjects. J Appl Physiol. 1998;85:1929–1940. doi: 10.1152/jappl.1998.85.5.1929. [DOI] [PubMed] [Google Scholar]
- 21.Skatrud JB, Dempsey JA. Interaction of sleep state and chemical stimuli in sustaining rhythmic ventilation. J Appl Physiol. 1983;55:813–822. doi: 10.1152/jappl.1983.55.3.813. [DOI] [PubMed] [Google Scholar]
- 22.Dempsey JA. Crossing the apnoeic threshold: causes and consequences. Exp Physiol. 2005;90:13–24. doi: 10.1113/expphysiol.2004.028985. [DOI] [PubMed] [Google Scholar]
- 23.White DP, Douglas NJ, Pickett CK, et al. Hypoxic ventilatory response during sleep in normal premenopausal women. Am Rev Respir Dis. 1982;126:530–533. doi: 10.1164/arrd.1982.126.3.530. [DOI] [PubMed] [Google Scholar]
- 24.Douglas NJ, White DP, Weil JV, et al. Hypoxic ventilatory response decreases during sleep in normal men. Am Rev Respir Dis. 1982;125:286–289. doi: 10.1164/arrd.1982.125.3.286. [DOI] [PubMed] [Google Scholar]
- 25.Wiegand L, Zwillich CW, White DP. Sleep and the ventilatory response to resistive loading in normal men. J Appl Physiol. 1988;64:1186–1195. doi: 10.1152/jappl.1988.64.3.1186. [DOI] [PubMed] [Google Scholar]
- 26.Skatrud JB, Dempsey JA, Badr S, et al. Effect of airway impedance on CO2 retention and respiratory muscle activity during NREM sleep. J Appl Physiol. 1988;65:1676–1685. doi: 10.1152/jappl.1988.65.4.1676. [DOI] [PubMed] [Google Scholar]
- 27.Gugger M, Bogershausen S, Schaffler L. Arousal responses to added inspiratory resistance during REM and non-REM sleep in normal subjects. Thorax. 1993;48:125–129. doi: 10.1136/thx.48.2.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Horner RL, Rivera MP, Kozar LF, et al. The ventilatory response to arousal from sleep is not fully explained by differences in CO(2) levels between sleep and wakefulness. J Physiol. 2001;534:881–890. doi: 10.1111/j.1469-7793.2001.00881.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Trinder J, Padula M, Berlowitz D, et al. Cardiac and respiratory activity at arousal from sleep under controlled ventilation conditions. J Appl Physiol. 2001;90:1455–1463. doi: 10.1152/jappl.2001.90.4.1455. [DOI] [PubMed] [Google Scholar]
- 30.Xie A, Wong B, Phillipson EA, et al. Interaction of hyper-ventilation and arousal in the pathogenesis of idiopathic central sleep apnea. Am J Respir Crit Care Med. 1994;150:489–495. doi: 10.1164/ajrccm.150.2.8049835. [DOI] [PubMed] [Google Scholar]
- 31.Bradley TD, McNicholas WT, Rutherford R, et al. Clinical and physiologic heterogeneity of the central sleep apnea syndrome. Am Rev Respir Dis. 1986;134:217–221. doi: 10.1164/arrd.1986.134.2.217. [DOI] [PubMed] [Google Scholar]
- 32.American Thoracic Society. Idiopathic congenital central hypoventilation syndrome: diagnosis and management. Am J Respir Crit Care Med. 1999;160:368–373. doi: 10.1164/ajrccm.160.1.16010. [DOI] [PubMed] [Google Scholar]
- 33.Weese-Mayer DE, Silvestri JM, Menzies LJ, et al. Congenital central hypoventilation syndrome: diagnosis, management, and long-term outcome in thirty-two children. J Pediatr. 1992;120:381–387. doi: 10.1016/s0022-3476(05)80901-1. [DOI] [PubMed] [Google Scholar]
- 34.Fleming PJ, Cade D, Bryan MH, et al. Congenital central hypoventilation and sleep state. Pediatrics. 1980;66:425–428. [PubMed] [Google Scholar]
- 35.Antic NA, Malow BA, Lange N, et al. PHOX2B mutation-confirmed congenital central hypoventilation syndrome: presentation in adulthood. Am J Respir Crit Care Med. 2006;174:923–927. doi: 10.1164/rccm.200605-607CR. [DOI] [PubMed] [Google Scholar]
- 36.Santiago TV, Edelman NH. Opioids and breathing. J Appl Physiol. 1985;59:1675–1685. doi: 10.1152/jappl.1985.59.6.1675. [DOI] [PubMed] [Google Scholar]
- 37.Shook JE, Watkins WD, Camporesi EM. Differential roles of opioid receptors in respiration, respiratory disease, and opiate-induced respiratory depression. Am Rev Respir Dis. 1990;142:895–909. doi: 10.1164/ajrccm/142.4.895. [DOI] [PubMed] [Google Scholar]
- 38.The use of opioids for the treatment of chronic pain: a consensus statement from the American Academy of Pain Medicine and the American Pain Society. Clin J Pain. 1997;13:6–8. [PubMed] [Google Scholar]
- 39.Farney RJ, Walker JM, Cloward TV, et al. Sleep-disordered breathing associated with long-term opioid therapy. Chest. 2003;123:632–639. doi: 10.1378/chest.123.2.632. [DOI] [PubMed] [Google Scholar]
- 40.Wang D, Teichtahl H, Drummer O, et al. Central sleep apnea in stable methadone maintenance treatment patients. Chest. 2005;128:1348–1356. doi: 10.1378/chest.128.3.1348. [DOI] [PubMed] [Google Scholar]
- 41.Elliott AM, Smith BH, Penny KI, et al. The epidemiology of chronic pain in the community. Lancet. 1999;354:1248–1252. doi: 10.1016/s0140-6736(99)03057-3. [DOI] [PubMed] [Google Scholar]
- 42.Luo X, Pietrobon R, Hey L. Patterns and trends in opioid use among individuals with back pain in the United States. Spine. 2004;29:884–890. doi: 10.1097/00007632-200404150-00012. discussion 891. [DOI] [PubMed] [Google Scholar]
- 43.Weil JV, McCullough RE, Kline JS, et al. Diminished ventilatory response to hypoxia and hypercapnia after morphine in normal man. N Engl J Med. 1975;292:1103–1106. doi: 10.1056/NEJM197505222922106. [DOI] [PubMed] [Google Scholar]
- 44.Teichtahl H, Wang D, Cunnington D, et al. Ventilatory responses to hypoxia and hypercapnia in stable methadone maintenance treatment patients. Chest. 2005;128:1339–1347. doi: 10.1378/chest.128.3.1339. [DOI] [PubMed] [Google Scholar]
- 45.Zwillich CW, Sutton FD, Pierson DJ, et al. Decreased hypoxic ventilatory drive in the obesity-hypoventilation syndrome. Am J Med. 1975;59:343–348. doi: 10.1016/0002-9343(75)90392-7. [DOI] [PubMed] [Google Scholar]
- 46.Olson AL, Zwillich C. The obesity hypoventilation syndrome. Am J Med. 2005;118:948–956. doi: 10.1016/j.amjmed.2005.03.042. [DOI] [PubMed] [Google Scholar]
- 47.Strumpf DA, Millman RP, Hill NS. The management of chronic hypoventilation. Chest. 1990;98:474–480. doi: 10.1378/chest.98.2.474. [DOI] [PubMed] [Google Scholar]
- 48.Sampson MG, Grassino K. Neuromechanical properties in obese patients during carbon dioxide rebreathing. Am J Med. 1983;75:81–90. doi: 10.1016/0002-9343(83)91171-3. [DOI] [PubMed] [Google Scholar]
- 49.Sharp JT, Henry JP, Sweany SK, et al. Effects of mass loading the respiratory system in man. J Appl Physiol. 1964;19:959–966. doi: 10.1152/jappl.1964.19.5.959. [DOI] [PubMed] [Google Scholar]
- 50.Shimura R, Tatsumi K, Nakamura A, et al. Fat accumulation, leptin, and hypercapnia in obstructive sleep apnea-hypopnea syndrome. Chest. 2005;127:543–549. doi: 10.1378/chest.127.2.543. [DOI] [PubMed] [Google Scholar]
- 51.O’Donnell CP, Schaub CD, Haines AS, et al. Leptin prevents respiratory depression in obesity. Am J Respir Crit Care Med. 1999;159:1477–1484. doi: 10.1164/ajrccm.159.5.9809025. [DOI] [PubMed] [Google Scholar]
- 52.Phipps PR, Starritt E, Caterson I, et al. Association of serum leptin with hypoventilation in human obesity. Thorax. 2002;57:75–76. doi: 10.1136/thorax.57.1.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Trinder J, Merson R, Rosenberg JI, et al. Pathophysiological interactions of ventilation, arousals, and blood pressure oscillations during Cheyne-Stokes respiration in patients with heart failure. Am J Respir Crit Care Med. 2000;162:808–813. doi: 10.1164/ajrccm.162.3.9806080. [DOI] [PubMed] [Google Scholar]
- 54.Szollosi I, Roebuck T, Thompson B, et al. Lateral sleeping position reduces severity of central sleep apnea/Cheyne-Stokes respiration. Sleep. 2006;29:1045–1051. doi: 10.1093/sleep/29.8.1045. [DOI] [PubMed] [Google Scholar]
- 55.Naughton M, Benard D, Tam A, et al. Role of hyperventilation in the pathogenesis of central sleep apneas in patients with congestive heart failure. Am Rev Respir Dis. 1993;148:330–338. doi: 10.1164/ajrccm/148.2.330. [DOI] [PubMed] [Google Scholar]
- 56.Solin P, Roebuck T, Johns DP, et al. Peripheral and central ventilatory responses in central sleep apnea with and without congestive heart failure. Am J Respir Crit Care Med. 2000;162:2194–2200. doi: 10.1164/ajrccm.162.6.2002024. [DOI] [PubMed] [Google Scholar]
- 57.Xie A, Skatrud JB, Puleo DS, et al. Apnea-hypopnea thresh- old for CO2 in patients with congestive heart failure. Am J Respir Crit Care Med. 2002;165:1245–1250. doi: 10.1164/rccm.200110-022OC. [DOI] [PubMed] [Google Scholar]
- 58.Hall MJ, Xie A, Rutherford R, et al. Cycle length of periodic breathing in patients with and without heart failure. Am J Respir Crit Care Med. 1996;154:376–381. doi: 10.1164/ajrccm.154.2.8756809. [DOI] [PubMed] [Google Scholar]
- 59.Xie A, Skatrud JB, Khayat R, et al. Cerebrovascular response to carbon dioxide in patients with congestive heart failure. Am J Respir Crit Care Med. 2005;172:371–378. doi: 10.1164/rccm.200406-807OC. [DOI] [PubMed] [Google Scholar]
- 60.Paintal AS. Vagal sensory receptors and their reflex effects. Physiol Rev. 1973;53:159–227. doi: 10.1152/physrev.1973.53.1.159. [DOI] [PubMed] [Google Scholar]
- 61.Solin P, Bergin P, Richardson M, et al. Influence of pulmonary capillary wedge pressure on central apnea in heart failure. Circulation. 1999;99:1574–1579. doi: 10.1161/01.cir.99.12.1574. [DOI] [PubMed] [Google Scholar]
- 62.Bradley TD, Phillipson EA. Central sleep apnea. Clin Chest Med. 1992;13:493–505. [PubMed] [Google Scholar]
- 63.Solin P, Jackson DM, Roebuck T, et al. Cardiac diastolic function and hypercapnic ventilatory responses in central sleep apnoea. Eur Respir J. 2002;20:717–723. doi: 10.1183/09031936.02.00742002. [DOI] [PubMed] [Google Scholar]
- 64.Xie A, Rutherford R, Rankin F, et al. Hypocapnia and increased ventilatory responsiveness in patients with idiopathic central sleep apnea. Am J Respir Crit Care Med. 1995;152:1950–1955. doi: 10.1164/ajrccm.152.6.8520761. [DOI] [PubMed] [Google Scholar]
- 65.Sleep-related breathing disorders in adults: recommendations for syndrome definition and measurement techniques in clinical research: the report of an American Academy of Sleep Medicine Task Force. Sleep. 1999;22:667–689. [PubMed] [Google Scholar]
- 66.Khoo MC, Anholm JD, Ko SW, et al. Dynamics of periodic breathing and arousal during sleep at extreme altitude. Respir Physiol. 1996;103:33–43. doi: 10.1016/0034-5687(95)00057-7. [DOI] [PubMed] [Google Scholar]
- 67.Dempsey JA, Smith CA, Harms CA, et al. Sleep-induced breathing instability: University of Wisconsin-Madison Sleep and Respiration Research Group. Sleep. 1996;19:236–247. [PubMed] [Google Scholar]
- 68.Hlavac MC, Catcheside PG, McDonald R, et al. Hypoxia impairs the arousal response to external resistive loading and airway occlusion during sleep. Sleep. 2006;29:624–631. [PubMed] [Google Scholar]
- 69.Eckert DJ, Catcheside PG, McDonald R, et al. Sustained hypoxia depresses sensory processing of respiratory resistive loads. Am J Respir Crit Care Med. 2005;172:1047–1054. doi: 10.1164/rccm.200505-699OC. [DOI] [PubMed] [Google Scholar]
- 70.Xu F, Gu QH, Zhou T, et al. Acute hypoxia prolongs the apnea induced by right atrial injection of capsaicin. J Appl Physiol. 2003;94:1446–1454. doi: 10.1152/japplphysiol.00767.2002. [DOI] [PubMed] [Google Scholar]
- 71.Morgenthaler TI, Kagramanov V, Hanak V, et al. Complex sleep apnea syndrome: is it a unique clinical syndrome. Sleep. 2006;29:1203–1209. doi: 10.1093/sleep/29.9.1203. [DOI] [PubMed] [Google Scholar]
- 72.Gastrointestinal surgery for severe obesity: National Institutes of Health Consensus Development Conference statement. Am J Clin Nutr. 1992;55:615S–619S. doi: 10.1093/ajcn/55.2.615s. [DOI] [PubMed] [Google Scholar]
- 73.Perez de Llano LA, Golpe R, Ortiz Piquer M, et al. Short-term and long-term effects of nasal intermittent positive pressure ventilation in patients with obesity-hypoventilation syndrome. Chest. 2005;128:587–594. doi: 10.1378/chest.128.2.587. [DOI] [PubMed] [Google Scholar]
- 74.Berger KI, Ayappa I, Chatr-Amontri B, et al. Obesity hypoventilation syndrome as a spectrum of respiratory disturbances during sleep. Chest. 2001;120:1231–1238. doi: 10.1378/chest.120.4.1231. [DOI] [PubMed] [Google Scholar]
- 75.Hida W, Okabe S, Tatsumi K, et al. Nasal continuous positive airway pressure improves quality of life in obesity hypoventilation syndrome. Sleep Breath. 2003;7:3–12. doi: 10.1007/s11325-003-0003-1. [DOI] [PubMed] [Google Scholar]
- 76.Shneerson JM, Simonds AK. Noninvasive ventilation for chest wall and neuromuscular disorders. Eur Respir J. 2002;20:480–487. doi: 10.1183/09031936.02.00404002. [DOI] [PubMed] [Google Scholar]
- 77.Storre JH, Seuthe B, Fiechter R, et al. Average volume-assured pressure support in obesity hypoventilation: a randomized crossover trial. Chest. 2006;130:815–821. doi: 10.1378/chest.130.3.815. [DOI] [PubMed] [Google Scholar]
- 78.McNicholas WT, Carter JL, Rutherford R, et al. Beneficial effect of oxygen in primary alveolar hypoventilation with central sleep apnea. Am Rev Respir Dis. 1982;125:773–775. doi: 10.1164/arrd.1982.125.6.773. [DOI] [PubMed] [Google Scholar]
- 79.Sutton FD, Jr, Zwillich CW, Creagh CE, et al. Progesterone for outpatient treatment of Pickwickian syndrome. Ann Intern Med. 1975;83:476–479. doi: 10.7326/0003-4819-83-4-476. [DOI] [PubMed] [Google Scholar]
- 80.Baylor P, Tayloe D, Owen D, et al. Cardiac failure presenting as sleep apnea: elimination of apnea following medical management of cardiac failure. Chest. 1988;94:1298–1300. doi: 10.1378/chest.94.6.1298. [DOI] [PubMed] [Google Scholar]
- 81.Dark DS, Pingleton SK, Kerby GR, et al. Breathing pattern abnormalities and arterial oxygen desaturation during sleep in the congestive heart failure syndrome: improvement following medical therapy. Chest. 1987;91:833–836. doi: 10.1378/chest.91.6.833. [DOI] [PubMed] [Google Scholar]
- 82.Walsh JT, Andrews R, Starling R, et al. Effects of captopril and oxygen on sleep apnoea in patients with mild to moderate congestive cardiac failure. Br Heart J. 1995;73:237–241. doi: 10.1136/hrt.73.3.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Bradley TD, Logan AG, Kimoff RJ, et al. Continuous positive airway pressure for central sleep apnea and heart failure. N Engl J Med. 2005;353:2025–2033. doi: 10.1056/NEJMoa051001. [DOI] [PubMed] [Google Scholar]
- 84.Teschler H, Dohring J, Wang YM, et al. Adaptive pressure support servo-ventilation: a novel treatment for Cheyne-Stokes respiration in heart failure. Am J Respir Crit Care Med. 2001;164:614–619. doi: 10.1164/ajrccm.164.4.9908114. [DOI] [PubMed] [Google Scholar]
- 85.Philippe C, Stoica-Herman M, Drouot X, et al. Compliance with and effectiveness of adaptive servoventilation versus continuous positive airway pressure in the treatment of Cheyne-Stokes respiration in heart failure over a six month period. Heart. 2006;92:337–342. doi: 10.1136/hrt.2005.060038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Pepperell JC, Maskell NA, Jones DR, et al. A randomized controlled trial of adaptive ventilation for Cheyne-Stokes breathing in heart failure. Am J Respir Crit Care Med. 2003;168:1109–1114. doi: 10.1164/rccm.200212-1476OC. [DOI] [PubMed] [Google Scholar]
- 87.Pittman S, Hill P, Malhotra A, et al. Stabilizing Cheyne Stokes respiration associated with congestive heart failure using computer assisted positive airway pressure. Comput Cardiol. 2000:201–204. [Google Scholar]
- 88.Lorenzi-Filho G, Rankin F, Bies I, et al. Effects of inhaled carbon dioxide and oxygen on Cheyne-Stokes respiration in patients with heart failure. Am J Respir Crit Care Med. 1999;159:1490–1498. doi: 10.1164/ajrccm.159.5.9810040. [DOI] [PubMed] [Google Scholar]
- 89.Khayat RN, Xie A, Patel AK, et al. Cardiorespiratory effects of added dead space in patients with heart failure and central sleep apnea. Chest. 2003;123:1551–1560. doi: 10.1378/chest.123.5.1551. [DOI] [PubMed] [Google Scholar]
- 90.Franklin KA, Eriksson P, Sahlin C, et al. Reversal of central sleep apnea with oxygen. Chest. 1997;111:163–169. doi: 10.1378/chest.111.1.163. [DOI] [PubMed] [Google Scholar]
- 91.Javaheri S, Ahmed M, Parker TJ, et al. Effects of nasal O2 on sleep-related disordered breathing in ambulatory patients with stable heart failure. Sleep. 1999;22:1101–1106. doi: 10.1093/sleep/22.8.1101. [DOI] [PubMed] [Google Scholar]
- 92.Hanly PJ, Millar TW, Steljes DG, et al. The effect of oxygen on respiration and sleep in patients with congestive heart failure. Ann Intern Med. 1989;111:777–782. doi: 10.7326/0003-4819-111-10-777. [DOI] [PubMed] [Google Scholar]
- 93.Krachman SL, D’Alonzo GE, Berger TJ, et al. Comparison of oxygen therapy with nasal continuous positive airway pressure on Cheyne-Stokes respiration during sleep in congestive heart failure. Chest. 1999;116:1550–1557. doi: 10.1378/chest.116.6.1550. [DOI] [PubMed] [Google Scholar]
- 94.Sinha AM, Skobel EC, Breithardt OA, et al. Cardiac resynchronization therapy improves central sleep apnea and Cheyne-Stokes respiration in patients with chronic heart failure. J Am Coll Cardiol. 2004;44:68–71. doi: 10.1016/j.jacc.2004.03.040. [DOI] [PubMed] [Google Scholar]
- 95.Gabor JY, Newman DA, Barnard-Roberts V, et al. Improvement in Cheyne-Stokes respiration following cardiac resynchronisation therapy. Eur Respir J. 2005;26:95–100. doi: 10.1183/09031936.05.00093904. [DOI] [PubMed] [Google Scholar]
- 96.Javaheri S, Parker TJ, Wexler L, et al. Effect of theophylline on sleep-disordered breathing in heart failure. N Engl J Med. 1996;335:562–567. doi: 10.1056/NEJM199608223350805. [DOI] [PubMed] [Google Scholar]
- 97.Javaheri S. Acetazolamide improves central sleep apnea in heart failure: a double-blind, prospective study. Am J Respir Crit Care Med. 2006;173:234–237. doi: 10.1164/rccm.200507-1035OC. [DOI] [PubMed] [Google Scholar]
- 98.Hoffstein V, Slutsky AS. Central sleep apnea reversed by continuous positive airway pressure. Am Rev Respir Dis. 1987;135:1210–1212. doi: 10.1164/arrd.1987.135.5.1210. [DOI] [PubMed] [Google Scholar]
- 99.Issa FG, Sullivan CE. Reversal of central sleep apnea using nasal CPAP. Chest. 1986;90:165–171. doi: 10.1378/chest.90.2.165. [DOI] [PubMed] [Google Scholar]
- 100.Xie A, Rankin F, Rutherford R, et al. Effects of inhaled CO2 and added dead space on idiopathic central sleep apnea. J Appl Physiol. 1997;82:918–926. doi: 10.1152/jappl.1997.82.3.918. [DOI] [PubMed] [Google Scholar]
- 101.Garrigue S, Bordier P, Jais P, et al. Benefit of atrial pacing in sleep apnea syndrome. N Engl J Med. 2002;346:404–412. doi: 10.1056/NEJMoa011919. [DOI] [PubMed] [Google Scholar]
- 102.Melzer C, Fietze I, Duru F, et al. Nocturnal overdrive pacing for the treatment of sleep apnea syndrome. Sleep. 2006;29:1197–1202. doi: 10.1093/sleep/29.9.1197. [DOI] [PubMed] [Google Scholar]
- 103.Luthje L, Unterberg-Buchwald C, Dajani D, et al. Atrial overdrive pacing in patients with sleep apnea with implanted pacemaker. Am J Respir Crit Care Med. 2005;172:118–122. doi: 10.1164/rccm.200409-1258OC. [DOI] [PubMed] [Google Scholar]
- 104.Unterberg C, Luthje L, Szych J, et al. Atrial overdrive pacing compared to CPAP in patients with obstructive sleep apnoea syndrome. Eur Heart J. 2005;26:2568–2575. doi: 10.1093/eurheartj/ehi448. [DOI] [PubMed] [Google Scholar]
- 105.Pepin JL, Defaye P, Garrigue S, et al. Overdrive atrial pacing does not improve obstructive sleep apnoea syndrome. Eur Respir J. 2005;25:343–347. doi: 10.1183/09031936.05.00132703. [DOI] [PubMed] [Google Scholar]
- 106.Krahn AD, Yee R, Erickson MK, et al. Physiologic pacing in patients with obstructive sleep apnea: a prospective, randomized crossover trial. J Am Coll Cardiol. 2006;47:379–383. doi: 10.1016/j.jacc.2005.09.026. [DOI] [PubMed] [Google Scholar]
- 107.Simantirakis EN, Schiza SE, Chrysostomakis SI, et al. Atrial overdrive pacing for the obstructive sleep apnea-hypopnea syndrome. N Engl J Med. 2005;353:2568–2577. doi: 10.1056/NEJMoa050610. [DOI] [PubMed] [Google Scholar]
- 108.White DP, Zwillich CW, Pickett CK, et al. Central sleep apnea: improvement with acetazolamide therapy. Arch Intern Med. 1982;142:1816–1819. [PubMed] [Google Scholar]
- 109.Mak S, Azevedo ER, Liu PP, et al. Effect of hyperoxia on left ventricular function and filling pressures in patients with and without congestive heart failure. Chest. 2001;120:467–473. doi: 10.1378/chest.120.2.467. [DOI] [PubMed] [Google Scholar]
- 110.Shigemitsu M, Nishio K, Kusuyama T, et al. Nocturnal oxygen therapy prevents progress of congestive heart failure with central sleep apnea. Int J Cardiol. 2007 doi: 10.1016/j.ijcard.2006.03.018. (in press) [DOI] [PubMed] [Google Scholar]
- 111.Sasayama S, Izumi T, Seino Y, et al. Effects of nocturnal oxygen therapy on outcome measures in patients with chronic heart failure and Cheyne-Stokes respiration. Circ J. 2006;70:1–7. doi: 10.1253/circj.70.1. [DOI] [PubMed] [Google Scholar]
- 112.Steens RD, Millar TW, Su X, et al. Effect of inhaled 3% CO2 on Cheyne-Stokes respiration in congestive heart failure. Sleep. 1994;17:61–68. doi: 10.1093/sleep/17.1.61. [DOI] [PubMed] [Google Scholar]
- 113.Szollosi I, Jones M, Morrell MJ, et al. Effect of CO2 inhalation on central sleep apnea and arousals from sleep. Respiration. 2004;71:493–498. doi: 10.1159/000080634. [DOI] [PubMed] [Google Scholar]
- 114.Andreas S, Weidel K, Hagenah G, et al. Treatment of Cheyne-Stokes respiration with nasal oxygen and carbon dioxide. Eur Respir J. 1998;12:414–419. doi: 10.1183/09031936.98.12020414. [DOI] [PubMed] [Google Scholar]
- 115.Krachman SL, Crocetti J, Berger TJ, et al. Effects of nasal continuous positive airway pressure on oxygen body stores in patients with Cheyne-Stokes respiration and congestive heart failure. Chest. 2003;123:59–66. doi: 10.1378/chest.123.1.59. [DOI] [PubMed] [Google Scholar]
- 116.Arzt M, Bradley TD. Treatment of sleep apnea in heart failure. Am J Respir Crit Care Med. 2006;173:1300–1308. doi: 10.1164/rccm.200511-1745PP. [DOI] [PubMed] [Google Scholar]
- 117.Hommura F, Nishimura M, Oguri M, et al. Continuous versus bilevel positive airway pressure in a patient with idiopathic central sleep apnea. Am J Respir Crit Care Med. 1997;155:1482–1485. doi: 10.1164/ajrccm.155.4.9105099. [DOI] [PubMed] [Google Scholar]
- 118.Thomas RJ, Daly RW, Weiss JW. Low-concentration carbon dioxide is an effective adjunct to positive airway pressure in the treatment of refractory mixed central and obstructive sleep-disordered breathing. Sleep. 2005;28:69–77. doi: 10.1093/sleep/28.1.69. [DOI] [PubMed] [Google Scholar]
- 119.Johnson KG, Johnson DC. Bilevel positive airway pressure worsens central apneas during sleep. Chest. 2005;128:2141–2150. doi: 10.1378/chest.128.4.2141. [DOI] [PubMed] [Google Scholar]
- 120.Masa JF, Celli BR, Riesco JA, et al. The obesity hypoventilation syndrome can be treated with noninvasive mechanical ventilation. Chest. 2001;119:1102–1107. doi: 10.1378/chest.119.4.1102. [DOI] [PubMed] [Google Scholar]
- 121.Collop NA. Cheyne-stokes ventilation converting to obstructive sleep apnea following heart transplantation. Chest. 1993;104:1288–1289. doi: 10.1378/chest.104.4.1288. [DOI] [PubMed] [Google Scholar]
- 122.Mansfield DR, Solin P, Roebuck T, et al. The effect of successful heart transplant treatment of heart failure on central sleep apnea. Chest. 2003;124:1675–1681. doi: 10.1378/chest.124.5.1675. [DOI] [PubMed] [Google Scholar]
- 123.Javaheri S, Abraham WT, Brown C, et al. Prevalence of obstructive sleep apnoea and periodic limb movement in 45 subjects with heart transplantation. Eur Heart J. 2004;25:260–266. doi: 10.1016/j.ehj.2003.10.032. [DOI] [PubMed] [Google Scholar]
- 124.Himmrich E, Przibille O, Zellerhoff C, et al. Proarrhythmic effect of pacemaker stimulation in patients with implanted cardioverter-defibrillators. Circulation. 2003;108:192–197. doi: 10.1161/01.CIR.0000080291.65638.CC. [DOI] [PubMed] [Google Scholar]
- 125.Nakayama H, Smith CA, Rodman JR, et al. Effect of ventilatory drive on carbon dioxide sensitivity below eupnea during sleep. Am J Respir Crit Care Med. 2002;165:1251–1260. doi: 10.1164/rccm.2110041. [DOI] [PubMed] [Google Scholar]
- 126.Zwillich CW, Natalino MR, Sutton FD, et al. Effects of progesterone on chemosensitivity in normal men. J Lab Clin Med. 1978;92:262–269. [PubMed] [Google Scholar]
- 127.Bittar G, Friedman HS. The arrhythmogenicity of theophylline: a multivariate analysis of clinical determinants. Chest. 1991;99:1415–1420. doi: 10.1378/chest.99.6.1415. [DOI] [PubMed] [Google Scholar]
- 128.Suissa S, Hemmelgarn B, Blais L, et al. Bronchodilators and acute cardiac death. Am J Respir Crit Care Med. 1996;154:1598–1602. doi: 10.1164/ajrccm.154.6.8970341. [DOI] [PubMed] [Google Scholar]