

Abstract
Sequence profile and fold recognition methods identified mammalian purple acid phosphatase (PAP), a member of a dimetal-containing phosphoesterase (DMP) family, as a remote homolog of human acid sphingomyelinase (ASM). A model of the phosphoesterase domain of ASM was built based on its predicted secondary structure and the metal-coordinating residues of PAP. Due to the low sequence identity between ASM and PAP (~15%), the highest degree of confidence in the model resides in the metal-binding motifs. The ASM model predicts residues Asp 206, Asp 278, Asn 318, His 425, and His 457 to be dimetal coordinating. A putative orientation for the phosphorylcholine head group of the ASM substrate, sphingomyelin (SM), was made based on the predicted catalysis of the phosphorus–oxygen bond in the active site of ASM and on a structural comparison of the PAP–phosphate complex to the C-reactive protein–phosphorylcholine complex. These complexes revealed similar spatial interactions between the metal-coordinating residues, the metals, and the phosphate groups, suggesting a putative orientation for the head group in ASM consistent with the mechanism considerations. A conserved sequence motif in ASM, NX3CX3N, was identified (Asn 381 to Asn 389) and is predicted to interact with the choline amine moiety in SM. The resulting ASM model suggests that the enzyme uses an SN2-type catalytic mechanism to hydrolyze SM, similar to other DMPs. His 319 in ASM is predicted to protonate the ceramide-leaving group in the catalysis of SM. The putative functional roles of several ASM Niemann-Pick missense mutations, located in the predicted phosphoesterase domain, are discussed in context to the model.
Keywords: acid sphingomyelinase, phosphoesterase, purple acid phosphatase, threading, structure predictions
Human acid sphingomyelinase (ASM) is an enzyme that catalyzes the hydrolysis of sphingomyelin (SM) into ceramide and phosphorylcholine (Fig. 1 ▶). This zinc-dependent, membrane-associated glycoprotein has a pH optimum of ~5.0, but has been shown to catalyze SM at both acidic and neutral pHs (Yamanaka and Suzuki 1982; Quintern et al. 1987; Schissel et al. 1998a). Two forms of ASM have been described, an intracellular form found in lysosomes and an extracellular secreted form (see Goni and Alonso2002 and references therein). Both forms are derived from the same ASM gene through differential protein trafficking of a common protein precursor (Schissel et al. 1996a, 1998b). The secreted form of ASM is typically activated by physiologic concentrations of Zn2+ (Schissel et al. 1996a, 1998b; Marathe et al. 1998; He et al. 1999).
Figure 1.

Comparison of the enzymatic phosphate ester cleavage reactions carried out by ASM and PAP. (A) Reaction catalyzed by ASM and (B) an example of an acid phosphatase reaction (glycerophosphatase) catalyzed by mammalian PAP.
Enzymatic activity of secreted ASM has been implicated in the promotion of atherogenesis (Schissel et al. 1996b, 1998a; Marathe et al. 1998, 1999; Tabas 1999). During atherogenesis, the engorgement of macrophages with a large amount of sphingomyelin-containing low-density lipoproteins (LDL) leads to foam cell formation in the sub-endothelium of arteries (Tabas et al. 1993; Williams and Tabas 1995). Cell culture studies suggest that secreted ASM catalyzes LDL-sphingomyelin into LDL-ceramide, which leads to subendothelial LDL aggregation and retention, followed subsequently by foam cell formation (Schissel et al. 1996b, 1998a). These processes catalyzed by extracellular ASM may be important both in the initiation and progression of atherosclerotic lesions.
The lysosomal form of ASM catalyzes the lysosomal degradation of SM, a major lipid constituent of the outer leaflet of eukaryotic membranes. A deficiency of ASM activity can lead to the neuropathic (type A) or non-neuropathic (type B) lysosomal storage disorder known as Niemann-Pick disease (Callahan and Khalil 1976; Kolodny 2000). Somatic cell hybridization studies have shown that type A and B Niemann-Pick disease occur due to allelic mutations in the ASM gene (Besley et al. 1980). The absence of ASM recapitulates the severe neurological disorder found in type A Niemann-Pick disease; however, expression of low levels of the enzyme often found in type B disease (3%–10%) appear to be sufficient to preserve CNS function (Levade et al. 1986; Marathe et al. 2000).
The ASM gene encodes a 629-amino-acid protein, consisting of a signal peptide, a predicted saposin domain, a phosphoesterase domain containing a predicted dimetal center, and a carboxy-terminal region (Schuchman et al. 1991; Koonin 1994; Ponting 1994; Zhuo et al. 1994; Klabunde et al. 1996). Studies have shown that the phosphoesterase domain of ASM is a member of a dimetal-containing phosphoesterase (DMP) family of proteins comprising five metal ion-binding sequence motifs (Koonin 1994; Zhuo et al. 1994; Klabunde et al. 1996). This protein family includes exonucleases, Ser/Thr phosphatases, 5′ nucleotidase, purple acid phosphatase (mammalian form is also known as TRAP, tartrate-resistant acid phosphatase; Vincent and Averill 1990), and other phosphoesterases. One of the best-characterized members of this family is the Fe3+-Zn2+ kidney bean purple acid phosphatase (PAP) (Klabunde et al. 1996).
In this study, we build on the work of Klabunde et al. (1996) to produce a model of the ASM phosphoesterase domain. Using sequence analysis and fold recognition methods that incorporate sequence information in combination with structural information, we rediscovered the relationship of ASM to the DMP family and identified the closest remote structural homolog of the ASM phosphoesterase domain to be PAP. The predicted secondary structure of ASM and the metal-coordinating residues of PAP (Klabunde et al. 1996) were used to optimize the alignments and build a model of the ASM phosphoesterase domain based on the pig PAP structure (PDB entry 1UTE; Guddat et al. 1999). The model was further refined to be consistent with the recently identified disulfide bond pattern in ASM (Lansmann et al. 2003). The highest degree of confidence in the model resides in the metal-coordinating motifs. The structure of ASM beyond this site could not be accurately determined due to the low sequence homology between ASM and PAP (~15%). The resultant model contains ASM residues Ile 201 through Leu 472, but does not include the saposin domain, a 30-residue insertion (residues 218–247) after the first metal-coordinating motif, and the carboxy-terminal domain region of the protein. Predictions of the residues involved in substrate binding and hydrolysis are discussed, as are the proposed mechanisms of action of a number of Niemann-Pick disease missense mutations. On the basis of the proposed catalytic mechanism of ASM and on the only known structure of phosphorylcholine bound to two metal ions in the Protein Data Bank (PDB), phosphorylcholine was oriented into our ASM model. This orientation predicts that a conserved sequence motif NX3CX3N is involved with phosphorylcholine head group recognition of the ASM substrate SM. We propose residues and regions in this ASM model that can be the focus of subsequent experimental evaluation.
Results
Identification of a structural template to build the ASM phosphoesterase domain
With the goal of identifying structural homologs of human ASM for use as a template to model the phosphoesterase domain of ASM, conventional sequence searches were initially performed (BLAST, Smith-Waterman, and FASTA). These analyses identified matches to ASM and ASM-like proteins from different species, as well as vacuolar polyphosphatases, but did not identify any structural templates with significant sequence homology to the phosphoesterase domain of ASM. It should be noted that the function of ASM-like proteins is not presently known.
Sequence profile searches using PSI-BLAST (Altschul et al. 1997) and the hidden Markov model (HMM) method (Eddy 1998; Sonnhammer et al. 1998) were then performed. As shown in Table 1, PSI-BLAST was successful in identifying PAP from the GenPept database with high confidence (E-value of 10−51). PSI-BLAST also identified a second phosphoesterase template, 5′ nucleotidase, with an Evalue of 10−2. Using the more sensitive HMM method, PAP was detected using both the GenPept and SwissProt databases, with a P-score of 10−5, but not in the PDB. The HMM method also identified other phosphoesterases that were previously shown to be homologous to PAP (Koonin 1994; Zhuo et al. 1994; Klabunde et al. 1996). The different Evalues for PAP, generated by the respective search methods, reflect the differences in the algorithms and the diversity and size of the databases used by the two independent methods. The search results also show the value of using a more robust HMM search method in addition to PSI-BLAST to identify remote homologs. PSI-BLAST can generate false positives with significant E-values. In this case, only two known members of the large phosphatase family were identified by PSI-BLAST, thus requiring a further confirmation of these matches.
Table 1.
Sequence profile search and fold recognition analysis
| A. Sequence Profile Search | |||||
| Method | GenPept | SwissProt | PDB | ||
| PSI-BLAST | Acid SMascb | 1 × 10−133 | NOc | NO | |
| PAP | 3 × 10−51 | ||||
| 5′ Nucleotidase | 1 × 10−2 | ||||
| HMMa | Acid SMaseb | 1 × 10−202 | Acid SMase | 1 × 10−202 | NO |
| PAP | 2.8 × 10−5 | PAP | 3.0 × 10−5 | ||
| Ser/Thr Phosphatase 1 | 1.2 × 10−4 | Exonuclease sbcd | 2.1 × 10−3 | ||
| 5′ Nucleotidase | 4.5 × 10−4 | Bis(5′-nucleosyl)-tetraphosphatase | 2.3 × 10−3 | ||
| Exonuclease sbcd | 2.1 × 10−3 | 2′ ,3′-cyclic-nucleotide 2′ Phosphodiesterase | 3.4 × 10−3 | ||
| Bis(5′-nucleosyl)-tetraphosphatase | 2.3 × 10−3 | 5′ Nucleotidase | 7.3 × 10−3 | ||
| 2′, 3′-cyclic-nucleotide 2′ Phosphodiesterase | 3.4 × 10−3 | Ser/Thr Phosphatase 2B | 9.0 × 10−3 | ||
| B. Fold Recognition Analysis | |||||
| Method | Fold Library | PDB ID | Rank | E-value | |
| a HMM built from a protein family that have ASM or ASM-like annotations only. | |||||
| b E-value or P-score from PSI-BLAST and HMM, respectively, corresponds to the first occurrence of the protein (independent of source) present in the motif alignment from the study of Klabunde et al. (1996). PSI-BLAST identified templates after five iterations using an E-value of 0.001 for the inclusion of the next pass. | |||||
| c Method was not able to identify a structural template with significant sequence homology (E-value or P score < 1). | |||||
| d E-values from 3D-PSSM below 0.05 are highly confident. | |||||
| 3D-PSSM | Pig PAP | (1UTE) | 1 | 0.02d | 95% confidence level |
| Kidney Bean PAP | (4KBP) | 2 | 1.03 | ||
| Kidney Bean PAP | (1KBP) | 3 | 2.06 | ||
| 5′ Nucleotidase | (1USH) | 15 | 6.32 | ||
| Z-score | |||||
| 123D+ | Phenol Hydrolase | 1 | 5.34 | ||
| Pig PAP | (1UTE) | 2 | 4.34 | ||
| Kidney Bean PAP | (4KBP) | 9 | 3.39 | ||
In addition to evaluating several sequence search methods, it was also beneficial to run fold recognition methods, because not all sequences in the sequence databases (i.e., GenPept, SwissProt) are annotated with structural information from the PDB. Moreover, fold analysis methods, which include secondary structure data, result in alignments that are more suitable for model building. Therefore, twofold recognition methods (3D-PSSM and 123D+) were used in an attempt to identify structural homologs of ASM. These methods also identified PAP among the top ranked folds (Table 1). The fold recognition method 3D-PSSM identified the pig PAP as the first ranked protein fold with a 95% confidence level (PDB entry 1UTE; Guddat et al. 1999). Two kidney bean PAPs (PDB entries 4KBP and 1KBP; Klabunde et al. 1996) and a 5′ nucleotidase (PDB entry 1USH, Knöfel and Sträter 1999) were identified as the 2nd, 3rd, and 15th ranked folds, respectively. In comparison, the program 123D+ identified pig and kidney bean PAPs as the second and ninth ranked folds, respectively.
The profile sequence searches and fold recognition methods identified several conserved sequence motifs, including the two metallophosphoesterase signature motifs A and B originally defined by Koonin (1994), Zhuo et al. (1994), and Klabunde et al. (1996). These motifs actually contained five smaller metal-coordinating motifs, which we refer to as MM1 through MM5 (see Fig. 2 ▶). The MM1 and MM5 metal-coordinating motifs are expanded in ASM and ASM-like proteins (DXHXDXXY, GHXHXD) when compared to the DMP family of proteins.
Figure 2.
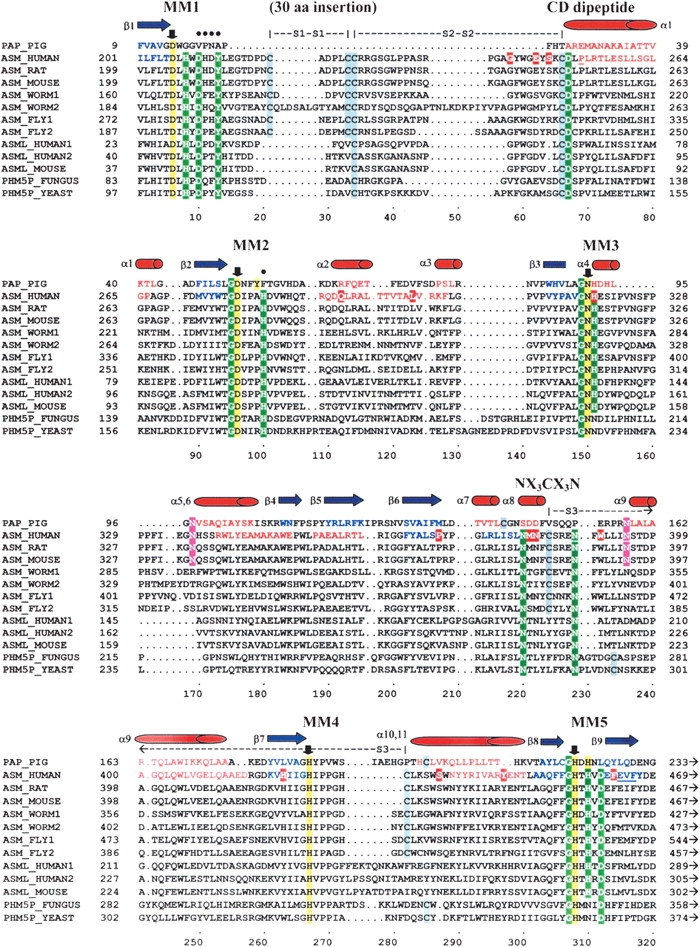
Sequence alignment of the PAP, ASM, and ASM-like phosphoesterase domains. Pig PAP was used as the structural template to build the ASM model. The metal-coordinating motifs 1 through 5 are labeled MM1 through MM5. Down arrows (↓) and yellow highlighted residues indicate known or predicted metal-coordinating residues that are conserved within MM1 to MM5 for the PAP and ASM protein families, respectively (Asp 206, Asp 278, Asn 318, His 425, and His 457 for human ASM). The Cys 250–Asp 251 conserved dipeptide sequence is labeled “CD peptide.” The predicted substrate recognition loop motif is labeled “NX3CX3N.” The residues included in the conserved hydrophilic/aromatic cluster in ASM are marked with solid circles (•). The three disulfide bonds are indicated, respectively, as “S1–S1,” “S2–S2,” and “S3–S3.” The 30-residue insertion (218–247) that could not be modeled is designated. The underlined residues indicate putative misfolded regions of the SMase model, based on an average window length of 10 residues. Highlighted regions indicate the following: Green indicates highly conserved residues in the ASM and ASM-like proteins that are residues often found in DMP active site structures; gray indicates cysteine residues; red indicates known Niemann-Pick mutations in human ASM; magenta indicates predicted N-glycosylation sites for the human and mouse ASM. Red residues and barrels (α1 through α11) are known and predicted α-helices in the PAP and human ASM, respectively. Blue residues and arrows (β1 through β9) are known and predicted β sheets of the PAP and human ASM, respectively. Sequence accession numbers: PAP_PIG–P09889, ASM_HUMAN–P17405, ASM_RAT–XP_219098, ASM_MOUSE–NP_035551, ASM_WORM1–AAC46756, ASML_WORM2–NP_509894, ASML_FLY1–NP_611904, ASML_FLY2–AAF57045, ASML_HUMAN1–NP_055289, ASML_HUMAN2–AH18999, ASML_MOUSE–NP_065586, PHM5P_FUNGUS–T50959, PHM5P_YEAST–NP_010740.
Structural alignment of ASM and PAP phosphoesterase domains
The four search methods, PSI-BLAST, HMM, 3D-PSSM, and 123D+, each generated sequence-structure alignments between the phosphoesterase domains of human ASM and pig PAP. Four of the five predicted phosphatase metal-coordinating residues in ASM (located in MM2–MM5) were aligned by all of these programs to the known metal-coordinating residues of pig PAP that had been previously identified by Klabunde et al. (1996) in kidney bean PAP (Fig. 2 ▶). The alignment of the first predicted phosphatase metal-coordinating motif, MM1, was taken from the original alignment of ASM to kidney bean PAP generated by Klabunde et al (1996).
To refine the ASM sequence-structure alignment, secondary structure predictions were used to minimize the disruptions of α-helices and β-sheets within the core of the template structure and the known secondary structure of PAP. Four independent methods (DSC, PHD, Predator, and PSIPred) were used to predict the secondary structure of ASM (data not shown). This alignment identified a region in ASM spanning nearly 50 residues between the predicted first and the second metal-coordinating motifs (Leu 204–Leu 252) that did not contain predicted α-helices or β-strands. Within this region, a 30-amino-acid insertion loop was identified that could not be modeled based on the PAP structure (Fig. 2 ▶).
The disulfide bond pattern for ASM has been recently identified (Lansmann et al. 2003). Three of the reported disulfide bonds lie in the phosphoesterase domain of ASM. A comparison of the ASM sequence-structure alignment to PAP revealed that there are no apparent cysteine residue pairs conserved between the two proteins (see Fig. 2 ▶). One of the disulfide bonds occurring between Cys 385 and -Cys 431 in ASM was used to refine the subsequent model. The two other disulfide bonds, corresponding to Cys 221–Cys 226 and Cys 227–Cys 250, are located in the 30-residue region (Asp 218–Tyr 247) described above that could not be modeled.
Several interesting motifs were identified in the sequence structure alignment and are illustrated in Figure 2 ▶. Two of the cysteine residues, Cys 227 and Cys 250, and an adjacent aspartic acid (Asp 251) are conserved among the ASM and ASM-like proteins as a conserved dipeptide sequence (Cys 250–Asp 251). There is also a cluster of hydrophilic and aromatic residues (see Figs. 2 ▶, 3A ▶) that are conserved among the mammalian ASMs (Asp 210, His 211, Asp/Glu 212, Tyr 213, and His 282). Of these, Asp 210, Tyr 213, and His 282 are conserved residues among both the ASM and the ASM-like proteins. Lastly, a sequence motif, NX3CX3N, was identified from Asn 381 to Asn 389 that is located in a loop in the model and is conserved in all of the ASM proteins (Figs. 2 ▶, 3A ▶). This motif contains Cys 385, which is known to form a disulfide bond with Cys 431. The two Asn residues are also conserved in the ASM-like proteins; however, Cys 385 is not.
Figure 3.

Secondary structure rendering of the model of the human ASM phosphodiesterase domain. Secondary structures are indicated with red barrels (α-helices) and green arrows (β-sheets). The amino and carboxyl termini of the domain are labeled with “N” and “C,” respectively. The dimetal center, indicated with a red arrow, is located within the pseudo twofold symmetry axis of this domain. The highest confidence region, consisting of the side chains of five predicted conserved metal-coordinating residues (D206, D278, N318, H425, and H457) are shown in red. The metal ions are indicated with pink spheres. The 30-residue insertion is indicated with a black arrow. (A) The side chains of the conserved residues with respect to the dimetal center are shown in purple: N381 and N389 from the NX3CX3N motif; Asp 210–Tyr 213 and His 282 from the cluster of hydrophilic/aromatic residues; and C250 and D251 from the CD dipeptide. (B) The side chains of the Niemann-Pick mutation residues are indicated in blue in context to the dimetal center: M382, N383, and W391 in or near the NX3CX3N motif; H319 in MM3; and L302, P371, and H421, predicted to lie outside of the dimetal center.
Model validation
The resulting model of the phosphoesterase domain of human ASM was checked using the Program PROFILES-3D. The scores throughout the ASM model range from -0.21 to 0.87. The sum total PROFILES-3D score for the ASM model is 77.5, which lies between the expected score of 109.9 and the “totally misfolded” model score of 49.5 for a domain of this size. No misfolded regions were identified in the ASM model using an averaging window length of 21 residues. However, one putative misfolded region was predicted when an averaging window length of 10 residues was used. This misfolded region (Phe 463–Phe 466, with scores of -0.02 to -0.12) is located immediately downstream of the fifth metal-coordinating motif MM5. The five predicted metal-coordinating motifs are not part of this misfolded region. Iterative modifications of the alignment corresponding to the misfolded regions did not improve the PROFILES-3D score of the model. Further energy refinement of the model resulted in additional stereochemical problems and mis-folded regions. The Ramachandran analysis of the ASM model assigned ~74% to the core allowed region (compared to 85% for the PAP structure). With exception of the insertions and deletions, the dihedral angles for the residues located in the core region are similar to the angles for residues from pig PAP, as the backbone coordinates were extracted from PAP and constrained during energy minimizations. The root-mean-squared deviation (RMSD) between the backbone conformations of the model (comprising 84% of the ASM residues) and PAP is 0.78 Å.
Model description
Sequence profile and fold recognition methods suggest a remote evolutionary relationship between the phosphoester-ase domain of ASM and PAP. The resulting model of the phosphoesterase domain of human ASM predicts that the general topological protein fold is an α β sandwich, similar to other DMP structures. The predicted dimetal center of ASM is located on the pseudo twofold symmetry axis (Guddat et al. 1999), with each half containing a βαβαβstructural motif (Fig. 3 ▶).
The 30-residue insertion (Asp 218–Tyr 247) described earlier could not be modeled based on the PAP structure and thus was not included. In addition, the orientations of the amino-terminal saposin domain and the carboxy-terminal domain region comprising more than 150 residues are not known. The predicted misfolded region (Phe 463–Phe 466) is adjacent to the carboxy-terminal domain and protein–protein interactions may occur between these regions. The loop region comprising the NX3CX3N motif was difficult to build due to lack of sequence homology. This loop contains Cys 385, which forms a disulfide bond to Cys 431. Figure 2 ▶ illustrates that this disulfide bond is not conserved in PAP or among the ASM-like proteins, and neither are the sequences adjacent to either of these cysteines. Although ASM is likely to adopt a PAP fold topology, the extent to which the spatial backbone conformation of the native ASM phosphoesterase domain differs from that of PAP cannot be assessed.
The five metal-coordinating sequence motifs in the DMP family are not entirely conserved in ASM (Klabunde et al. 1996). However, within each of the five motifs, there is at least one conserved metal-coordinating residue among the known phosphoesterases. For ASM, the residues are Asp 206, Asp 278, Asn 318, His 425, and His 457, whose side chains are structurally equivalent to Asp 14, Asp 52, Asn 91, His 186, and His 221 in PAP (Figs. 4 ▶, 5 ▶). The average RMSD of the heavy atoms for these five conserved metal-coordinating residues, when superpositioned onto the known phosphoesterase structures (PDB entries 1UTE, 4KBP, 1USH, 1FJM, and 1AUI), is ~0.5 Å. Therefore, the positions of the predicted conserved metal-coordinating residues are considered to be the regions of highest confidence within our ASM model. Within this superposition, the pair of dimetal atoms is also found to be similarly positioned compared to the known phosphoesterase structures, despite different metal types (Fe2+, Fe3+, Zn2+, Mn2+). Due to the fact that ASM is a zinc-dependent enzyme (Schissel et al. 1998b), the dimetal center was modeled with two Zn2+ ions.
Figure 4.

Superposition of the dimetal-coordinating sites of PAP–phosphate, CRP–phosphorylcholine, and ASM–phosphorylcholine complexes. Comparison of ASM to the known PAP–phosphate and the CRP–phos-phorylcholine complexes reveal similar spatial interactions between the respective metal-coordinating residues and the metal ions through the phosphate group. Stick-and-ball colors: Red represents the human ASM-phosphorylcholine model showing the predicted structurally equivalent metal-ligating residues; purple represents the pig PAP–phosphate complex (1UTE); and green represents the CRP–phosphorylcholine complex (1B09). The metal ions (Ca2+ in CRP, Fe2+ in PAP, and Zn2+ in ASM) are indicated with the corresponding colored large spheres. The RMSD between the atoms involved in interactions in ASM (two Zn ions, phosphorus atom, Asp 278, Asn 318, His 457, and His 459) or PAP (two Fe ions, phosphorus atom, Asp 52, Asn 91, His 221, and His 223) and the respective atoms in CRP (two Ca ions, phosphorus atom, Asp 140, Asp 60, Asn 61, and Gln 150) is 1.1 Å. The quaternary nitrogen atom of phosphorylcholine (shown in blue) is ~4 Å from the side chain of Glu 81 in the CRP–phosphorylcholine complex.
Figure 5.

Proposed catalytic mechanism for the hydrolysis of phosphodi-esters by ASM. The active site region of human ASM is displayed with the metal-coordinating residues. Zinc atoms are indicated in red. Phosphate is indicated in green. The bridging hydroxide ion is indicated in blue.
The invariant tyrosine in the second metal-coordinating motif of PAP, responsible for the purple color of this enzyme, is not conserved in the ASM or ASM-like proteins (Tyr 55 in Fig. 4 ▶). In PAP, the phenolic oxygen atom of Tyr 55 is the fourth metal coordination atom to one of the two iron ions in the active site. A hydroxide ion and a phosphate oxygen, occupying the fifth and sixth metal coordination sites, complete the coordination sphere. In our ASM model, this tyrosine is replaced by an alanine, resulting in the loss of the fourth coordinating residue to the second zinc ion (Zn2 in Fig. 5 ▶). We speculate that the fourth coordination atom in ASM may be fulfilled by a water molecule. Alternatively, the conserved histidine residue His 208 may play this role, but to accomplish this, significant movement of the backbone would be required.
Modeling of the phosphorylcholine head group portion of the ASM substrate
To address the modeling of the phosphorylcholine head group portion of the SM substrate in our ASM model, both structural precedents and mechanistic considerations were taken into consideration. As outlined in the Materials and Methods section, a model of ASM was initially built in complex with phosphate, based on the corresponding PAP-phosphate complex structure (PDB entry 1UTE). Thereafter, a search was made in the PDB for proteins in complex with phosphorylcholine to evaluate whether these structures had some similarity to our model of the ASM–phosphate complex. This search identified the C-reactive protein (CRP)–phosphorylcholine complex (PDB entry 1B09; Thompson et al. 1999) as the only phosphorylcholine complex in the PDB having its phosphate head group bound to two metal ions. Human CRP is a member of the pentraxin family (Pepys et al. 1978) and is characterized by its high calcium-dependent binding affinity for phosphorylcholine-containing lipids (Schwalbe et al. 1992). Structural analysis of the ASM–phosphate and the CRP–phosphorylcholine complexes revealed similar spatial interactions between the metal-coordinating residues and the metal ions through the phosphate group, despite having different fold-topologies. Figure 4 ▶ highlights the superposition of the metal ligating atoms (Asp 278:OD2, Asn 318:OD1, His 457:ND1, His 459:NE2 in ASM [or Asp 52:OD2, Asn 91:OD1, His 221:ND1, His 223:NE2 in PAP] and Asp 140:OD1, Asp 60:OD1, Asn 61:ND2, Gln 150:NE2 in CRP), the phosphorus atoms in the phosphates, and the metal ions (Zn 501, Zn 502 in ASM [or Fe1, Fe2 in PAP]) and Ca1, Ca2 in CRP), resulting in a RMSD of 1.1 Å. ASM and CRP could be another example of two proteins with different folds that have common structural motifs for ligand recognition (Denessiouk and Johnson 2000; Schmitt et al. 2002).
An evaluation of our proposed catalytic mechanism for ASM predicts the involvement of His 319 in the hydrolysis of a phosphorus–oxygen bond (detailed in the Discussion section and Fig. 5 ▶). This putative mechanism suggests that the phosphate oxygen atom that is neither bound to the two zinc atoms nor within hydrogen-bonding distance of His 319 will form a phosphoester bond to choline when bound to ASM. This prediction is consistent with the final orientation of the phosphorylcholine in the ASM model.
Discussion
Proposed catalytic mechanism of ASM
Both ASM and PAP are DMP enzymes that catalyze the hydrolysis of phosphoesters (see Fig. 1 ▶). With the goal of identifying residues that may be essential for the catalysis of SM and developing a catalytic mechanism of ASM, we initially studied the proposed catalytic mechanisms of the DMPs (Klabunde et al. 1996; Guddat et al. 1999; Knöfel and Sträter 1999; Uppenberg et al. 1999). This evaluation identified a number of common mechanistic features. In general, after substrate binding, the phosphorus atom of the phosphoester is attacked by a hydroxide ion bound to the dimetal center, resulting in a pentacoordinate phosphate transition state. A proton is then donated from the enzyme to the oxygen atom bridging the phosphorus atom and the alcohol, resulting in the hydrolysis of the phosphorus–oxygen bond. The process is completed by the departure of the two products. The molecular details are slightly different between different DMP enzymes, as described later.
Klabunde et al. (1996) proposed that the phosphate leaving group of the kidney bean PAP substrate is bound to the Zn2+ ion through one of its oxygen atoms. This binding displaces a water molecule occupying the sixth coordination site of the Zn2+ ion. A hydroxide ion occupying the sixth coordination site of the second metal ion (Fe3+) subsequently makes a nucleophilic attack on the phosphorus atom, resulting in a pentacoordinate phosphate transition state. In comparison, Egloff et al. (1995) proposed that the phosphate group of the protein phosphatase 1 (PP1) substrate is bound to both the metal ions (Mn2+ and Fe3+) in PP1. Simultaneously, the hydroxide ion, which is also bound to both metal ions, makes a nucleophilic attack that again results in a pentacoordinate phosphate transition state. Interestingly, the crystal structure of pig PAP has a hydroxide ion/water molecule bridging the two metal ions, consistent with a catalytic mechanism similar to PP1 rather than kidney bean PAP.
After the nucleophilic attack in the DMP catalytic cycle, a His residue donates a proton to a substrate phosphate oxygen atom to facilitate the cleavage of the phosphorus–oxygen bond. During hydrolysis, the phosphate undergoes an inversion (Mueller et al. 1993). These findings suggest that the DMP mechanism uses an SN2-type hydrolysis reaction. The His proton donor described previously is hydrogen bonded to the phosphate, and is most often part of the third metal-coordinating region of the metallophosphoester-ase signature motif A (GNH-D/E) in the DMPs (Koonin 1994; Zhuo et al. 1994). The majority of the DMPs use a catalytic mechanism involving an Asp that enhances the general acid character of the His residue. For pig PAP, this is a role played by Asp 146, which is within 3.7 Å of the donating His 92 residue and is located 54 residues downstream from His 92. In comparison, Asp 95 in Ser/Thr phos-phatase 1 (PP1) is located three residues after the GlyAsp in the second signature motif and forms a salt bridge to the downstream His 125 proton donor (Egloff et al. 1995). Asp 120 in 5′-nucleotidase is located in the third signature motif and forms a salt bridge to the His 117 proton donor (Knöfel and Sträter 1999). Finally, Asp 169 in kidney bean PAP is located five residues after the GlyAsp in the second signature motif and is hydrogen bonded to His 202. His 202, in turn, is hydrogen bonded to the substrate phosphate. However, an alternative mechanism has also been proposed for kidney bean PAP (Klabunde et al. 1996). This alternative mechanism uses two His residues (His 295 and His 296) that are hydrogen bonded to the substrate phosphate and may be involved in the general acid attack of the substrate.
On the basis of this evaluation, we propose that the catalytic mechanism of ASM is similar to those of other DMPs. ASM carries out its catalysis on the surface of a phospho-lipid membrane bilayer and therefore, only the head group of SM is predicted to be available for initial substrate recognition. We hypothesize that the SM substrate initially binds to ASM with each of its two free phospho-oxygen atoms bound to the Zn2+ ions, as seen in PP1 and pig PAP, and its positively charged tertiary amine interacting with a negatively charged residue, possibly Glu 388 (see Fig. 6 ▶). As illustrated in Figure 5 ▶, a hydroxide ion is simultaneously bound as a bridging ligand between the two Zn2+ ions. This hydroxide ion launches a nucleophilic attack on the phosphorus atom of SM, producing a pentacoordinate phosphate transition state. Our model predicts that the cleavage of the phosphorus–oxygen bond is catalyzed by the donation of a proton from His 319. The enzymatic reaction is completed by the release of the products, ceramide and phosphoryl-choline. A neighboring carboxylate-containing residue is likely to assist His 319; however, due to the limitations of the model we have not been able to identify this acid side chain. This important residue may reside in a region of the ASM protein that was not modeled. As described, different DMPs use different Asp residues located throughout the protein to assist the His residue in catalysis. Alternatively, it is possible that this carboxylate-containing residue may not play a role in the ASM catalytic mechanism. Regardless, given that ASM is predicted to use an SN2-type mechanism as do other DMPs, the substrate phosphorus atom would experience stereochemical inversion during substrate cleavage.
Figure 6.
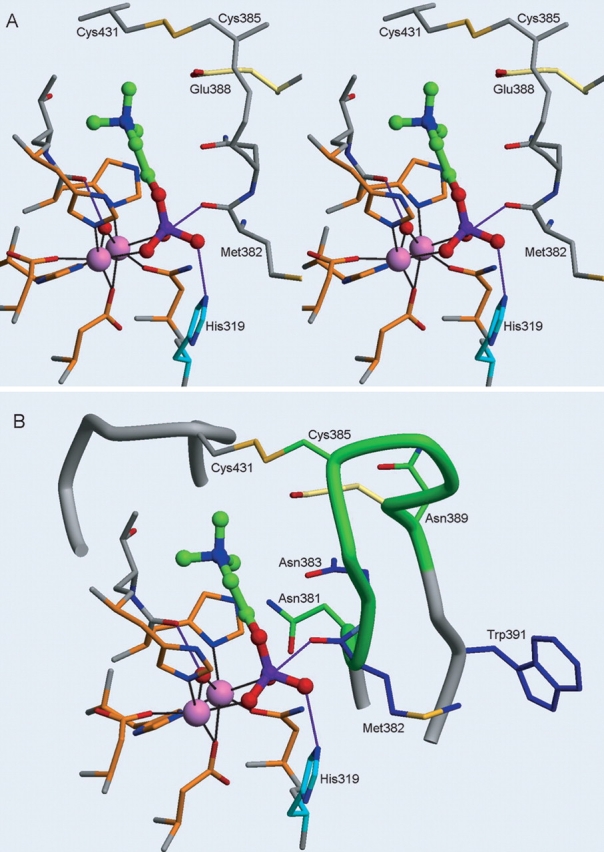
Active site region of the ASM model showing phosphorylcholine, the NX3CX3N loop, and the location of the Niemann-Pick mutations. (A) Relaxed eye stereo view of phosphorylcholine in the ASM model shown in a ball-and-stick representation (carbon atoms, green; oxygen atoms, red; nitrogen atom, blue; and phosphorus atom, purple). The coordinating residues (Asp 206, Asp 278, Asn 318, His 425, His 457, and His 459) to the zinc ions (pink spheres) are shown with orange carbon atoms, and the coordination shown as black lines. Catalytic His 319 is shown in blue, and Glu 388 is shown in yellow. The hydroxide proposed to attach the phosphate is depicted as a red sphere and hydrogen-bonds are shown as purple lines. (B) Similar orientation to A with the addition of the ASM NX3CX3N loop motif shown as a green tube, with carbon atoms of Asn 381, Cys 385, and Asn 389 shown in green, and the adjacent loop (residues 427–434) containing Cys 431 involved in disulfide bond formation shown in gray. Niemann-Pick mutation residues in or near the NX3CX3N motif (Met 382, Asn 383, and Trp 391) are depicted with blue carbon atoms.
A number of differences between the predicted catalytic mechanism of ASM and many of the DMPs should be noted. Most of the DMPs hydrolyze phosphomonoester substrates, in contrast to ASM, whose substrate SM is a phos-phodiester. Another potential dissimilarity lies in the dimetal center of ASM. DMPs can contain two different metal ions in their dimetal center, including Fe3+, Zn2+, and Mn2+ ions. We have placed two Zn2+ ions in the dimetal center of our ASM model because ASM is known to be a zinc-dependent enzyme (Schissel et al. 1998b) and our studies have confirmed that purified human ASM is most active in the presence of 10–50 μM Zn2+. However, further experimentation is required to identify the exact metals in the ASM phosphoesterase domain.
Evaluation of the kinetic properties of ASM in context to model predictions
To further evaluate potential structure/function relationships of residues in our ASM model, the kinetic characteristics of ASM were studied. Callahan and colleagues (1983) performed pH rate studies on ASM to determine the kinetic properties of the enzyme. These studies revealed that the maximal velocity for the hydrolysis of SM is independent of pH between 3.5 and 6.2, but declined at pH 6.5. Interestingly, the PAPs catalyze the hydrolysis of activated phosphoric acid esters and anhydrides at a similar pH range of 4.0 to 7.0 (Dietrich et al. 1991). The evaluation of log(Vmax/Km) and pKm, plotted against pH, identified two proton dissociation constants for the binding of SM to ASM, resulting in pK values of 4.1 and 5.5. It was proposed that the two dissociation constants affecting the Km most likely represent ionic groups involved in the formation of the enzyme–substrate complex. Our model and proposed mechanism of hydrolysis by ASM are consistent with these data. We predict that the effect of pH (at 6.5 and above) on Vmax is most likely due to His 319, given its postulated key role in the catalytic mechanism (Fig. 5 ▶). With respect to the pH dependence of Km, Glu 388 is predicted to interact with the amide moiety on SM in our model and this interaction may be responsible for the pK 4.1 effect. Alternatively, dissociation of the highly conserved Asp 210 residue may be responsible for the pK 4.1 effect through its potential interaction with the glycerol moiety on ceramide. The pK 5.5 effect on Km is most likely represented by the protonation of a histidine residue. In the ASM model, it is more difficult to predict which histidine residue may be responsible for this pK 5.5 effect. It is possible that either the highly conserved His 208 or His 282 residue could interact with the glycerol moiety in SM and when protonated at pH 5.5, would result in a reduced affinity for SM.
Key residues and motifs identified in the ASM model
Several potentially important residues and motifs in ASM were identified during this study. The NX3CX3N motif that was identified in our analyses is a sequence motif that is highly conserved within the ASM proteins. In our model, this motif lies in a loop that is predicted to be involved in substrate recognition. Interestingly, several Niemann-Pick mutations (M382I, N383S, and W391G) lie in or near to this loop (see Fig. 6 ▶ and the Niemann-Pick discussion later). The first Asn in the motif, Asn 381, is predicted to be near the metal-coordinating residues Asn 318 and His 457 from motifs MM3 and MM5, respectively (see Figs. 3A ▶, 6B ▶). The other Asn residue from the motif, Asn 389, does not appear to have a critical interaction with any of the metal-coordinating residues or the substrate; however, it is near Asn 383 and may interact with this residue. Regarding substrate recognition, Glu 388, which lies within the motif, is predicted to interact with the positively charged amide moiety of SM (Figs. 4 ▶, 6 ▶), serving a similar role as Glu 81 in CRP. Figure 2 ▶ illustrates that Glu 388 is conserved in the mouse, rat, and human ASM, but is not conserved in the other ASM or ASM-like proteins. Therefore, it is possible that the amine in the choline moiety is stabilized in a different manner in those proteins. Conversely, the ASM-like proteins may bind phosphorylcholine-containing substrates with lower affinity or not at all. The sequence diversity of the residues found between the two conserved Asn residues in this motif may help define the substrate-binding specificity of this enzyme family. As mentioned earlier, however, this loop proved difficult to model, resulting in low PROFILES-3D scores. Given the lack of sequence identity with PAP and the other known DMP structures, as well as the existence of several possibilities when modeling this loop region, the structure/ function roles of the residues within the NX3CX3N motif are speculative.
We also speculate that a cluster of hydrophilic and aromatic residues conserved among the mammalian ASMs (Asp 210, His 211, Asp/Glu 212, Tyr 213, and His 282) could represent the ceramide head group binding site. Our model suggests that Asp 210 and His 282 could make hydrogen bonds to the glycerol moiety of ceramide in SM. Alternatively, because ASM carries out its enzymatic hydrolysis on the surface of membrane lipid bilayers, this cluster of residues may be a divalent cation-binding site involved in membrane association, which has been observed in other proteins (Swairjo et al. 1995; Perisic et al. 1999; Verdaguer et al. 1999).
Evaluation of Niemann-Pick mutations in context to the ASM model
A deficiency in ASM activity in humans results in type A or B Niemann-Pick disease (Callahan and Khalil 1976; Kolodny 2000; Marathe et al. 2000). More than 30 mutations associated with both types of Niemann-Pick disease have been identified and are located throughout the ASM gene (Schuchman and Miranda 1997; Takahashi et al. 1997; Simonaro et al. 2002 and references therein; Sikora et al. 2003). It should be noted that none of the Niemann-Pick mutations characterized to date occur at the predicted metal-coordinating residues. It is possible that a homozygous mutation in one of these key residues leads to fetal death or miscarriage. Preliminary mutagenesis studies were performed in our laboratory on two of the five predicted metal-coordinating residues in ASM. The mutants made, Asp 206 → Ala and Asn 318 → Ala, resulted in altered enzymes with no residual ASM activity (data not shown), consistent with the severity of mutations in these metal-ligating residues. Several Niemann-Pick missense mutations, located in the predicted phosphoesterase domain, are discussed in context to our model (outlined in Table 2, Fig. 3B ▶). These mutations are either homozygous or heterozygous, which have been confirmed by mutagenesis studies. The type A mutation, His 319 → Tyr, results in childhood death (Sikora et al. 2003) and is predicted to affect the catalytic activity of ASM. On the basis of our ASM model and the mechanism of other DMPs, we predict that His 319 is the catalytic residue that donates a proton to a substrate phosphate oxygen atom and causes the cleavage of the phosphorus–oxygen bond. Any mutation of this residue should reduce the kcat of the mutant ASM by several orders of magnitude. The severity of the His 319 → Tyr mutation seen in the type A Niemann-Pick patients supports this hypothesis. Two other mutations that are predicted to affect the catalytic mechanism of ASM are the Met 382 → Ile and Asn 383 → Ser mutations characterized by Takahashi and colleagues (1992). Each of these was shown by mutagenesis studies to result in an ASM protein with no residual catalytic activity. Analysis of the Met 382 → Ile and Asn 383 → Ser mutations in our model reveals that they are located in the NX3CX3N motif (Figs. 3B ▶, 6B ▶). As described earlier, our ASM–phosphorylcholine model predicts that the NX3CX3N loop interacts with the SM substrate. Met 382 is near the metal-coordinating residue Asn 318 and is part of a hydrophobic patch that includes Leu 380, Phe 384, Phe 390, and Trp 391. Asn 383 is predicted to be near the metal-coordinating residue His 457. Thus, the lack of ASM catalytic activity resulting from these mutations is consistent with the prediction from our model that these residues lie in a region critical to the catalytic activity of ASM.
Table 2.
Niemann-Pick mutations in the phosphoesterase domain of ASM
| Mutation | N-P type | Character of mutation | Model interpretation | Reference |
| L302P | A | Homozygous; no measurable ASM activity in patients. Lack of activity confirmed by mutagenesis studies. May result in misfolded protein. | Not in the proximity of the dimetal center in the hydrophobic core. Predicted to result in a kink in the helix or loop where it resides and possibly result in an unstable protein. | Levran et al. 1992 |
| H319Y | A | Homozygous; lethal at 1 year | His319 is predicted to be directly involved in substrate hydrolysis. Mutation of this residue is predicted to significantly reduce the Kcat of ASM. | Sikora et al. 2003 |
| P371S | B | Homozygous; mild disease | Not in the proximity of the dimetal center. Appears to be solvent-exposed and may affect subdomain orientation, resulting in decreased catalytic efficiency of ASM. | Sikora et al. 2003 |
| M382I | A | Heterozygous; no residual ASM activity seen in mutagenesis studies | Located in the loop containing the NX3CX3N motif predicted to be involved in SM recognition | Takahashi et al. 1992 |
| N383S | B | Heterozygous; no residual ASM activity seen in mutagenesis studies | Located in the loop containing the NX3CX3N motif predicted to be involved in SM recognition | Takahashi et al. 1992 |
| W391G | B | Homozygous; slow SM degradation rates; results in an unstable protein | Predicted to sit in a hydrophobic pocket that contains residues from the NX3CX3N motif. Mutation may destabilize ASM by disrupting important hydrophobic interactions | Sperl et al. 1994; Ferlinz et al. 1995 |
| H421Y | B | Homozygous; childhood death, predicted to sit in putative catalytic site and possibly interact with zinc | Not in the proximity of the dimetal center or the zinc ions. Sits between two central beta sheets. Predictions of mutation cannot be made due to limitations of the model. | Simonaro et al. 2002 |
A number of Niemann-Pick mutations are predicted by our model to lie outside of the core of the catalytic domain of ASM or within regions that could not be modeled. These mutations include Gly 242 → Arg, Glu 246 → Gln, Ser 248 → Arg, Gln 292 → Lys, Leu 302 → Pro, Pro 371 → Ser, Trp 391 → Gly, His 421 → Tyr, Ser 436 → Arg, and Tyr 446 → Cys (see Table 2, Fig. 3B ▶). The Leu 302 → Pro mutation, often associated with type A Nie-mann-Pick disease, has been shown by mutagenesis studies to produce an altered ASM protein with no detectable catalytic activity (Levran et al. 1992). However, this study noted that the proline amino acid substitution in the Leu 302 → Pro mutation may result in the production of an unstable residual enzyme that is rapidly degraded within the cell due to incorrect folding of ASM. Our model is consistent with this hypothesis in that it also predicts that a leucine to proline substitution within the α-helix where Leu 302 resides should result in a kink in the helix and possibly lead to protein destabilization. The Pro 371 → Ser mutation is predicted to produce an altered ASM with some residual activity because it was identified in a patient who had mild type B Niemann-Pick disease and who was homozygous for the Pro 371 → Ser mutation (Sikora et al. 2003). According to our model, Pro 371 is predicted to sit at the end of a β-strand between the two central β sheets, but is not near the catalytic center. A conservative serine mutation of this residue might cause a slight change in the orientation of the two subdomains in the ASM phosphoesterase domain and thereby affect the catalytic efficiency of ASM. Studies by Sperl and colleagues (1994) on cultured skin fibroblasts from Niemann-Pick patients homoallelic for the Trp 391 → Gly mutation showed slow hydrolysis rates (8% ± 3%). Expression of ASM containing the Trp 391 → Gly mutation resulted in a protein that was properly synthesized, processed, and active, but unstable and degraded once in the lysosome (Ferlinz et al. 1995). These finding are consistent with our model, which predicts that Trp 391 is part of a hydrophobic pocket that includes residues from the NX3CX3N motif predicted to be involved in substrate recognition (Figs. 3B ▶, 6B ▶). A mutation to Gly in this hydrophobic pocket would likely destabilize the ASM by reducing important hydrophobic interactions. Lastly, the His 421 → Tyr mutation leads to an early onset, severe form of type B Niemann-Pick disease in patients homoallelic for the mutation (Simonaro et al. 2002). It was proposed that this mutation may be involved in zinc binding, given that it may sit within the putative active site of ASM (He et al. 1999; Simonaro et al. 2002). Our model predicts that His 421 is not near the catalytic center or involved in zinc binding but rather sits in the middle of a β-strand in the phos-phoesterase domain between the two central β sheets. However, due to the model limitations, we are unable to make a definitive hypothesis to explain the adverse effect that this mutation has on ASM activity.
Comparison of the model of neutral sphingomyelinase to ASM
In addition to ASM, several other classes of sphingomy-elinases have been characterized including neutral Mg2+-dependent, neutral Mg2+-independent, and alkaline sphin-gomyelinase enzymes (for review, see Goni and Alonso 2002). Interestingly, using advanced sequence recognition methods including Psi-BLAST, HMM, and threading, we were unable to show any homology between ASM and any of the other characterized sphingomyelinases. Protein fold recognition methods were used by Matsuo and colleagues (1996) to predict the three-dimensional structure of the bacterial Mg2+-dependent sphingomyelinase (bNSM) based on its high compatibility with the mammalian DNase I structure. bNSM, like ASM, catalyzes the hydrolysis of SM into ceramide and phosphorylcholine, but this enzyme is predicted to contain a single metal ion in its catalytic center in contrast to the dimetal center of ASM. A comparison of the bNSM and the ASM models reveals that their fold topologies are not similar, although the secondary structural classes of the two enzymes are both predicted to be α/β four-layer sandwiches (Murzin et al. 1995; Orengo et al. 1997). The predicted active site of ASM is located between the two βαβαβ units on one side of the enzyme, whereas the predicted active site of bNSM is located on the opposite side of the α/β four-layer sandwich.
Based on their predicted model, Matsuo et al. (1996) identified and confirmed His 151 and His 296 of bNSM as key residues involved in the hydrolysis of the phosphodi-ester bond of SM. Subsequently, the corresponding histidine residues in mammalian neutral Mg2+-dependent sphingo-myelinase (NSM) were also confirmed by mutagenesis studies (Rodrigues-Lima et al. 2000). The proposed catalytic mechanism of bNSM, based on its distant relationship to DNase I, is predicted to involve a general base activation of a water molecule to a hydroxide ion by His 296. The hydroxide ion subsequently makes a nucleophilic attack on the phosphoryl group of SM (Weston et al. 1992; Matsuo et al. 1996). Both ASM and bNSM are predicted to use a hydroxide ion that attacks a phosphorus atom to produce a pentacoordinate phosphoryl group. In ASM, however, the hydroxide anion is generated on the dimetal center, whereas in bNSM it is generated by His 296. A second histidine in bNSM (His 151) is predicted to function as a general acid to protonate the leaving oxygen of the ceramide alcohol, similar to the predicted function of His 319 in ASM. The Mg2+ ion in bNSM is predicted to be involved in the stabilization of the pentacoordinate transition state, whereas we hypothesize that this function in ASM is accomplished by the dimetal center. In summary, despite the absence of any sequence homology between ASM and bNSM, their predicted structures share a number of similarities including secondary structure class and features of the catalytic mechanism.
Summary
Purple acid phosphatase was identified by sequence profile and fold recognition methods as a remote structural homolog of ASM. A model of the ASM phosphoesterase domain was built based on its predicted secondary structure and on the corresponding conserved metal-ligating residues of PAP. A model of the ASM–phosphorylcholine complex was also built, based on the structural information from the PAP–phosphate and CRP–phosphorylcholine complexes. Residues that may be important for substrate recognition and catalysis were predicted from this complex model.
Although the highest degree of confidence in the resulting ASM model resides in the position of metal-coordinating residues, additional experimental data are available that support our ASM model beyond this region. First, based on our model of ASM, the hydrolysis of SM is predicted to be carried out through an SN2-type mechanism and involves a His proton donor, which is similar to other DMPs. Second, the severity of the His 319 → Tyr, Met 382 → Ile, and Asn 383 → Ser Niemann-Pick mutations in ASM corroborate our predictions from the model that these residues are functionally important for either substrate recognition involving the NX3CX3N motif or for catalysis. Third, ASM contains the five metal-coordinating motifs (MM1 through MM5) that are conserved in the DMP family of proteins. We predict that the ASM phosphoesterase domain shares the PAP topological fold; however, the details of the spatial backbone conformation of the native ASM phosphoesterase structure is likely to be different from that of PAP. Nevertheless, the predicted functional residues identified in our ASM model will be useful in guiding future experimental studies on ASM.
Materials and methods
Sequence analysis and alignments
The human ASM protein sequence, corresponding to the predicted phosphoesterase domain (amino acid residues 201–472), was used to search for homologs. Three common search algorithms, BLAST, Smith-Waterman, and FASTA (Smith and Waterman 1981; Altschul et al. 1990; Pearson 1990), were used to search for possible templates in three protein databases—GenPept (database dated October 2000) (Benson et al. 2000), SwissProt (August 1999 version) (Bairoch and Boeckmann 1994), and Protein Data Bank (PDB; August 1999 version) (Berman et al. 2000).
ClustalW (Thompson et al. 1994) generated a multiple sequence alignment of the ASM protein family, consisting of ASM or ASM-like annotation only. A hidden Markov model was built based on this sequence alignment and was used to score sequences in the three protein databases using HMMER (Eddy 1998; Sonnhammer et al. 1998). The sequence profile search method PSI-BLAST (Altschul et al. 1997) was also used to search the same protein databases. With the exception of FASTA, all database searches were run using TimeLogic DeCypher (version 2000).
Fold recognition and secondary structure prediction
The fold recognition methods 3D-PSSM (Kelley et al. 2000) and 123D+ (Alexandrov et al. 1996) were used to identify potential folds that ASM may adopt. To optimize the sequence-structure alignments, the following secondary structure programs were used: DSC (King and Sternberg 1996), PHD (Rost and Sander 1993), Predator (Frishman and Argos 1997), and PSI-Pred (Jones 1999). Each of these secondary structure programs has a prediction accuracy above 70%.
Model of ASM phosphoesterase domain
The model of the ASM phosphoesterase domain was built using the program HOMOLOGY (InsightII, Accelrys), based on both the pig PAP structure (PDB entry 1UTE; Guddat et al. 1999) and the sequence-structure alignment shown in Figure 2 ▶. The coordinates for the insertions and loop conformations that differ from those of PAP were extracted from the PDB (Berman et al. 2000). Two zinc ions and a phosphate ion were placed into the active site of ASM model based on the PAP–phosphate complex. The model was refined by energy minimization using DISCOVER (Accelrys), with constraints on the predicted conserved metal-coordinating residues, the metal ions, the phosphate ion, and the backbone, except where deletions and insertions occur. The AMBER force field was used with a nonbond cutoff of 12 Å, a switching distance of 1.5 Å, and a dielectric constant of 4. ProStat from HOMOLOGY was used to evaluate the stereochemical properties of the model. The model was validated with program PROFILES-3D (Lüthy et al. 1992) to identify putative misfolded regions using average window lengths of 10 and 21 residues. The model building was performed on a Silicon Graphics Indigo workstation running IRIX 6.5.
Model of the ASM–phosphorylcholine complex
To identify a putative orientation for the substrate-containing phosphorylcholine in the ASM model, a model of ASM was initially generated in complex with phosphate. This was accomplished by keeping the corresponding metal-binding residues and metal ions, conserved in the pig PAP structure, fixed throughout the building and refinement of the ASM model. Thereafter, the phosphate group and water (Mu-oxo-diiron) in the PAP structure could be superpositioned onto the ASM model. This resulted in the production of a model of ASM bound to phosphate.
A search was then made in the PDB for structures that had phosphorylcholine bound by its phosphate to a dimetal ion center. This search identified only one structure, the CRP–phosphorylcho-line complex (PDB entry 1B09; Thompson et al. 1999). This structure was used to identify a putative orientation for phosphorylcho-line in the ASM model, with respect to the dimetal ion center and the predicted metal-coordinating residues. The phosphorylcholine from the CRP complex was placed into the ASM model by superimposing its phosphate group, the two calcium ions, and the contact atoms from the metal ligating residues (Asp 278, Asn 318, His 457, His 459 in ASM and Asp 140, Asp 60, Asn 61, Gln 150 in CRP) onto the corresponding atoms in the ASM model, providing an initial orientation for the choline moiety head group. The position of the phosphorylcholine was adjusted such that the phosphorus atom and the two oxygen atoms bound to the metal ions were superimposed on the corresponding atoms of the phosphate in the ASM–phosphate model described previously. Finally, a water molecule bridging the metal ions in the PAP structure (PDB entry 1UTE) was transferred into the ASM model. This model has been deposited in the PDB (entry 1X9O).
Acknowledgments
We thank Dr. Marc Adler, Dr. Tom Morgan, Dr. Mark Polokoff, Dr. Sofia Ribeiro, and Dr. Ira Tabas for their helpful scientific discussions, suggestions, and expertise.
Abbreviations
ASM, acid sphingomyelinase
CRP, C-reactive protein
DMP, dimetal-containing phosphoesterase
HMM, hidden Markov model
LDL, low-density lipoproteins
MM1–MM5, predicted phosphatase metal-coordinating motifs 1 through 5
PAP, purple acid phosphatase
RMSD, root mean square deviation
SM, sphingomyelin
TRAP, tartrate-resistant acid phosphatase
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.04966204.
References
- Alexandrov, N.N., Nussinov, R., and Zimmer, R.M. 1996. Fast protein fold recognition via sequence to structure alignment and contact capacity potentials. Pac. Symp. Biocomput. 53–73. [PubMed]
- Altschul, S.F., Gish, W., Miller, W., Myers, E.W., and Lipman, D.J. 1990. Basic local alignment search tool. J. Mol. Biol. 215 403–410. [DOI] [PubMed] [Google Scholar]
- Altschul, S.F., Madden, T.L., Schaffer, A.A., Zhang, J., Zhang, Z., Miller, W., and Lipman, D. J. 1997. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 25 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bairoch, A. and Boeckmann, B. 1994. The SWISS-PROT protein sequence data bank: Current status. Nucleic Acids Res. 22 3578–3580. [PMC free article] [PubMed] [Google Scholar]
- Benson, D.A., Karsch-Mizrachi, I., Lipman, D.J., Ostell, J., Rapp, B.A., and Wheeler, D.L. 2000. GenBank. Nucleic Acids Res. 28 15–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman, H.M., Westbrook, J., Feng, Z., Gilliland, G., Bhat, T.N., Weissig, H., Shindyalov, I.N., and Bourne, P.E. 2000. The Protein Data Bank. Nucleic Acids Res. 28 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Besley, G.T., Hoogeboom, A.J., Hoogeveen, A., Kleijer, W.J., and Galjaard, H. 1980. Somatic cell hybridization studies showing different gene mutations in Niemann-Pick variants. Hum. Genet. 54 409–412. [DOI] [PubMed] [Google Scholar]
- Callahan, J.W. and Khalil, M. 1976. Sphingomyelinases and the genetic defects in Niemann-Pick disease. Adv. Exp. Med. Biol. 68 367–378. [DOI] [PubMed] [Google Scholar]
- Callahan, J.W., Jones, C.S., Davidson, D.J., and Shankaran, P. 1983. The active site of lysosomal sphingomyelinase: Evidence for the involvement of hydrophobic and ionic groups. J. Neurosci. Res. 10 151–163. [DOI] [PubMed] [Google Scholar]
- Denessiouk, K.A. and Johnson, M.S. 2000. When fold is not important: A common structural framework for adenine and AMP binding in 12 unrelated protein families. Proteins 38 310–326. [PubMed] [Google Scholar]
- Dietrich, M., Munstermann, D., Suerbaum, H., and Witzel, H. 1991. Purple acid phosphatase from bovine spleen. Interactions at the active site in relation to the reaction mechanism. Eur. J. Biochem. 199 105–113. [DOI] [PubMed] [Google Scholar]
- Eddy, S.R. 1998. Profile hidden Markov models. Bioinformatics 14 755–763. [DOI] [PubMed] [Google Scholar]
- Egloff, M.P., Cohen, P.T., Reinemer, P., and Barford, D. 1995. Crystal structure of the catalytic subunit of human protein phosphatase 1 and its complex with tungstate. J. Mol. Biol. 254 942–959. [DOI] [PubMed] [Google Scholar]
- Ferlinz, K., Hurwitz, R., Weiler, M., Suzuki, K., Sandhoff, K., and Vanier, M.T. 1995. Molecular analysis of the acid sphingomyelinase deficiency in a family with an intermediate form of Niemann-Pick disease. Am. J. Hum. Genet. 56 1343–1349. [PMC free article] [PubMed] [Google Scholar]
- Frishman, D. and Argos, P. 1997. Seventy-five percent accuracy in protein secondary structure prediction. Proteins 27 329–335. [DOI] [PubMed] [Google Scholar]
- Goni, F.M. and Alonso, A. 2002. Sphingomyelinases: Enzymology and membrane activity. FEBS Letters 531 38–46. [DOI] [PubMed] [Google Scholar]
- Guddat, L.W., McAlpine, A.S., Hume, D., Hamilton, S., de Jersey, J., and Martin, J.L. 1999. Crystal structure of mammalian purple acid phosphatase. Structure Fold. Des. 7 757–767. [DOI] [PubMed] [Google Scholar]
- He, X., Miranda, S.R., Xiong, X., Dagan, A., Gatt, S., and Schuchman, E.H. 1999. Characterization of human acid sphingomyelinase purified from the media of overexpressing Chinese hamster ovary cells. Biochim. Biophys. Acta 1432 251–264. [DOI] [PubMed] [Google Scholar]
- Jones, D.T. 1999. Protein secondary structure prediction based on position-specific scoring matrices. J. Mol. Biol. 292 195–202. [DOI] [PubMed] [Google Scholar]
- Kelley, L.A., MacCallum, R.M., and Sternberg, M.J. 2000. Enhanced genome annotation using structural profiles in the program 3D-PSSM. J. Mol. Biol. 299 499–520. [DOI] [PubMed] [Google Scholar]
- King, R.D. and Sternberg, M.J. 1996. Identification and application of the concepts important for accurate and reliable protein secondary structure prediction. Protein Sci. 5 2298–2310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klabunde, T., Strater, N., Frohlich, R., Witzel, H., and Krebs, B. 1996. Mechanism of Fe(III)–Zn(II) purple acid phosphatase based on crystal structures. J. Mol. Biol. 259 737–748. [DOI] [PubMed] [Google Scholar]
- Knöfel, T. and Sträter, N. 1999. X-ray structure of the Escherichia coli periplas-mic 5′-nucleotidase containing a dimetal catalytic site. Nat. Struct. Biol. 6 448–453. [DOI] [PubMed] [Google Scholar]
- Kolodny, E.H. 2000. Niemann-Pick disease. Curr. Opin. Hematol. 7 48–52. [DOI] [PubMed] [Google Scholar]
- Koonin, E.V. 1994. Conserved sequence pattern in a wide variety of phos-phoesterases. Protein Sci. 3 356–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lansmann, S., Schuette, C.G., Bartelsen, O., Hoernschemeyer, J., Linke, T., Weisgerber, J., and Sandhoff, K. 2003. Human acid sphingomyelinase: Assignment of the disulfide bond pattern. Eur. J. Biochem. 270 1076–1088. [DOI] [PubMed] [Google Scholar]
- Levade, T., Salvayre, R., and Douste-Blazy, L. 1986. Sphingomyelinases and Niemann-Pick disease. J. Clin. Chem. Clin. Biochem. 24 205–220. [PubMed] [Google Scholar]
- Levran, O., Desnick, R.J., and Schuchman, E.H. 1992. Identification and expression of a common missense mutation (L302P) in the acid sphingomy-elinase gene of Ashkenazi Jewish type A Niemann-Pick disease patients. Blood 80 2081–2087. [PubMed] [Google Scholar]
- Lüthy, R., Bowie, J.U., and Eisenberg, D. 1992. Assessment of protein models with three-dimensional profiles. Nature 356 83–85. [DOI] [PubMed] [Google Scholar]
- Marathe, S., Schissel, S.L., Yellin, M.J., Beatini, N., Mintzer, R., Williams, K.J., and Tabas, I. 1998. Human vascular endothelial cells are a rich and regu-latable source of secretory sphingomyelinase. Implications for early athero-genesis and ceramide-mediated cell signaling. J. Biol. Chem. 273 4081–4088. [DOI] [PubMed] [Google Scholar]
- Marathe, S., Kuriakose, G., Williams, K. J., and Tabas, I. 1999. Sphingomy-elinase, an enzyme implicated in atherogenesis, is present in atherosclerotic lesions and binds to specific components of the subendothelial extracellular matrix. Arterioscler. Thromb. Vasc. Biol. 19 2648–2658. [DOI] [PubMed] [Google Scholar]
- Marathe, S., Miranda, S.R., Devlin, C., Johns, A., Kuriakose, G., Williams, K.J., Schuchman, E.H., and Tabas, I. 2000. Creation of a mouse model for non-neurological (type B) Niemann-Pick disease by stable, low level expression of lysosomal sphingomyelinase in the absence of secretory sphin-gomyelinase: Relationship between brain intra-lysosomal enzyme activity and central nervous system function. Hum. Mol. Genet. 9 1967–1976. [DOI] [PubMed] [Google Scholar]
- Matsuo, Y., Yamada, A., Tsukamoto, K., Tamura, H., Ikezawa, H., Nakamura, H., and Nishikawa, K. 1996. A distant evolutionary relationship between bacterial sphingomyelinase and mammalian DNase I. Protein Sci. 5 2459–2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller, E.G., Crowder, M.W., Averill, B.A., and Knowles, J.R. 1993. Purple acid phosphatase: Diiron enzyme that catalyzes a direct phospho group transfer to water. J. Am. Chem. Soc. 115 2974–2975. [Google Scholar]
- Murzin, A.G., Brenner, S.E., Hubbard, T., and Chothia, C. 1995. SCOP: A structural classification of proteins database for the investigation of sequences and structures. J. Mol. Biol. 247 536–540. [DOI] [PubMed] [Google Scholar]
- Orengo, C.A., Michie, A.D., Jones, S., Jones, D.T., Swindells, M.B., and Thorn-ton, J.M. 1997. CATH—A hierarchic classification of protein domain structures. Structure 5 1093–1108. [DOI] [PubMed] [Google Scholar]
- Pearson, W.R. 1990. Rapid and sensitive sequence comparison with FASTP and FASTA. Methods Enzymol. 183 63–98. [DOI] [PubMed] [Google Scholar]
- Pepys, M.B., Dash, A.C., Fletcher, T.C., Richardson, N., Munn, E.A., and Feinstein, A. 1978. Analogues in other mammals and in fish of human plasma proteins, C-reactive protein and amyloid P component. Nature 273 168–170. [DOI] [PubMed] [Google Scholar]
- Perisic, O., Paterson, H.F., Mosedale, G., Lara-Gonzalez, S., and Williams, R.L. 1999. Mapping the phospholipid-binding surface and translocation determinants of the C2 domain from cytosolic phospholipase A2. J. Biol. Chem. 274 14979–14987. [DOI] [PubMed] [Google Scholar]
- Ponting, C.P. 1994. Acid sphingomyelinase possesses a domain homologous to its activator proteins: Saposins B and D. Protein Sci. 3 359–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintern, L.E., Weitz, G., Nehrkorn, H., Tager, J.M., Schram, A.W., and Sand-hoff, K. 1987. Acid sphingomyelinase from human urine: Purification and characterization. Biochim. Biophys. Acta. 922 323–336. [DOI] [PubMed] [Google Scholar]
- Rodrigues-Lima, F., Fensome, A.C., Josephs, M., Evans, J., Veldman, R.J., and Katan, M. 2000. Structural requirements for catalysis and membrane targeting of mammalian enzymes with neutral sphingomyelinase and lyso-phospholipid phospholipase C activities. Analysis by chemical modification and site-directed mutagenesis. J. Biol. Chem. 275 28316–28325. [DOI] [PubMed] [Google Scholar]
- Rost, B. and Sander, C. 1993. Prediction of protein secondary structure at better than 70% accuracy. J. Mol. Biol. 232 584–599. [DOI] [PubMed] [Google Scholar]
- Schissel, S.L., Schuchman, E.H., Williams, K.J., and Tabas, I. 1996a. Zn2+-stimulated sphingomyelinase is secreted by many cell types and is a product of the acid sphingomyelinase gene. J. Biol. Chem. 271 18431–18436. [DOI] [PubMed] [Google Scholar]
- Schissel, S.L., Tweedie-Hardman, J., Rapp, J.H., Graham, G., Williams, K.J., and Tabas, I. 1996b. Rabbit aorta and human atherosclerotic lesions hydro-lyze the sphingomyelin of retained low-density lipoprotein. Proposed role for arterial-wall sphingomyelinase in subendothelial retention and aggregation of atherogenic lipoproteins. J. Clin. Invest. 98 1455–1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schissel, S.L., Jiang, X., Tweedie-Hardman, J., Jeong, T., Camejo, E.H., Najib, J., Rapp, J.H., Williams, K.J., and Tabas, I. 1998a. Secretory sphingomy-elinase, a product of the acid sphingomyelinase gene, can hydrolyze ath-erogenic lipoproteins at neutral pH. Implications for atherosclerotic lesion development. J. Biol. Chem. 273 2738–2746. [DOI] [PubMed] [Google Scholar]
- Schissel, S.L., Keesler, G.A., Schuchman, E.H., Williams, K.J., and Tabas, I. 1998b. The cellular trafficking and zinc dependence of secretory and lyso-somal sphingomyelinase, two products of the acid sphingomyelinase gene. J. Biol. Chem. 273 18250–18259. [DOI] [PubMed] [Google Scholar]
- Schmitt, S., Kuhn, D., and Klebe, G. 2002. A new method to detect related function among proteins independent of sequence and fold homology. J. Mol. Biol. 323 387–406. [DOI] [PubMed] [Google Scholar]
- Schuchman, E.H. and Miranda, S.R. 1997. Niemann-Pick disease: Mutation update, genotype/phenotype correlations, and prospects for genetic testing. Genet. Test. 1 13–19. [DOI] [PubMed] [Google Scholar]
- Schuchman, E.H., Suchi, M., Takahashi, T., Sandhoff, K., and Desnick, R.J. 1991. Human acid sphingomyelinase. Isolation, nucleotide sequence and expression of the full-length and alternatively spliced cDNAs. J. Biol. Chem. 266 8531–8539. [PubMed] [Google Scholar]
- Schwalbe, R.A., Dahlback, B., Coe, J.E., and Nelsestuen, G.L. 1992. Pentraxin family of proteins interact specifically with phosphorylcholine and/or phos-phorylethanolamine, Biochemistry 31 4907–4915. [DOI] [PubMed] [Google Scholar]
- Sikora, J., Pavlu-Pereira, H., Elleder, M., Roelofs, H., and Wevers, R.A. 2003. Seven novel acid sphingomyelinase gene mutations in Niemann-Pick type A and B patients. Ann. Hum. Genet. 67 63–70. [DOI] [PubMed] [Google Scholar]
- Simonaro, C.M., Desnick, R.J., McGovern, M.M., Wasserstein, M.P., and Schuchman, E.H. 2002. The demographics and distribution of type B Niemann-Pick disease: Novel mutations lead to new genotype/phenotype correlations. Am. J. Hum. Genet. 71 1413–1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith, T.F. and Waterman, M.S. 1981. Identification of common molecular subsequences. J. Mol. Biol. 147 195–197. [DOI] [PubMed] [Google Scholar]
- Sonnhammer, E.L., Eddy, S.R., Birney, E., Bateman, A., and Durbin, R. 1998. Pfam: Multiple sequence alignments and HMM-profiles of protein domains. Nucleic Acids Res. 26 320–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperl, W., Bart, G., Vanier, M.T., Christomanou, H., Baldissera, I., Steichen-Gersdorf, E., and Paschke, E. 1994. A family with visceral course of Nie-mann-Pick disease, macular halo syndrome and low sphingomyelin degradation rate. J. Inherit. Metab. Dis. 17 93–103. [DOI] [PubMed] [Google Scholar]
- Swairjo, M.A., Concha, N.O., Kaetzel, M.A., Dedman, J.R., and Seaton, B.A. 1995. Ca(2+)-bridging mechanism and phospholipid head group recognition in the membrane-binding protein annexin V. Nat. Struct. Biol. 2 968–974. [DOI] [PubMed] [Google Scholar]
- Tabas, I. 1999. Secretory sphingomyelinase. Chem. Phys. Lipids 102 123–130. [DOI] [PubMed] [Google Scholar]
- Tabas, I., Li, Y., Brocia, R.W., Xu, S.W., Swenson, T.L., and Williams, K.J. 1993. Lipoprotein lipase and sphingomyelinase synergistically enhance the association of atherogenic lipoproteins with smooth muscle cells and extra-cellular matrix. A possible mechanism for low density lipoprotein and li-poprotein(a) retention and macrophage foam cell formation. J. Biol. Chem. 268 20419–20432. [PubMed] [Google Scholar]
- Takahashi, T., Suchi, M., Desnick, R.J., Takada, G., and Schuchman, E.H. 1992. Identification and expression of five mutations in the human acid sphingo-myelinase gene causing types A and B Niemann-Pick disease. Molecular evidence for genetic heterogeneity in the neuronopathic and non-neurono-pathic forms. J. Biol. Chem. 267 12552–12558. [PubMed] [Google Scholar]
- Takahashi, T., Akiyama, K., Tomihara, M., Tokudome, T., Nishinomiya, F., Tazawa, Y., Horinouchi, K., Sakiyama, T., and Takada, G. 1997. Heterogeneity of liver disorder in type B Niemann-Pick disease. Hum. Pathol. 28 385–388. [DOI] [PubMed] [Google Scholar]
- Thompson, D., Pepys, M.B., and Wood, S.P. 1999. The physiological structure of human C-reactive protein and its complex with phosphocholine. Structure Fold. Des. 7 169–177. [DOI] [PubMed] [Google Scholar]
- Thompson, J.D., Higgins, D.G., and Gibson, T.J. 1994. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22 4673–4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uppenberg, J., Lindqvist, F., Svensson, C., Ek-Rylander, B., and Andersson, G. 1999. Crystal structure of a mammalian purple acid phosphatase. J. Mol. Biol. 290 201–211. [DOI] [PubMed] [Google Scholar]
- Verdaguer, N., Corbalan-Garcia, S., Ochoa, W.F., Fita, I., and Gomez-Fernan-dez, J.C. 1999. Ca(2+) bridges the C2 membrane-binding domain of protein kinase Cα directly to phosphatidylserine. EMBO J. 18 6329–6338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent, J.B. and Averill, B.A. 1990. An enzyme with a double identity: Purple acid phosphatase and tartrate-resistant acid phosphatase. FASEB J. 4 3009–3014. [DOI] [PubMed] [Google Scholar]
- Weston, S.A., Lahm, A., and Suck, D. 1992. X-ray structure of the DNase I-d(GGTATACC)2 complex at 2.3 A resolution. J. Mol. Biol. 226 1237–1256. [DOI] [PubMed] [Google Scholar]
- Williams, K.J. and Tabas, I. 1995. The response-to-retention hypothesis of early atherogenesis. Arterioscler. Thromb. Vasc. Biol. 15 551–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamanaka, T. and Suzuki, K. 1982. Acid sphingomyelinase of human brain: Purification to homogeneity. J. Neurochem. 38 1753–1764. [DOI] [PubMed] [Google Scholar]
- Zhuo, S., Clemens, J.C., Stone, R.L., and Dixon, J.E. 1994. Mutational analysis of a Ser/Thr phosphatase. Identification of residues important in phos-phoesterase substrate binding and catalysis. J. Biol. Chem. 269 26234–26238. [PubMed] [Google Scholar]