

Abstract
α-Synuclein filaments are the major component of intracytoplasmic inclusion bodies characteristic of Parkinson’s disease and related disorders. The process of α-synuclein filament formation proceeds via intermediate or protofibrillar species, each of which may be cytotoxic. Because high levels of calcium(II) and other metal ions may play a role in disease pathogenesis, we investigated the influence of calcium and other metals on α-synuclein speciation. Here we report that calcium(II) and cobalt(II) selectively induce the rapid formation of discrete annular α-synuclein oligomeric species. We used atomic force microscopy to monitor the aggregation state of α-synuclein after 1 d at 4°C in the presence of a range of metal ions compared with the filament formation pathway in the absence of metal ions. Three classes of effect were observed with different groups of metal ions: (1) Copper(II), iron(III), and nickel(II) yielded 0.8–4 nm spherical particles, similar to α-synuclein incubated without metal ions; (2) magnesium(II), cadmium(II), and zinc(II) gave larger, 5–8 nm spherical oligomers; and, (3) cobalt(II) and calcium(II) gave frequent annular oligomers, 70–90 nm in diameter with calcium(II) and 22–30 nm in diameter with cobalt(II). In the absence of metal ions, annular oligomers ranging 45–90 nm in diameter were observed after 10 d incubation, short branched structures appeared after a further 3 wk and extended filaments after 2–3 mo. Previous studies have shown that α-synuclein calcium binding is mediated by the acidic C terminus. We found that truncated α-synuclein (1–125), lacking the C-terminal 15 amino acids, did not form annular oligomers upon calcium addition, indicating the involvement of the calcium-binding domain.
Keywords: Parkinson’s disease, α-synuclein, amyloid, calcium, fibrillogenesis, atomic force microscopy, Lewy body
α-Synuclein is a small (140 amino acid), acidic protein, highly concentrated in presynaptic nerve terminals where it is associated with the cytosolic side of small synaptic vesicles (Iwai et al. 1995; Totterdell et al. 2004). Although its precise function is unknown, α-synuclein is associated with neuronal plasticity (Clayton and George 1998; Lykkebo and Jensen 2002; Norris et al. 2004) and α-synuclein depletion causes a reduction in neurotransmitter vesicle recycling (Murphy et al. 2000; Cabin et al. 2002). At low concentrations in vitro, monomeric α-synuclein has no defined secondary structure (Weinreb et al. 1996; Uversky 2003). However, interactions between α-synuclein and a variety of factors, including proteins, polyamines, lipids, oxidants, polycations, and metal ions, have been shown to promote α-synuclein folding, oligomerization, and aggregation (Souza et al. 2000; Perrin et al. 2001; Uversky et al. 2001; Golts et al. 2002; Goers et al. 2003a).
A major subgroup of adult-onset neurodegenerative diseases, including Parkinson’s disease (PD), Multiple System Atrophy (MSA), and Dementia with Lewy Bodies (DLB) is characterised by abnormal filamentous neural intracytoplasmic inclusion bodies composed largely of α-synuclein, leading to the collective designation, α-synucleinopathy (Goedert 1999; Jensen and Gai 2001; Trojanoswki and Lee 2003). A substantial body of evidence points to a central role of α-synuclein in disease pathogenesis. Triplication of the α-synuclein locus and missense point mutations in the α-synuclein gene (G209A, G88C, and G188A) are linked to rare, early-onset familial PD and DLB cases (Polymeropoulos et al. 1997; Kruger et al. 1998; Singleton et al. 2003; Zarranz et al. 2004). Human wild-type or mutant α-synuclein overexpressed in animal models results in the formation of α-synuclein immunoreactive cytoplasmic inclusions that correlate with neuropathology and neurological deficit (Hashimoto et al. 2003; Maries et al. 2003). Wild-type and mutant (A53T, A30P) α-synucleins have been shown to form filaments in vitro (Conway et al. 2000a) that closely resemble filaments in disease-affected brains, and both A53T (G209A) and A30P (G88C) mutations confer an increased rate of α-synuclein oligomerization, although only the A53T mutant shows a significant increase in the rate of filament formation compared to wild-type (Conway et al. 2000b).
Although there is good correlation between the frequency and regional distribution of pathological inclusion bodies in sporadic α-synucleinopathies and the severity and nature of clinical presentation (Braak et al. 2002; Armstrong et al. 2004; Iseki 2004), recent evidence suggests that an intermediate or protofibrillar oligomeric species in the α-synuclein fibrillation pathway rather than α-synuclein filaments themselves may be the principal pathogenic species. Coexpression of the chaperone HSP70 with α-synuclein in Drosophila slows the loss of dopaminergic neurons, but does not significantly decrease the number of fibrillar inclusions (Auluck et al. 2002). Transgenic mice that overexpress human α-synuclein and develop Parkinsonian phenotypes are characterized by nonfibrillar aggregates (Masliah et al. 2000) and HSP70 coexpression results in reduced detergent-resistant α-synuclein oligomers concomitant with decreased toxicity (Klucken et al. 2004).
Detergent resistant α-synuclein oligomers are a consistent pathological hallmark of α-synucleinopathies (Campbell et al. 2001; Pountney et al. 2004). In vitro, α-synuclein oligomers have been reported with spherical, chain-like, and annular morphologies (Conway et al. 2000b; Ding et al. 2002; Lashuel et al. 2002). Annular α-synuclein oligomers have an increased propensity to form with α-synuclein A30P and A53T mutants, topologically resemble bacterial pore-forming toxins, are able to permeabilize lipid membranes and can be released from pathological inclusion bodies, leading to their implication as potential pathogenic species (Volles et al. 2001; Ding et al. 2002; Lashuel et al. 2002; Pountney et al. 2004). Several recent studies also implicate a role of calcium ions and calcium-dependent processes in α-synucleinopathy. Calcium(II) binds with high affinity to the C terminus of α-synuclein, thereby promoting oligomer formation (Nielsen et al. 2001). The calcium-activated protease, calpain, has been shown to cleave α-synuclein at specific sites in vitro (Mishizen-Eberz et al. 2003), resulting in loss of the calcium-binding domain, and mediates cytotoxicity in animal and cell culture models of PD (Crocker et al. 2003; Kim et al. 2003).
In this work, we have investigated the pathway of α-synuclein oligomerization at 4°C using atomic force microscopy, and have examined the effects of calcium and a range of other metal ions on oligomer formation. We show that annular α-synuclein oligomer formation immediately precedes filament growth, and that rapid formation of annular α-synuclein oligomers is selectively induced by calcium(II) and cobalt(II) ions.
Results
Time-dependent α-synuclein oligomerization to annular and fibrillar structures
To examine intermediate steps in the pathway of α-synuclein aggregation, samples of highly purified monomeric protein were incubated at 4°C without stirring for time periods of between 1 d and 3 mo. Aliquots were taken at successive time points, dried onto freshly cleaved mica support, and imaged by tapping mode atomic force microscopy. Progressively larger particles were detected as a function of incubation time, with the appearance of some annular structures after 8–10 d incubation, branched structures after a further 3 wk, and abundant filaments after 2–3 mo (Fig. 1 ▶). In protein samples imaged immediately following purification of the monomer by gel filtration chromatography, exclusively spherical particles were observed (Fig. 2A ▶) ranging between 0.7–0.9 nm in height (mean height, 0.8 ± 0.2 nm; n = 16). Cytochrome c, which occurs as a monomer of 11 kDa, was also imaged yielding particle sizes of between 1.1–2.0 nm (mean height, 1.4 ± 0.4 nm; n = 10; not shown). This indicates the 0.8-nm spherical particles are α-synuclein monomers.
Figure 1.

Annular α-synuclein oligomers form at 4°C after 10 d. Tapping mode AFM height images of α-synuclein particles after incubation for 2 d (A), 10 d (B), 1 mo (C), and 3 mo (D). Scan rate, 1.001 Hz; z-scale, 50.0 nm; cantilever length, 225 μm; tip frequency, 77–94 kHz; set point, 1.5 V.
Figure 2.
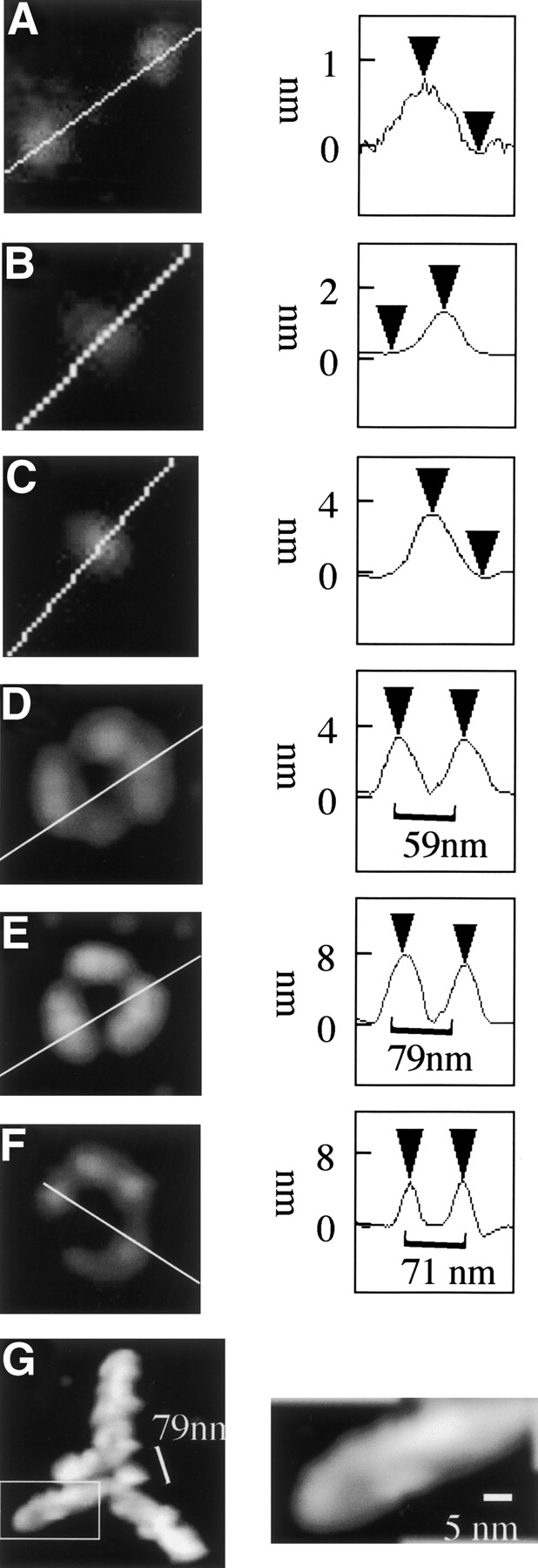
α-Synuclein filaments grow from annular oligomers at 4°C. (Left) Tapping mode AFM height image of representative α-synuclein particle(s) immediately after Superdex 75 chromatography (A), after incubation at 4°C for 2 d (B,C), 10 d (D–F), and 1 mo (G). (Right) Height cross-section of α-synuclein particle. Scan rate, 1.001 Hz; z-scale, 50.0 nm; cantilever length, 225 μm; tip frequency, 77–94 kHz; set point, 1.5 V.
After 2 d incubation the average particle height had increased to 2 nm (Fig. 2B ▶), with two distinct populations of spherical particles 1.6 ± 0.3 nm (n = 10) and 2.4 ± 0.3 nm (n = 10) in height, respectively. After 10 d incubation at 4°C, spherical particles were observed with two distinct particle heights with mean particle heights of 2 ± 0.6 nm (n = 10) and 4 ± 0.8 nm (n = 10) (Fig. 2C ▶), respectively. In addition to spherical particles, frequent annular structures were also observed (Fig. 2D ▶), equivalent to 5% of total particles. The perimeter of the annular particles was not of even height with the topology, typically indicating a ring made of six spherical particles grouped into three sets of two particles with overall threefold symmetry (Fig. 2D,E ▶). The maximum height of the annular particles ranged between 3–9 nm, and they could be grouped into two subsets with mean height 4 ± 0.8 nm (n = 12) (Fig. 2D ▶) and 7.9 ± 1.6 nm (n = 5) (Fig. 2E ▶), respectively. An estimate of the diameter of each annulus was obtained by measuring the peak-to-peak distance of a cross-section bisecting two of the component particles from opposite sides across the symmetry axis. The 4 nm-height annular oligomers ranged between 45–60 nm in diameter and the 7.9 nm-height aggregates ranged between 70–90 nm in diameter. In the case of the 7.9 nm-height particles, the grouped pairs of spherical particles were often merged, resulting in three ovoid particles, but preserving the threefold symmetry of the annulus. We also observed some open rings composed of a chain of five spherical 4 nm-height particles (Fig. 2F ▶).
After samples were incubated at 4°C for a further 3 wk, trigonal branched structures were observed, as shown in Figures 1C ▶ and 2G ▶, which preserve the threefold symmetry axis of the annular structures observed after 10 d (Fig. 2E ▶). The mean maximum height of these structures was 9.1 ± 0.9 nm (n = 12). These structures also exhibit a central annulus with mean diameter estimated from the peak-to-peak distance of the height cross-section indicated in Figure 2G ▶, ranging between 70–100 nm. 8–10 nm-height filaments extended between 200–300 nm from each of the three peripheral segments of the central annulus. These radial filaments comprised twisted double strands with a pitch of 30–50 nm (Fig. 2G ▶, right) coated with occasional spherical particles. In addition to the branched structures, discrete spherical species were also observed with mean height 5.6 ± 0.4 nm (n = 10). Incubation for 3 mo at 4°C yielded 8–10-nm filaments, including some branched filaments (Fig. 1D ▶).
Mg(II), Cd(II), and Zn(II) induce spherical α-synuclein oligomers
To examine the effects of metal ions on α-synuclein aggregation, the metal ions Co(II), Ca(II), Mg(II), Cd(II), Zn(II), Cu(II), Ni(II), and Fe(III) were added to freshly purified protein to a 10-fold molar excess over protein. Samples were then incubated overnight at 4°C, and AFM images acquired the next day. Magnesium(II), cadmium(II), and zinc(II) ions induced the aggregation of α-synuclein to form large oligomeric species (Fig. 3A–C ▶). Magnesium induced oligomers were 6.1 ± 0.9 nm in height (n = 10) (Fig. 3A ▶) while both cadmium and zinc each exhibited two distinct species, 4.0 ± 0.8 nm (n = 10) and 6.9 ± 0.9 nm in height (n = 10), respectively (Fig. 3B,C ▶). The addition of copper yielded spherical species 1.4 ± 0.4 nm in height (n = 10) (Fig. 3D ▶), indicating that aggregation is inhibited compared to no metal added. The presence of iron(III) or nickel(II) had no influence on the aggregation of α-synuclein within the short time scale of these experiments (Fig. 3E,F ▶, respectively), with particle sizes closely similar to those observed without metal addition.
Figure 3.
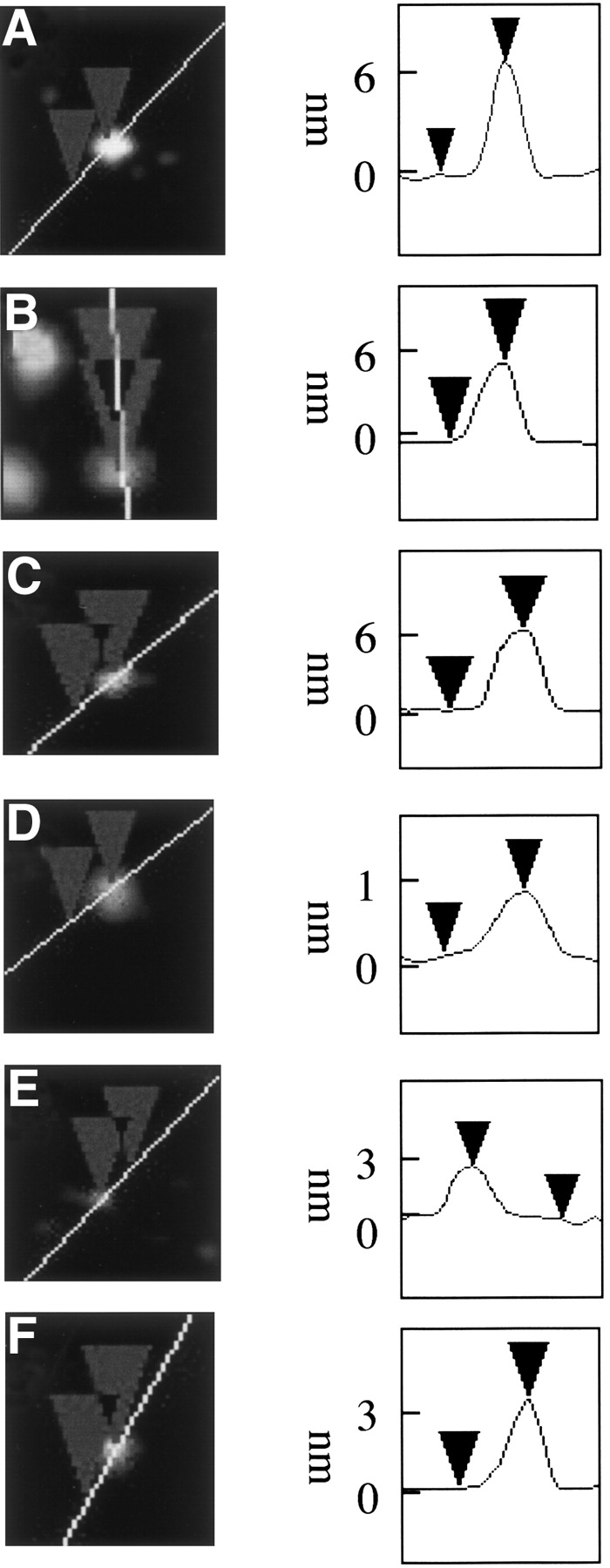
Cadmium(II), zinc(II), and magnesium(II) induce spherical α-synuclein oligomers, copper(II) inhibits oligomerization and iron(III) and nickel(II) do not influence α-synuclein particle size. (Left) Tapping mode AFM height images of representative particles after incubation at 4°C for 1 d with Cd(II) (A), Zn(II) (B), Mg(II) (C), Cu(II) (D), Fe(III) (E), and Ni(II) (F). (Right) Height cross-sections of α-synuclein particles. Cantilever length, 160 μm; tip frequency, 267–302 kHz; set point 2 V.
Co(II) and Ca(II) specifically induce α-synuclein annular species
After overnight incubation in the presence of Co(II) and Ca(II), distinctive annular particles were observed (Fig. 4 ▶) that closely resemble annular particles observed after 10 d incubation of the protein without metal ion addition. Control samples with no metal ions added contained only 2–5 nm height spherical particles as in Figure 1B ▶. Calcium addition resulted in 5% of the total particles detected having annular morphology (Fig. 4A ▶), with the remaining particles being spherical and 2–5 nm in height. Measurement of the peak-to-peak cross-sectional distance of the calcium-induced annular species revealed a diameter range of 70–95nm, and a mean maximum height of 3.9 ± 1.2 nm (n = 10) (Fig. 4B,C ▶). Each particle appeared to be composed of a ring of six spherical particles (Fig. 4C ▶) with occasional open-ring structures composed of a chain of five 4 nm-height spherical particles (Fig. 4B ▶). In the presence of cobalt(II) ions, smaller annular particles were observed (Fig. 4D–F ▶) with mean maximum height 2 ± 0.8 nm (n = 7) and diameter ranging 22–30 nm.
Figure 4.

Calcium(II) and cobalt(II) induce α-synuclein annular particles. (Left) Tapping mode AFM height images of particles formed after incubation at 4°C for 1 d in the presence of calcium(II) (A–C) and cobalt(II) (D–F). (Right) Height cross-sections of α-synuclein particles. Cantilever length, 160 μm; tip frequency, 267–302 kHz; set point, 1.5 V.
The binding of calcium(II) ions with high affinity to α-synuclein is dependent on the C terminus of the protein (aa 126–140), which contains six aspartic/glutamic acid residues, thought to mediate metal coordination (Nielsen et al. 2001). Nielsen et al. (2001) showed that a truncated protein comprising amino acids 1–125 is deficient in calcium binding, and that a synthetic peptide representing amino acids 109–140 binds calcium(II) with equal affinity to the full-length protein (see Fig. 5A ▶). To determine whether specific binding of calcium(II) was required to induce annular oligomer formation, we acquired atomic force microscope images of truncated α-synuclein (aa 1–125) incubated overnight at 4°C with and without a 10-fold excess of calcium(II) (Fig. 5B,C ▶). In contrast to the full-length protein, the truncated protein showed only 2–6-nm spherical particles both in the presence and absence of Ca(II) ions.
Figure 5.

C-terminal α-synuclein calcium binding domain is involved in annular oligomer formation. (A) Domain model of α-synuclein structure, illustrating protease cleavage sites, lipid and calcium binding domains, and C-terminally truncated protein. (B,C) Tapping mode AFM height images of truncated α-synuclein incubated 1 d at 4°C without (B) and with (C) calcium(II). Cantilever length, 160 μm; tip frequency, 267–302 kHz; set point, 1.5 V.
Discussion
In the present study, we investigated the polymerization pathway of α-synuclein and the effects of metal ions on α-synuclein oligomer formation at 4°C in the absence of mixing. Previous studies on α-synuclein fibrillation have focussed on incubation with mixing at 37°C, conditions under which some intermediate steps in the filament formation pathway are difficult to observe. We show that α-synuclein has a complex polymerization pathway leading to amyloid-like filaments, involving spherical and annular intermediates. The formation of filaments proceeds via sequential nonfibrillar spherical particles to annular particles and then on to branched filaments, demonstrating that the annular oligomer is the immediate precursor to filament formation and not an alternative aggregation pathway. Furthermore, the formation of α-synuclein oligomers is accelerated in the presence of magnesium(II), cadmium(II), zinc(II), cobalt(II), and calcium(II), with calcium(II) and cobalt(II) specifically inducing the rapid formation of annular α-synuclein oligomers.
Annular α-synuclein oligomers nucleate filament formation
Fibrillation of α-synuclein in vitro has been shown previously to be a nucleation-dependent process (Wood et al. 1999) involving the assembly of intermediate oligomeric protofibrillar species that are freely diffusible and available for molecular interactions. There is evidence with other amyloidogenic proteins to suggest that monomer to protofibril conversion and protofibril to filament conversion are step-wise processes (Harper et al. 1999; Serio et al. 2000; Bucciantini et al. 2002; Poirier et al. 2002; Hoyer et al. 2004). Filament formation occurs when the protofibrils are concentrated and interact with one another. Our data indicate that α-synuclein filament formation in vitro is driven by protofibril–protofibril interactions, with spherical oligomers first interacting to form annular oligomers from which three linear fibrils then extend by further addition of spherical oligomers (or monomers) about the threefold symmetry axis of the central annulus. Earlier AFM studies on α-synuclein oligomerization, principally by Lansbury and coworkers, have shown 32–180 nm-diameter annular oligomers, 3–7 nm in height and annular particles with diameters of 18 nm to 27 nm and 3 nm height upon interaction with microsomal vesicles (Ding et al. 2002). In electron micrograph reconstructions, Lashuel et al. (2002) showed that the A53T and A30P α-synuclein mutants readily form annular particles 10–20 nm and 8–24 nm in diameter, respectively. Previously, we have observed annular α-synuclein-positive particles released by mild detergent dissociation of purified α-synucleinopathy aggregates (Pountney et al. 2004). These particles were 19–39 nm in diameter with sarcosine detergent and 39–57 nm in diameter with CHAPS detergent, ~12 nm in height in both cases. In the current work, we observed 4 nm-height annular oligomers, 45–60 nm in diameter and 7.9 nm-height oligomers, 70–90 nm in diameter in the absence of metal ions and 3.9-nm height particles, 70–95nm in diameter with calcium and 2 nm-height particles, 22–30 nm in diameter with cobalt. There is a close correlation between the subset of 7–12 nm-height annular oligomers formed in vitro and annular particles derived from pathological samples and the 8–12 nm height of branched and linear filaments either in vitro or in disease tissue (Conway et al. 2000a; Crowther et al. 2000; Gai et al. 2003). This is consistent with the progression from monomer to ~4 nm-height spherical oligomer, to ~4-nm height annular oligomer, then to ~8 nm-height annular oligomer, from which ~8–10 nm-height linear filaments then extend. The wide range of diameters (8–180 nm) observed here and in previous work (Ding et al. 2002; Lashuel et al. 2002; Pountney et al. 2004) indicates that annular α-synuclein oligomers are likely not functionally defined quaternary structures. Rather, they are formed as a result of the competing self-association processes of either linear extension or ring closure, consistent with studies of equine lysosyme and apolipoprotein C-II (Hatters et al. 2003; Malisauskas et al. 2003).
Mechanism of calcium-induced annular α-synuclein oligomerization
We found that incubation of α-synuclein monomer for 1 d at 4°C with the metal ions Ca(II) and Co(II) induced the formation of 2–4 nm-height annular particles, whereas Mg(II), Cd(II), and Zn(II) led to 4–7 nm-height spherical oligomeric species. Cu(II) appeared to inhibit oligomerization, whereas incubation with Fe(III) and Ni(II) yielded 2–4 nm-height particles similar to incubation in the absence of metal ions. These results demonstrate that both the morphology and rate of formation of α-synuclein oligomers are influenced by the presence of metal ions. Previous studies have shown that the overall rate of α-synuclein aggregation and the morphology of aggregates is highly dependent on changes in pH or salt concentration (Hoyer et al. 2002, 2004), suggesting a role for electrostatic interactions in the oligomerization mechanism. With a pI of 4.7, α-synuclein is an acidic protein; thus, neutralization of negative charges may cause changes in aggregation rate and morphology. Similarly to protonation of the acidic residues, metal ion binding would result in charge pairing and may balance the negative charges in the C terminus of the protein by electrostatic shielding. However, in this case, we would expect to see annular oligomers formed from the C-terminally truncated protein that lacks the acidic residues that likely mediate calcium binding. Alternatively, calcium coordination by the C-terminal domain could induce a conformational transition in the protein that favors annular oligomer formation. Several metal ions, including Cu(II) and Al(III), have been shown to induce α-helix-rich conformations in α-synuclein (Weinreb et al. 1996; Uversky et al. 2001); however, calcium(II) binding has no effect on the secondary structure, as determined by circular dichroism spectroscopy (Weinreb et al. 1996; D.L. Pountney, unpubl.). The dimerization of α-synuclein stabilized by intramolecular oxidative cross-linking of tyrosine residues has been shown to be a rate-limiting step in the filament formation pathway (Krishnan et al. 2003). Nielsen et al. (2001) determined that calcium binds to α-synuclein via the C-terminal 15 residues with a stoichiometry of 0.5 Ca(II) per protein molecule. It is tempting to speculate that the initial formation of metal-linked dimers similar to covalently cross-linked dimers could provide a mechanism thereby enabling rapid progression toward higher order oligomers. The formation of annular oligomers could then be further catalyzed by interactions with charged surfaces, such as biological phospholipid membranes, the mica support used in AFM experiments or the plastic walls of incubation vessels.
Recent studies have implicated calcium in the pathogenesis of synucleinopathies. In the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) mouse model of PD, neuronal cell death is associated with activation of the calcium-dependent protease, calpain, that can be partially abrogated by the specific calpain inhibitor, calpastatin (Crocker et al. 2003). The role of calpain in disease pathology is further supported by the upregulation of the specific calpain proteolysis product of α-spectrin in the substantia nigra in PD brain and the observation of calpain cleavage products of α-synuclein in the A53T α-synuclein transgenic mouse (Mishizen-Eberz et al. 2003). Interestingly, in these latter studies, calpain-mediated digestion of α-synuclein filaments in vitro resulted in specific cleavage at amino acids 114 and 122, yielding C-terminally truncated protein lacking the calcium-binding domain. Similar proteolytic cleavage of the C-terminal region of α-synuclein in filaments has also been reported with the nonspecific protease, proteinase K, consistent with the accessibility of this region to proteases (Miake et al. 2002). Moreover, it has been shown that α-synuclein binds to a variety of other proteins, in particular cytoskeleton-associated proteins, proteasome components, histones, and enzymes (Jensen et al. 2000; Payton et al. 2001; Power et al. 2002; Goers et al. 2003b; Snyder et al. 2003). The C-terminal calcium-binding domain has been implicated in mediating both α-synuclein interactions with other proteins and α-synuclein self-association. Interaction with the C-terminal domain of α-synuclein is required for inhibition of phospholipase D2 (Payton et al. 2004). Nielsen et al. (2001) showed that binding of the MAP 1A protein to the C-terminal α-synuclein domain is regulated by calcium and recent studies have demonstrated a critical role of Tyr-125 in oxidative stabilization of α-synuclein dimers (Takahasi et al. 2002; Zhou and Freed 2004). Furthermore, Eliezer et al. (2001) have demonstrated that when α-synuclein is bound to reconstituted lipid vesicles or SDS micelles, the N terminus of the protein adopts a folded conformation embedded in the liposomal or micellar membrane, and the C terminus from about residue 100 is solvent accessible and highly mobile, and suggest that this domain could also mediate intramolecular interactions of the vesicle-associated protein. Thus, the rapid induction of annular oligomers by calcium binding to the C-terminal domain of α-synuclein could provide another mechanistic link between the regulation of intracellular free calcium and the pathogenesis of α-synucleinopathies. Features of the domain structure of α-synuclein are illustrated in Figure 5 ▶.
In conclusion, we have shown that α-synuclein is a structurally versatile molecule, which can be induced rapidly to form oligomers with spherical or annular structures by metal–ligand interactions. By providing a nucleation center for filament growth, the annular oligomers promote filament formation, a process influenced by metal ion binding. Recently, it was reported that aggregates formed from nondisease-related amyloid-forming proteins are substantially cytotoxic, depending on their morphology, with granular and amorphous aggregates having a much higher potential to impair cell viability in comparison to mature fibrils (Bucciantini et al. 2002). Thus, studies of other amyloidogenic proteins support the view that the structural diversity of α-synuclein is a key element in the pathology of the α-synucleinopathies. Further elucidation of whether the formation of fibrils is a protective mechanism of the cell or the process that causes cell death will be critical.
Materials and methods
Expression and purification of recombinant α-synuclein
Recombinant full-length and truncated (amino acids 1–125) human α-synuclein was expressed in Escherichia coli BL21 (DE3)/ pLysS from the pET11d plasmid containing human α-synuclein cDNA, and purified as described previously (Nielsen et al. 2001). Protein expression was induced with 1 mM IPTG, and the pellet was lysed in 50 mM Tris HCl (pH 7.6), 5 mM EDTA, 1 mM DTT in the presence of protease inhibitor cocktail (Sigma). The clarified extract was then heated (100°C, 10 min) and centrifuged (10,000g, 5 min). The supernatant was applied to a Mono Q column (Pharmacia), had been equilibrated with 20 mM Tris HCl (pH 7.6), 5 mM EDTA, 1 mM DTT, and was eluted with 0–1 M NaCl using a Pharmacia AKTA chromatography system. The α-synuclein peak fraction was determined by dot blot using an anti-α-synuclein antibody. The purity of this fraction was >90%, as judged by SDS-PAGE. The anion exchange peak fraction was then further purified by gel permeation chromatography (Superdex 75 HR [10/ 30]; Pharmacia) in 0.02 M Tris, 0.15 M NaCl (pH 7.4). The α-synuclein–containing fraction by dot blot corresponding to an apparent molecular weight of 50 KDa was then either used immediately or snap frozen in liquid nitrogen and stored at −20°C prior to use. All column steps were at 15°C. Protein concentration was determined by Biorad protein assay versus bovine serum albumin.
α-Synuclein incubations
Identical samples of purified recombinant α-synuclein (10 μM) in 0.02 M Tris-HCl, 0.15 M NaCl (pH 7.4) were incubated at 4°C without agitation either with or without the addition of metal ions. Stock metal solutions (10 mM) were prepared in 1 mM HCl and diluted immediately prior to addition to give a final metal ion concentration of 100 μM.
AFM analysis
Aliquots of each sample (3 μL) were pipetted onto freshly cleaved mica and allowed to dry (5 min). The mica was then rinsed with 800 μL of ultrapure water (Labconco) and dried under a stream of compressed air. Images were acquired using a Nanoscope IV Multimode SPM (Digital Instruments) atomic force microscope in tapping mode in air using etched silicon probes (Digital Instruments) with cantilever length 160 μm, tip frequency 267–302 kHz, or cantilever length 225 μm, tip frequency 77–94 kHz, as indicated. A set point of 1.5–2 V was used.
Acknowledgments
We acknowledge financial support from the Australian National Health and Medical Research Council, Flinders Foundation, Flinders Institute of Health, and Medical Research and Flinders Institute for Research in Science and Technology.
Abbreviations
PD, Parkinson’s disease
DLB, dementia with Lewy bodies
MSA, multiple system atrophy
AFM, atomic force microscopy
MPTP, 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine
Article published online ahead of print. Article and publication date are at http://www.proteinscience.org/cgi/doi/10.1110/ps.04879704.
References
- Armstrong, R.A., Lantos, P.L., and Cairns, N.J. 2004. Spatial patterns of α-synuclein positive glial cytoplasmic inclusions in multiple system atrophy. Mov. Disord. 19 109–112. [DOI] [PubMed] [Google Scholar]
- Auluck, P.K., Chan, H.Y., Trojanowski, J.Q., Lee, V.M., and Bonini, N.M. 2002. Chaperone suppression of α-synuclein toxicity in a Drosophila model for Parkinson’s disease. Science 295 865–868. [DOI] [PubMed] [Google Scholar]
- Braak, H., Del Tredici, K., Bratzke, H., Hamm-Clement, J., Sandmann-Keil, D., and Rub, U. 2002. Staging of the intracerebral inclusion body pathology associated with idiopathic Parkinson’s disease (preclinical and clinical stages). J. Neurol. 249 (Suppl. 3) 1–5. [DOI] [PubMed] [Google Scholar]
- Bucciantini, M., Giannoni, E., Chiti, F., Baroni, F., Formigli, L., Zurdo, J., Taddei, N., Ramponi, G., Dobson, C.M., and Stefani, M. 2002. Inherent toxicity of aggregates implies a common mechanism for protein misfolding diseases. Nature 416 507–511. [DOI] [PubMed] [Google Scholar]
- Cabin, D.E., Shimazu, K., Murphy, D., Cole, N.B., Gottschalk, W., McIlwain, K.L., Orrison, B., Chen, A., Ellis, C.E., Paylor, R., et al. 2002. Synaptic vesicle depletion correlates with attenuated synaptic responses to prolonged repetitive stimulation in mice lacking α-synuclein. J. Neurosci. 22 8797–8807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell, B.C., McLean, C.A., Culvenor, J.G., Gai, W.P., Blumbergs, P.C., Jakala, P., Beyreuther, K., Masters, C.L., and Li, Q.X. 2001. The solubility of α-synuclein in multiple system atrophy differs from that of dementia with Lewy bodies and Parkinson’s disease. J. Neurochem. 76 87–96. [DOI] [PubMed] [Google Scholar]
- Clayton, D.F. and George, J.M. 1998. The synucleins: A family of proteins involved in synaptic function, plasticity, neurodegeneration and disease. Trends Neurosci. 21 249–254. [DOI] [PubMed] [Google Scholar]
- Conway, K.A., Harper, J.D., and Lansbury Jr., P.T. 2000a. Fibrils formed in vitro from α-synuclein and two mutant forms linked to Parkinson’s disease are typical amyloid. Biochemistry 39 2552–2563. [DOI] [PubMed] [Google Scholar]
- Conway, K.A., Lee, S.J., Rochet, J.C., Ding, T.T., Williamson, R.E., and Lansbury Jr., P.T. 2000b. Acceleration of oligomerization, not fibrillization, is a shared property of both α-synuclein mutations linked to early-onset Parkinson’s disease: Implications for pathogenesis and therapy. Proc. Natl. Acad. Sci. 97 571–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crocker, S.J., Smith, P.D., Jackson-Lewis, V., Lamba, W.R., Hayley, S.P., Grimm, E., Callaghan, S.M., Slack, R.S., Melloni, E., Przedborski, S., et al. 2003. Inhibition of calpains prevents neuronal and behavioral deficits in an MPTP mouse model of Parkinson’s disease. J. Neurosci. 23 4081–4091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowther, R.A., Daniel, S.E., and Goedert, M. 2000. Characterisation of isolated α-synuclein filaments from substantia nigra of Parkinson’s disease brain. Neurosci. Lett. 292 128–130. [DOI] [PubMed] [Google Scholar]
- Ding, T.T., Lee, S.J., Rochet, J.C., and Lansbury Jr., P.T. 2002. Annular α-synuclein protofibrils are produced when spherical protofibrils are incubated in solution or bound to brain-derived membranes. Biochemistry 41 10209–10217. [DOI] [PubMed] [Google Scholar]
- Eliezer, D., Kutluay, E., Bussell Jr., R., and Browne, G. 2001. Conformational properties of α-synuclein in its free and lipid-associated states. J. Mol. Biol. 307 1061–1073. [DOI] [PubMed] [Google Scholar]
- Gai, W.P., Pountney, D.L., Power, J.H., Li, Q.X., Culvenor, J.G., McLean, C.A., Jensen, P.H., and Blumbergs, P.C. 2003. α-Synuclein fibrils constitute the central core of oligodendroglial inclusion filaments in multiple system atrophy. Exp. Neurol. 181 68–78. [DOI] [PubMed] [Google Scholar]
- Goedert, M. 1999. Filamentous nerve cell inclusions in neurodegenerative diseases: Tauopathies and α-synucleinopathies. Philos. Trans. R. Soc. Lond. B Biol. Sci. 354 1101–1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goers, J., Manning-Bog, A.B., McCormack, A.L., Millett, I.S., Doniach, S., Di Monte, D.A., Uversky, V.N., and Fink, A.L. 2003a. Nuclear localization of α-synuclein and its interaction with histones. Biochemistry 42 8465–8471. [DOI] [PubMed] [Google Scholar]
- Goers, J., Uversky, V.N., and Fink, A.L. 2003b. Polycation-induced oligomerization and accelerated fibrillation of human α-synuclein in vitro. Protein Sci. 12 702–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golts, N., Snyder, H., Frasier, M., Theisler, C., Choi, P., and Wolozin, B. 2002. Magnesium inhibits spontaneous and iron-induced aggregation of α-synuclein. J. Biol. Chem. 277 16116–16123. [DOI] [PubMed] [Google Scholar]
- Harper, J.D., Wong, S.S., Lieber, C.M., and Lansbury Jr., P.T. 1999. Assembly of A β amyloid protofibrils: An in vitro model for a possible early event in Alzheimer’s disease. Biochemistry 38 8972–8980. [DOI] [PubMed] [Google Scholar]
- Hashimoto, M., Rockenstein, E., and Masliah, E. 2003. Transgenic models of α-synuclein pathology: Past, present, and future. Ann. N. Y. Acad. Sci. 991 171–188. [PubMed] [Google Scholar]
- Hatters, D.M., MacRaild, C.A., Daniels, R., Gosal, W.S., Thomson, N.H., Jones, J.A., Davis, J.J., MacPhee, C.E., Dobson, C.M., and Howlett, G.J. 2003. The circularization of amyloid fibrils formed by apolipoprotein C-II. Biophys. J. 85 3979–3990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoyer, W., Antony, T., Cherny, D., Heim, G., Jovin, T.M., and Subramaniam, V. 2002. Dependence of α-synuclein aggregate morphology on solution conditions. J. Mol. Biol. 322 383–393. [DOI] [PubMed] [Google Scholar]
- Hoyer, W., Cherny, D., Subramaniam, V., and Jovin, T.M. 2004. Rapid self-assembly of α-synuclein observed by in situ atomic force microscopy. J. Mol. Biol. 340 127–139. [DOI] [PubMed] [Google Scholar]
- Iseki, E. 2004. Dementia with Lewy bodies: Reclassification of pathological subtypes and boundary with Parkinson’s disease or Alzheimer’s disease. Neuropathology 24 72–78. [DOI] [PubMed] [Google Scholar]
- Iwai, A., Masliah, E., Yoshimoto, M., Ge, N., Flanagan, L., de Silva, H.A., Kittel, A., and Saitoh, T. 1995. The precursor protein of non-A β component of Alzheimer’s disease amyloid is a presynaptic protein of the central nervous system. Neuron 14 467–475. [DOI] [PubMed] [Google Scholar]
- Jensen, P.H. and Gai, W.P. 2001. α-synuclein. Axonal transport, ligand interaction and neurodegeneration. Adv. Exp. Med. Biol. 487 129–134. [PubMed] [Google Scholar]
- Jensen, P.H., Islam, K., Kenney, J., Nielsen, M.S., Power, J., and Gai, W.P. 2000. Microtubule-associated protein 1B is a component of cortical Lewy bodies and binds α-synuclein filaments. J. Biol. Chem. 275 21500–21507. [DOI] [PubMed] [Google Scholar]
- Kim, S.J., Sung, J.Y., Um, J.W., Hattori, N., Mizuno, Y., Tanaka, K., Paik, S.R., Kim, J., and Chung, K.C. 2003. Parkin cleaves intracellular α-synuclein inclusions via the activation of calpain. J. Biol. Chem. 278 41890–41899. [DOI] [PubMed] [Google Scholar]
- Klucken, J., Shin, Y., Masliah, E., Hyman, B.T., and McLean, P.J. 2004. Hsp70 reduces α-synuclein aggregation and toxicity. J. Biol. Chem. Mar 25 [Epub ahead of print]. [DOI] [PubMed]
- Krishnan, S., Chi, E.Y., Wood, S.J., Kendrick, B.S., Li, C., Garzon-Rodriguez, W., Wypych, J., Randolph, T.W., Narhi, L.O., Biere, A.L., et al. 2003. Oxidative dimer formation is the critical rate-limiting step for Parkinson’s disease α-synuclein fibrillogenesis. Biochemistry 42 829–837. [DOI] [PubMed] [Google Scholar]
- Kruger, R., Kuhn, W., Muller, T., Woitalla, D., Graeber, M., Kosel, S., Przuntek, H., Epplen, J.T., Schols, L., and Riess, O. 1998. Ala30Pro mutation in the gene encoding α-synuclein in Parkinson’s disease. Nat. Genet. 18 106–108. [DOI] [PubMed] [Google Scholar]
- Lashuel, H.A., Petre, B.M., Wall, J., Simon, M., Nowak, R.J., Walz, T., and Lansbury Jr., P.T. 2002. α-synuclein, especially the Parkinson’s disease-associated mutants, forms pore-like annular and tubular protofibrils. J. Mol. Biol. 322 1089–1102. [DOI] [PubMed] [Google Scholar]
- Lykkebo, S. and Jensen, P.H. 2002. α-synuclein and presynaptic function: Implications for Parkinson’s disease. Neuromol. Med. 2 115–129. [DOI] [PubMed] [Google Scholar]
- Malisauskas, M., Zamotin, V., Jass, J., Noppe, W., Dobson, C.M., and Morozova-Roche, L.A. 2003. Amyloid protofilaments from the calcium-binding protein equine lysozyme: Formation of ring and linear structures depends on pH and metal ion concentration. J. Mol. Biol. 330 879–890. [DOI] [PubMed] [Google Scholar]
- Maries, E., Dass, B., Collier, T.J., Kordower, J.H., and Steece-Collier, K. 2003. The role of α-synuclein in Parkinson’s disease: Insights from animal models. Nat. Rev. Neurosci. 4 727–738. [DOI] [PubMed] [Google Scholar]
- Masliah, E., Rockenstein, E., Veinbergs, I., Mallory, M., Hashimoto, M., Takeda, A., Sagara, Y., Sisk, A., and Mucke, L. 2000. Dopaminergic loss and inclusion body formation in α-synuclein mice: Implications for neurode-generative disorders. Science 287 1265–1269. [DOI] [PubMed] [Google Scholar]
- Miake, H., Mizusawa, H., Iwatsubo, T., and Hasegawa, M. 2002. Biochemical characterization of the core structure of α-synuclein filaments. J. Biol. Chem. 277 19213–19219. [DOI] [PubMed] [Google Scholar]
- Mishizen-Eberz, A.J., Guttmann, R.P., Giasson, B.I., Day III, G.A., Hodara, R., Ischiropoulos, H., Lee, V.M., Trojanowski, J.Q., and Lynch, D.R. 2003. Distinct cleavage patterns of normal and pathologic forms of α-synuclein by calpain I in vitro. J. Neurochem. 86 836–847. [DOI] [PubMed] [Google Scholar]
- Murphy, D.D., Rueter, S.M., Trojanowski, J.Q., and Lee, V.M. 2000. Synucleins are developmentally expressed, and α-synuclein regulates the size of the presynaptic vesicular pool in primary hippocampal neurons. J. Neurosci. 20 3214–3220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen, M.S., Vorum, H., Lindersson, E., and Jensen, P.H. 2001. Ca2+ binding to α-synuclein regulates ligand binding and oligomerization. J. Biol. Chem. 276 22680–22684. [DOI] [PubMed] [Google Scholar]
- Norris, E.H., Giasson, B.I., and Lee, V.M. 2004. α-Synuclein: Normal function and role in neurodegenerative diseases. Curr. Top. Dev. Biol. 60 17–54. [DOI] [PubMed] [Google Scholar]
- Payton, J.E., Perrin, R.J., Clayton, D.F., and George, J.M. 2001. Protein–protein interactions of α-synuclein in brain homogenates and transfected cells. Brain Res. Mol. Brain Res. 95 138–145. [DOI] [PubMed] [Google Scholar]
- Payton, J.E., Perrin, R.J., Woods, W.S., and George, J.M. 2004. Structural determinants of PLD2 inhibition by α-synuclein. J. Mol. Biol. 337 1001–1009. [DOI] [PubMed] [Google Scholar]
- Perrin, R.J., Woods, W.S., Clayton, D.F., and George, J.M. 2001. Exposure to long chain polyunsaturated fatty acids triggers rapid multimerization of synucleins. J. Biol. Chem. 276 41958–41962. [DOI] [PubMed] [Google Scholar]
- Poirier, M.A., Li, H., Macosko, J., Cai, S., Amzel, M., and Ross, C.A. 2002. Huntingtin spheroids and protofibrils as precursors in polyglutamine fibrilization. J. Biol. Chem. 277 41032–41037. [DOI] [PubMed] [Google Scholar]
- Polymeropoulos, M.H., Lavedan, C., Leroy, E., Ide, S.E., Dehejia, A., Dutra, A., Pike, B., Root, H., Rubenstein, J., Boyer, R., et al. 1997. Mutation in the α-synuclein gene identified in families with Parkinson’s disease. Science 276 2045–2047. [DOI] [PubMed] [Google Scholar]
- Pountney, D.L., Lowe, R., Quilty, M., Vickers, J.C., Voelcker, N.H., and Gai, W.P. 2004. Annular α-synuclein species from purified multiple system atrophy inclusions. J. Neurochem. 90 502–512. [DOI] [PubMed] [Google Scholar]
- Power, J.H., Shannon, J.M., Blumbergs, P.C., and Gai, W.P. 2002. Nonselenium glutathione peroxidase in human brain: Elevated levels in Parkinson’s disease and dementia with lewy bodies. Am. J. Pathol. 161 885–894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singleton, A.B., Farrer, M., Johnson, J., Singleton, A., Hague, S., Kachergus, J., Hulihan, M., Peuralinna, T., Dutra, A., Nussbaum, R., et al. 2003. α-Synuclein locus triplication causes Parkinson’s disease. Science 302 841. [DOI] [PubMed] [Google Scholar]
- Serio, T.R., Cashikar, A.G., Kowal, A.S., Sawicki, G.J., Moslehi, J.J., Serpell, L., Arnsdorf, M.F., and Lindquist, S.L. 2000. Nucleated conformational conversion and the replication of conformational information by a prion determinant. Science 289 1317–1321. [DOI] [PubMed] [Google Scholar]
- Snyder, H., Mensah, K., Theisler, C., Lee, J., Matouschek, A., and Wolozin, B. 2003. Aggregated and monomeric α-synuclein bind to the S6′ proteasomal protein and inhibit proteasomal function. J. Biol. Chem. 278 11753–11759. [DOI] [PubMed] [Google Scholar]
- Souza, J.M., Giasson, B.I., Chen, Q., Lee, V.M., and Ischiropoulos, H. 2000. Dityrosine cross-linking promotes formation of stable α-synuclein polymers. Implication of nitrative and oxidative stress in the pathogenesis of neurodegenerative synucleinopathies. J. Biol. Chem. 275 18344–18349. [DOI] [PubMed] [Google Scholar]
- Takahashi, T., Yamashita, H., Nakamura, T., Nagano, Y., and Nakamura, S. 2002. Tyrosine 125 of α-synuclein plays a critical role for dimerization following nitrative stress. Brain Res. 938 73–80. [DOI] [PubMed] [Google Scholar]
- Totterdell, S., Hanger, D., amd Meredith, G.E. 2004. The ultrastructural distribution of α-synuclein-like protein in normal mouse brain. Brain Res. 1004 61–72. [DOI] [PubMed] [Google Scholar]
- Trojanowski, J.Q. and Lee, V.M. 2003. Parkinson’s disease and related α-synucleinopathies are brain amyloidoses. Ann. N. Y. Acad. Sci. 991 107–110. [DOI] [PubMed] [Google Scholar]
- Uversky, V.N. 2003. A protein-chameleon: Conformational plasticity of α-synuclein, a disordered protein involved in neurodegenerative disorders. J. Biomol. Struct. Dyn. 21 211–234. [DOI] [PubMed] [Google Scholar]
- Uversky, V.N., Li, J., and Fink, A.L. 2001. Metal-triggered structural transformations, aggregation, and fibrillation of human α-synuclein. A possible molecular NK between Parkinson’s disease and heavy metal exposure. J. Biol. Chem. 276 44284–44296. [DOI] [PubMed] [Google Scholar]
- Volles, M.J., Lee, S.J., Rochet, J.C., Shtilerman, M.D., Ding, T.T., Kessler, J.C., and Lansbury Jr., P.T. 2001. Vesicle permeabilization by protofibrillar α-synuclein: Implications for the pathogenesis and treatment of Parkinson’s disease. Biochemistry 40 7812–7819. [DOI] [PubMed] [Google Scholar]
- Weinreb, P.H., Zhen, W., Poon, A.W., Conway, K.A., and Lansbury Jr., P.T. 1996. NACP, a protein implicated in Alzheimer’s disease and learning, is natively unfolded. Biochemistry 35 13709–13715. [DOI] [PubMed] [Google Scholar]
- Wood, S.J., Wypych, J., Steavenson, S., Louis, J.C., Citron, M., and Biere, A.L. 1999. α-Synuclein fibrillogenesis is nucleation-dependent. Implications for the pathogenesis of Parkinson’s disease. J. Biol. Chem. 274 19509–19512. [DOI] [PubMed] [Google Scholar]
- Zarranz, J.J., Alegre, J., Gomez-Esteban, J.C., Lezcano, E., Ros, R., Ampuero, I., Vidal, L., Hoenicka, J., Rodriguez, O., Atares, B., et al. 2004. The new mutation, E46K, of α-synuclein causes Parkinson and Lewy body dementia. Ann. Neurol. 55 164–173. [DOI] [PubMed] [Google Scholar]
- Zhou, W. and Freed, C.R. 2004. Tyrosine-to-cysteine modification of human α-synuclein enhances protein aggregation and cellular toxicity. J. Biol. Chem. 279 10128–10135. [DOI] [PubMed] [Google Scholar]