

Abstract
Infrequent structural fluctuations of a globular protein is seldom detected and studied in detail. One tyrosine ring of HPr from Staphylococcus carnosus, an 88-residue phosphocarrier protein with no disulfide bonds, undergoes a very slow ring flip, the pressure and temperature dependence of which is studied in detail using the on-line cell high-pressure nuclear magnetic resonance technique in the pressure range from 3 MPa to 200 MPa and in the temperature range from 257 K to 313 K. The ring of Tyr6 is buried sandwiched between a β-sheet and α-helices (the water-accessible area is less than 0.26 nm2), its hydroxyl proton being involved in an internal hydrogen bond. The ring flip rates101~105 s−1 were determined from the line shape analysis of Hδ1, δ2 and Hɛ1,ɛ2 of Tyr6, giving an activation volume ΔV‡ of 0.044 ± 0.008 nm3 (27 mL mol−1), an activation enthalpy ΔH‡ of 89 ± 10 kJ mol−1, and an activation entropy ΔS‡ of 16 ± 2 JK−1 mol−1. The ΔV‡ and ΔH‡ values for HPr found previously for Tyr and Phe ring flips of BPTI and cytochrome c fall within the range of ΔV‡ of 28 to 51 mL mol−1 and ΔH‡ of 71 to 155 kJ mol−1. The fairly common ΔV‡ and ΔH‡ values are considered to represent the extra space or cavity required for the ring flip and the extra energy required to create a cavity, respectively, in the core part of a globular protein. Nearly complete cold denaturation was found to take place at 200 MPa and 257 K independently from the ring reorientation process.
Keywords: high pressure, NMR spectroscopy, ring flips, HPr, PTS
The intimate relationship between the dynamics and function of a protein has been extensively discussed in the past and is still an exciting field in protein science. Spin relaxation in NMR spectroscopy has proven to be useful for investigating the average microscopic dynamics of a protein occurring in the frequency range normally of 109 sec−1 or higher. Various lines of evidence derived from hydrogen exchange experiments (Woodward et al. 1982, 2004; Englander and Moyne 1992) and advanced spin relaxation analysis (Ishima and Torchia 2000; Frans et al. 2001; Meiler et al. 2003) indicate that slower motions in the time range of milliseconds to microseconds are widely involved in protein dynamics under conditions that stabilize the folded state. In many cases, slower and rare fluctuations in microseconds to milliseconds or even second range may play more crucial roles in protein function than the average fluctuation of the structure in the picosecond-to-nanosecond range.
Slow motions in proteins are generally associated with cooperative motions involving significant changes in energy and volume, and therefore are sensitive to temperature and pressure. Particularly straightforward and important from the view of NMR spectroscopy are the 180° flip motions of rings of aromatic amino acids about their Cβ–Cγ axes (Campbell et al. 1975, 1976; Wüthrich and Wagner 1975, 1978; Wagner 1980, 1983; Li et al. 1999). Intrinsically, the flip rate of a tyrosine or phenylalanine ring is rapid (>>106 sec−1) when the rings are well exposed on the surface of the protein, so that the NMR signals for the δ1, δ2 protons and the ɛ1, ɛ2 protons of a tyrosine or phenalylalanine ring merge into one because they feel the same time-averaged local magnetic fields. However, when the rings are buried in the interior of the folded structure, the flip rates can become much slower (<<106 sec−1), giving significant changes in line shape of the signals of the δ1, δ2, or the ɛ1, ɛ2 ring protons.
The flip rates of the side chains serve as probes reporting the fluctuation of the interior of the folded structure, accurately and site-specifically. Temperature and pressure dependence of the flip rate is expected to bring crucial information on energy and volume fluctuations in the interior of the folded protein. Temperature dependence of the rate of ring flip has been reported for phenylalanine and tyrosine residues of BPTI (Wagner et al. 1976; Wüthrich and Wagner 1978), cytochrome c (Dobson et al. 1975; Campbell et al. 1976), and hen lysozyme (Campbell et al. 1975). On the other hand, pressure dependence of the rate of ring flip has been reported only for BPTI using 1H one-dimensional (Wagner 1980, 1983) and two-dimensional (Li et al. 1999) high-pressure NMR techniques.
In the present work, we study the slow conformational dynamics of HPr from Staphylococcus carnosus using the reorientation of a tyrosine ring as a probe. The pressure and temperature dependence of the tyrosine reorientation rate in the interior of the protein is measured using the on-line cell variable-pressure NMR technique at 750 MHz (Akasaka and Yamada 2001; Arnold et al. 2003). HPr from S. carnosus is a small globular protein (88 amino acids), which unlike BPTI, has no disulfide bonds. It is an essential constituent of the phosphoenolpyruvate-dependent phosphotransferase system, a transport system for carbohydrates in bacteria (Postma et al. 1993), and transfers a phosphoryl group from Enzyme I (EI) to Enzyme IIA (EIIA). The 1H and 15N NMR spectra of HPr from S. carnosus have been completely assigned and the folded three-dimensional structure in solution has been elucidated (Görler et al. 1999a; Fig. 1 ▶). As is true for all HPr proteins, the basic structure of HPr from S. carnosus consists of four antiparallel β-strands and three α-helices facing the β-pleated sheet. It contains three tyrosine residues: one phenylalanine and one histidine residue; the 1H NMR spectra of all the aromatic residues are well separated and fully assigned (Görler et al. 1999). In our previous study of 15N-enriched HPr from S. carnosus by two-dimensional heteronuclear NMR spectroscopy under varying pressure (3 to 200 MPA) (Kalbitzer et al. 2000), large pressure-induced 1H and 15N shifts were found, suggesting relatively large amplitude motions within the folded manifold of this protein (Kalbitzer et al. 2000; Akasaka and Li 2001).
Figure 1.

The 3D structure of HPr from S. carnosus. The lowest energy structure of HPr from S. carnosus (Görler et al. 1999a) is depicted and the aromatic residues are highlighted. In addition, NOE contacts of the hydroxyl proton of Tyr6 are indicated.
Results
The microscopic environment of the aromatic residues in HPr and the internal hydrogen bonding of Tyr6
Figure 1 ▶ shows the average three-dimensional structure of HPr from S. carnosus determined by NMR spectroscopy (Görler et al. 1999a). We note that all aromatic residues are located close to or at the surface of HPr with the only exception, Tyr6. Tyr6 is embedded in the core of the protein between the four-stranded β-pleated sheet and helices b and c. The contacts of its ring atoms with other atoms were calculated from the bundle of structures and are deposited in the database (Görler et al. 1999a). The ring of Tyr6 is surrounded by the side chains of Ile61, Thr78, and Leu81 (Table 1). The protection factors of the amide groups of Tyr6 and Thr78 are quite high with 5.0 105 and 2.1 105 at 298 K. However, for Ile61 and Leu81, the protection factors are with 2.4 104 and 9.5 103 rather small compared to the maximum value of 3.7 106 found for Ser27 (data not shown).
Table 1.
Environment of Tyr6a
| Atom of Tyr6 | Amino acid j | Atom of j | Average distance 〈dij〉/nm |
| Cγ | Leu 81 | Hδ1 | 0.43 |
| Cδ1 | Ile 61 | Hδ1 | 0.44 |
| Hδ1 | Ile 61 | Hγ13 | 0.43 |
| Cδ2 | Ile 61 | Hδ1 | 0.46 |
| Hδ2 | Ile 61 | Hδ1 | 0.43 |
| Cɛ1 | Thr 78 | Hγ2 | 0.42 |
| Hɛ1 | Thr 78 | Hα | 0.47 |
| Cɛ2 | Leu 81 | Hδ1 | 0.41 |
| Hɛ2 | Thr 78 | Hα | 0.45 |
| Cζ | Thr 78 | Hγ2 | 0.39 |
| Oζ | Thr 78 | Oγ1 | 0.39 |
| Hζ | Thr 78 | Oγ1 | 0.34 |
a Only pairs of atoms i,j with average distance 〈dij〉≤ 0.5 nm are listed. For contacts with methyl hydrogens the closest distance was used.
Normally, the hydroxyl proton of a tyrosine ring is not observable by NMR spectroscopy since it exchanges rapidly with the bulk water. Surprisingly, there is a group of 16 otherwise unassigned NOE cross-peaks that can be explained by back calculation of the data under the assumption that the hydroxyl proton of Tyr6 is slowly exchanging with water and has a resonance frequency at 8.034 ppm relative to the DSS (see Materials and Methods). Figure 2 ▶ shows a comparison between a part of the experimental spectrum and the spectrum back calculated from the three-dimensional structure of HPr. In Figure 1 ▶, the NOE contacts following from such an assignment of the hydroxyl group are depicted in red.
Figure 2.

NOE contacts of the hydroxyl hydrogen of Tyr6. A screen shot of the program AUREMOL is shown where the experimental NOESY spectrum (left) of HPr from S. carnosus is compared with a NOESY spectrum (right) back calculated from the first NMR structure deposited in the PDB database. The experimental spectrum was recorded at 298 K, 800.13 MHz, and 0.1 MPa, with a mixing time of 150 msec. The signal-to-noise ratio was improved by exponential multiplication with a line broadening of 12 Hz in ν1 and 2.4 Hz in ν2. The back calculation was performed with identical parameters using an overall rotational correlation time of 5.62 nsec and allowing for internal motions (see Materials and Methods).
In Figure 2 ▶, the motions of all atom pairs except methyl groups and aromatic rings were taken into account by using the so-called model-free approach (Lipari and Szabo 1982a,b). When a structure and the resonance assignments are known, NOESY spectra can be back calculated with rather high confidence. For a better description the internal motions have to be considered. For all atom pairs containing protons from a methyl group a fast-jump model was used, for atom pairs involving aromatic rings a slow jump approximation was selected. Order parameters were taken from experimental data where available or taken from literature (see Materials and Methods).
In the majority of the NMR structures the hydroxyl proton of Tyr6 is close to Oγ1 of Thr78. The H–O distance in the 10 structures deposited in the protein database is 0.34 nm ±0.09 (at a confidence level of 99%) and the O–O distance is 0.39 nm ±0.09 (at a confidence level of 99%). This is somewhat larger than the typical hydrogen bond length. However, taking into account that the hydrogen bond was not stabilized by a hydrogen bonding restraint in the calculations and electrostatic terms were switched off the experimental distance is in line with the formation of a hydrogen bond between the hydroxyl proton of Tyr6 and the Oγ1 of Thr78. The analysis of the water accessibility of the hydroxyl proton by the program MOLMOL (Koradi et al. 1996) calculated for the first of the 10 submitted structures leads to the conclusion that the hydroxyl proton is shielded nearly completely from the bulk water (water accessible surface <0.003 nm2). The same is true for the ring of Tyr6, the water accessible area is less than 0.26 nm2 where the main contribution comes from Hδ1 with 0.14 nm2.
Temperature-dependent spectral changes at 3 MPa
Figure 3 ▶ shows the aromatic region of the 1H NMR spectrum of HPr from S. carnosus in D2O at 3 MPa at temperatures varying between 275 K and 313 K. The spectrum is dominated by the signals of the ring protons of the three tyrosines, the only phenylalanine and the only histidine residue of HPr. In addition, residual weak signals from amide protons can be observed that are still visible because the 1H content of the heavy water is 0.15% and because the freeze-dried protein still contains some bound 1H2O. The signals of the aromatic residues are well separated at high temperature (e.g., 313 K); the signals of the δ1, δ2 protons as well as the ɛ1, ɛ2 protons are averaged by the fast ring flip around the Cβ–Cγ bond. By referring to the literature (Görler et al. 1999a), all the signals of the aromatic ring protons are assigned as labeled in the spectrum at 313 K. They are the singlet signals of Hδ2 and Hɛ1 of His15 (at 7.25 ppm and 7.77 ppm, respectively), the doublet signals of Hδ1,δ2 and Hɛ1,ɛ2 of Tyr6 (6.86 ppm and 6.53 ppm, respectively), Tyr37 (7.26 ppm and 6.97 ppm, respectively), and Tyr64 (6.98 ppm and 6.69 ppm, respectively), and the Hδ1,δ2, Hɛ1,ɛ2, and Hζ signals of Phe29 (7.38 ppm, 7.16 ppm, and 7.07 ppm, respectively).
Figure 3.

Temperature effects in the aromatic region of the 1H NMR spectrum of HPr from S. carnosus in D2O at ambient pressure. The assignments of the resonance lines were taken from Görler et al. (1999a) by adapting the chemical shifts published to the standard TSP. Data were recorded at 3 MPa; only a selection of the recorded data is shown.
With decreasing temperature, the resonance positions and intensities of most aromatic ring protons remain nearly invariant, but the Hδ1,δ2 and Hɛ1,ɛ2 signals of Tyr6 become selectively broadened and disappear nearly completely at 275 K. The extensive broadening observed for the ring protons of Tyr6 at lower temperatures can only be explained by exchange broadening. It is caused by a decreased ring flipping rate with decreasing temperature. The differences of the magnetic environments (and, hence, of the resonance frequencies) are only insufficiently averaged for the protons in symmetry-related positions (Hδ1 and Hδ2, Hɛ1 and Hɛ2 of Tyr6) and thus contribute to the line widths.
Pressure-dependent spectral changes at ambient and low temperatures
Figure 4 ▶ shows the aromatic region of the 1H-NMR spectrum of HPr from S. carnosus in D2O at 298 K at pressures varying between 3 MPa and 200 MPa. With increasing pressure, the signals for Tyr37, Tyr64, and Phe29 exhibit only minor changes both in resonance positions and intensity, while the Hδ1,δ2 and Hɛ1,ɛ2 signals of Tyr6 become much broader and disappear nearly completely at 200 MPa. The Hδ2 and Hɛ1 signals of His15 also show significant down-field shifts with increasing pressure due most probably to the increased protonation of the histidine ring caused by the increased pKa.
Figure 4.

Pressure effects in the aromatic region of the 1H NMR spectrum of HPr from S. carnosus in D2O at ambient temperature. Data were recorded at 298 K in the pressure range from 3 MPa to 200 MPa.
Figure 5 ▶ shows the pressure effects on the aromatic region of the spectrum of HPr at low temperature (275 K). At 3 MPa, the signals of Tyr6 are already broadened beyond detection due to slow reorientation of the tyrosine ring. At higher temperatures the averaged resonances of the ring protons of Tyr6 can be observed easily (Fig. 2 ▶). They shift only slightly with temperature (Δ δ/Δt is 0.95 ppb/K and 0.60 ppb/K for the Hδ1,δ2 and Hɛ1,ɛ2 signals of Tyr6, respectively). An extrapolation of the temperature dependent shifts to 275 K leads to expected averaged shifts 6.825 ppm and 6.560 ppm for the Hδ1,δ2 and Hɛ1,ɛ2 signals of Tyr6, respectively. At 298 K the linear pressure coefficients Δ δ/Δp are 0.032 ppm/GPa and 0.058 ppm/GPa for the Hδ1,δ2 and Hɛ1,ɛ2 signals, respectively. By assuming that the pressure coefficient is approximately the same at 275 K, average resonance line positions at 275 K and 200 MPa can be predicted to be at 6.80 ppm and 6.52 ppm for Hδ1,δ2 and Hɛ1,ɛ2 of Tyr6. At 275 K, two new signals appear at 6.24 ppm and 6.47 ppm with increasing pressure, which must arise from the ring protons of Tyr6 at a slow exchange condition. Since the two signals are not symmetrically arranged about either of the predicted average resonance positions for the δ and ɛ protons of Tyr6, each of them must represent one component of a pair of signals expected either for the δ or ɛ protons of Tyr6 at slow exchange conditions. The position of the other components of a given pair of Hδ and Hɛ resonances can then be calculated to be at 7.13 ppm and 6.80 ppm, respectively (Fig. 5 ▶). Although the signals at 6.80 and 7.13 ppm occur in a crowded part of the spectrum and therefore cannot be identified in the spectrum, the experimental data are consistent with the predicted positions of the resonance lines.
Figure 5.

Pressure effects in the aromatic region of the 1H NMR spectrum of HPr from S. carnosus in D2O at low temperature. Data were recorded at 275 K in the pressure range from 3 MPa to 200 MPa.
Figure 6 ▶ shows further spectral changes at 200 MPa when the temperature is decreased stepwise to 257 K by taking advantage of a low freezing point of water (251 K) at 200 MPa. In the top spectrum recorded at 283 K the resonances of Tyr6 are broadened beyond detection. As already described above the upfield shifted components of the now inequivalent Hδ and Hɛ resonances become visible at 275 K and get more narrow at lower temperatures.
Figure 6.

Ring dynamics and cold denaturation of HPr. Spectra were recorded at 200 MPa at varying temperatures. The signal of formiate present as impurity in the sample is labeled by a star.
At 200 MPa and 283 K, additional resonances are observed, whose intensities increase with decreasing temperature (Fig. 6 ▶). Since the signals appear at positions expected for the ring protons of tyrosines, phenylalanines, and histidines in denatured, random coil-like proteins (Bundi and Wüthrich 1979; Arnold et al. 2002), they can be ascribed to the denatured fraction of the HPr protein. At 257 K HPr is found nearly completely denatured, meaning the cold denaturation under pressure.
Quantitative analysis of the ring flip dynamics
With the known chemical shifts of the individual ring protons of Tyr6, the line shapes of these signals can be fitted to the life time of the exchange τe using the density matrix formalism including J-coupling as it is implemented in the program WinDNMR (Reich 1995). Figure 7A ▶ shows such a fit to the temperature-dependent changes of the well-resolved Hδ1,ɛ2 signal(s) of Tyr6 at 3 MPa in the temperature range from 313 K to 275 K. Figure 7b ▶ shows a similar fit to the pressure-dependent changes of the Hδ1,ɛ2 signal of Tyr6 at 275 K. In each case, the good fits indicate that the assumptions made for the spectral parameters are reasonable under the experimental conditions.
Figure 7.

Simulation of the ring dynamics of Tyr6. The line shape of the Hδ1,ɛ2-resonance(s) of Tyr6 were fitted with the program WinDNMR. (A) Fit of the experimental data recorded at 3 MPa at various temperatures, and (B) at 275 K and various pressures.
The temperature and pressure dependence of the exchange rates k1, −1 (=1/τe) deduced above allows the calculation of the activation parameters ΔH‡, ΔS‡, and ΔV‡ for the ring flip motion using equation 6. Figure 8A ▶ shows the semilogarithmic plot of the temperature dependence of the rates at 3 MPa and 200 MPa (constant pressure), and 8B shows the semilogarithmic plot of the pressure dependence of the rates at 275 K and 298 K (constant temperature). The surprisingly good fits of the data to the straight lines indicate that the ΔH‡, ΔS‡, and ΔV‡ values may be considered practically constant in the pressure range between 3 MPa and 200 MPa and in the temperature range between 275 K and 298 K. Note that a larger set of data was used for the calculation of the parameters than those depicted in the figures. The values of ΔH‡ and ΔS‡ obtained at 3 MPa and 200 MPa for the Hδ and Hɛ resonances are separately listed in Table 2, as well as the values of ΔV‡ obtained at 275 K and 298 K. Since the ΔH‡, ΔS‡, and ΔV‡ values for the ring flip of Tyr6 as monitored by the Hδ resonances and by the Hɛ resonances should be equal, their averages are also given in Table 2.
Figure 8.

Temperature and pressure dependence of the flip rates k of Tyr6. (A) Plot of ln k (with k the rate constant for reorientation of Tyr6) as a function of 1/T (with T the absolute temperature). (B) Plot of ln k as function of the pressure p. Data were fitted with equation 6 neglecting (A) a possible temperature dependence of the activation volume ΔV‡, and (B) a possible pressure dependence of the activation enthalpy ΔH‡ and the activation entropy ΔS‡. The obtained values are listed in Table 2.
Table 2.
Thermodynamics and NMR parameters for aromatic residues in HPr, BPTI, and cytochrome c
| Protein | Amino acid | Proton | Δ δ (ppm) | ΔH‡ (kJ mol−1) | ΔS‡ (J mol−1 K−1) | ΔV‡ (mL mol−1) | ΔV‡ (nm3) |
| HPr | Tyr6 | Hδ1,δ2 | 0.66 | 87 ± 17 | 16 ± 3 | 26 ± 6 | 0.043 ± 0.009 |
| (3 MPa/275 K) | Hɛ1, ɛ2 | 0.56 | 87 ± 17 | 16 ± 3 | 28 ± 6 | 0.046 ± 0.009 | |
| HPr | Tyr6 | Hδ1,δ2 | 0.66 | 89 ± 18 | 15 ± 3 | 25 ± 6 | 0.041 ± 0.02 |
| (200 MPa/298 K) | Hɛ1, ɛ2 | 0.56 | 92 ± 18 | 16 ± 3 | 27 ± 6 | 0.045 ± 0.02 | |
| HPr average | Tyr6 | 89 ± 10 | 16 ± 2 | 27 ± 3 | 0.044 ± 0.008 | ||
| BPTI | Tyr23 | Hδ1,δ2 | 109a,c | 146a,c | |||
| (0.1 MPa/329 K)a,b,c | Hɛ1, ɛ2 | ||||||
| (0.1 MPa/329 K)a,b,c | Tyr35 | Hδ1,δ2 | 1.10 | 155a,c | 284a,c | 36 ± 12b,c | 0.06 ± 0.02b,c |
| (0.1 MPa/330 K)d | 139e | 223e | 51 ± 12d | 0.084 ± 0.019d | |||
| Hɛ1, ɛ2 | 0.11 | ||||||
| (0.1 MPa/329 K)a,b,c | Phe45 | Hδ1,δ2 | 0.65 | 71c | 46a,c | 30 ± 6b,c | 0.05 ± 0.01b,c |
| (0.1 MPa/330 K)d | Hɛ1, ɛ2 | 28 ± 5d | 0.046 ± 0.008d | ||||
| Cytochrome cf | Tyrf | Hδ1,δ2 | 0.86 | 97 | 96 | — | |
| (0.1 MPa) | Hɛ1, ɛ2 | 1.19 |
a Data from Wagner et al. (1976).
b Data from Wagner (1980).
c Data from Wagner (1983).
d Data from Li et al. (1999).
e Data from Otting et al. (1993).
f Data from Campbell et al. (1976), tyrosine residue not assigned.
Discussion
The peculiar microenvironment of Tyr6
Tyr 6 in HPr is located in the hydrophobic core of the protein (Fig. 1 ▶; Table 1) and is additionally stabilized by a hydrogen bond of its hydroxyl group with the Oγ1 of Thr78. Tyr 6 is conserved in HPr proteins of some other species such as Staphylococcus aureus or replaced by other hydrophobic residues as phenylalanine (Enterococcus faecalis, Bacillus subtilis) or valine (Escherichia coli). In S. aureus a similar hydrogen bonding pattern as in S. carnosus is observed, in which the threonine residue is replaced by a serine residue whose hydroxyl group serves now as acceptor. The two factors, namely the shielding of the hydroxyl groups from the solvent and the involvement of its hydroxyl proton in hydrogen bonding, leads to a remarkable slow down of the hydrogen exchange with bulk water, so that the hydroxyl proton can be directly observed by NMR.
The amide protons of Tyr6 and the amino acids whose side chains are in the close environment of the aromatic ring of Tyr 6 (Ile61, Thr78, and Leu81) are characterized by intermediate to high protection factors for hydrogen exchange ranging from 9.5 103 to 5.0 105. This suggests that these amino acids are part of a structural arrangement for the high stability of the hydroxyl proton. The amide group of Tyr6 is engaged in a hydrogen bond to the carbonyl group of Ile61 with an intermediate hydrogen bond energy of −6.3 ± 1.1 kJ mol−1 (Kalbitzer et al. 1999). Its protection factor is with 1.1 105 in the upper 15% of the highest protection factors observed for the exchangeable amide protons of HPr from S. carnosus, but is clearly smaller than 3.7 106 found for Ser27 (hydrogen bonded to Val23 in helix a of HPr).
In HPr from S. aureus, closely related HPr from carnosus, Tyr37 can easily be chemically modified by tetranitro-methane at one ring Cɛ, and Tyr64 is also modified after prolonged incubation with the reagent (Rösch et al. 1981). However, the modification of Tyr6 leads to irreversible denaturation of the protein. Apparently the Tyr6 ring of HPr from S. aureus is tightly packed by tertiary contacts in the folded state that modification is only possible when the Tyr6 is exposed to the solvent by global unfolding.
The temperature dependence of the flip rate: ΔH‡ at two pressures
Plotting ln k as function of 1/T (with T the temperature and k the flip rate constant) gives an apparently linear dependence both at 3 MPa and at 200 MPa (Fig. 8A ▶), suggesting that activation enthalpies ΔH‡ are constant over the temperature range studied. Furthermore, the difference between the ΔH‡ values at 3 (87 ± 17 kJ/mol) and 200 MPa (91 ± 18 kJ/mol are quite small (Table 2). This would mean that the compression at 200 MPa does not significantly affect the local environment, which modifies the rate of the Tyr6 ring flip within a pressure range from 3 MPa to 200 MPa. However, a slight increase in ΔH‡ at high pressure appears to be consistent with the relatively subtle general compression effect on the folded protein represented by an average shrinkage of hydrogen bonds by approximately 1% at 200 MPa (Li et al. 1998; Akasaka and Yamada 2001).
The pressure dependence of the flip rate: ΔV‡ at two temperatures
The dependence of ln k on pressure for HPr from S. carnosus can be satisfactorily represented by a straight line at 298 K (Fig. 8B ▶). This supports the assumption that the activation volume, beside the activation enthalpy and the activation entropy, is nearly independent of pressure for 3 MPa to 200 MPa at this temperature. The data at 275 K could be less accurately determined, and is not so clear, that the activation parameters are pressure independent. Within the (rather large) limits of error (Table 2) the activation volume is independent of pressure. The values of the activation volume ΔV‡ must be supplied by local volume fluctuations around the rings. The positive value ΔV‡ = 27 mL/mol (0.045 nm3) at 275 K (Table 2) indicates that the flip requires the opening of the space surrounding the Tyr6 ring by as much as 0.045 nm3 at the activation state. The ΔV‡ value is almost the same at 298 K, showing that it is relatively insensitive to temperature.
Cold denaturation
A reversible cold denaturation was observed at 200 MPa (Fig. 6 ▶). The local environment of Tyr6 is fully destroyed, and the ring would be free to rotate in the cold denatured state. This indicates a general possibility that a fluctuation leading to cold denaturation may contribute to the ring flip. However, since the flip rate decreases with decreasing temperature and increasing pressure, it is obvious that there is no direct connection between the ring flip and the cold denaturation. At 3 MPa where the ring flip takes place in the range of 103 to 105 sec−1, no cold denaturation is evident. At medium to high temperature, a possibility of contribution from the heat denaturation may be considered. However, the fraction of the heat denatured protein in solution has a parabolic profile with a maximum at an intermediate temperature, whereas the flip rate is increasing monotonously with temperature, denying such a possibility for Tyr6 of the HPr. Thus, the ring flip of Tyr6 is a phenomenon occurring entirely in the folded state of HPr and refers to the dynamics of the folded protein.
Amide proton exchange rates are usually slowest for hydrogen bonded amides in elements of tightly packed secondary structure. Amide exchange rates are also coupled to structural transitions involved in global unfolding and in fluctuations of the native state (Woodward et al. 1982). When exchange is entirely via global unfolding, the protection factor pi calculated for the proton i with the smallest exchange rate is directly related to the Gibbs free energy of global stabilization. Under conditions strongly favoring the folded state, the value of piis the reciprocal of the equilibrium constant for global unfolding, Ki, and ΔGstab by ΔGstab = −RT ln Ki (Woodward et al. 2004). From the protection factor of Ser27, the amino acid with the slowest exchanging amide proton, one obtains a ΔGstab of −37 kJ mol−1 at 298 K. For Tyr6, Thr78, Ile61, and Leu81 the values of ΔGstab calculated in the same way are −32 kJ mol−1, −30 kJ mol−1, −25 kJ mol−1, and −23 kJ mol−1, respectively, meaning that they exchange by subglobal motions of the folded state, or by a mixture of global unfolding and subglobal motions of the folded state. The latter internal fluctuations of the folded state are often viewed as due to one of two types of processes: local unfolding or penetration (Woodward et al. 1982, 2004). A simple local unfolding mechanism for residues 6, 61, 78, and 81 does not explain the motions involved in Tyr6 ring flips as the “open” form would be too rare to allow ring reorientation. However, a penetration model, which involves penetration of water and catalyst to the site of the exchanging amide, is consistent with the pressure dependence of Tyr6 ring flips. The observed activation volume indicates a cavity corresponding to two to four waters (see below) is transiently created in the vicinity of Tyr6.
Comparison of the ring flip in HPr with those in previously studied proteins
Slowly reorienting ring systems are seldom observed in proteins because the reorientation must be hindered considerably by the atoms located above and below the ring plane. So far, only a few examples have been described in the literature and analyzed in detail. The first and the best studied case for slowly exchanging aromatic rings is found in BPTI (Wüthrich and Wagner 1975,1978; Wagner 1980, 1983; Li et al. 1999), a small globular protein stabilized by disulfide bridges and thus potentially more restricted than proteins with no disulfide bridges, e.g., cytochrome c (Campbell et al. 1975, 1976) and HPr from S. carnosus. The rate of reorientation is two orders of magnitude larger for Tyr6 of HPr S. carnosus (at 298 K, 200 MPa) or for Phe45 of BPTI (at 330 K, 200 MPa; 1190 sec−1) than for Tyr35 of BPTI (at 330 K, 200 MPa; 12 sec−1) (Li et al. 1999). Our result (Fig. 7A ▶) shows that the ring flip of Tyr6 of HPr at pH 7.6 is quite slow at 3 MPa, occurring in the time range of milliseconds to approximately microseconds, a time domain considered likely to be coupled with function, although, at present, we do not know how this fluctuation is coupled to the function of HPr.
The activation enthalpies for ring flips in the two previously studied proteins are BPTI (71 and 139 kJ mol−1) and cytochrome c (Table 2). The activation enthalpy ΔH‡ 89 ± 10 kJ mol−1 found for HPr is close to that for cytochrome c (97 kJ mol−1) and falls between 71 and 155 kJ mol−1 for BPTI. The differences among residues of these proteins are fairly modest when we consider different numbers of disulfide bridges in different proteins and their thermal stabilities.
There are even less differences in activation volume among the two proteins BPTI and HPr. The only existing reported pressure dependence of the flip rate of aromatic rings is BPTI (Wagner 1980; Li et al. 1999) before the present report for HPr. The ΔV‡ values found for the Tyr and Phe ring flips of BPTI (at 353 K) lie between 28 and 51 mL/mol. The positive ΔV‡ values are commonly found for the two proteins, which indicates that a volume expansion is commonly involved in the activated state of the flip process. The value found for HPr (27 ± 3 mL/mol) falls within the relatively small range of 28 to 51 mL/mol found for the aromatic residues of BPTI.
Mechanistic implications for protein fluctuation
Two models may be considered about the mechanism and the consequent interpretation of the activation volume ΔV‡ for the aromatic ring flip in the interior of a globular protein. One is a diffusion model based on Kramers’s theory (Kramers 1940) in which the rotation of the ring is to take place diffusively in the viscous media of the surrounding (Karplus and McCammon 1981; Wagner 1983). The other is a cavity model in which the flip of the ring is to take place instantly upon spontaneous formation of a cavity that creates a sufficient free volume for the rotation of the ring (Wagner 1980) and is optimally described by the transition state theory. The size of the cavity can be estimated as the difference between the volume Vtotal necessary for a full rotation of the aromatic ring and the volume Vring of the ring itself. Assuming a sphere of 0.34 nm radius for calculating Vtotal one obtains 0.164 nm3. With a Vring of 0.082 nm3 a ΔV‡ of 0.082 nm3 was predicted by Wagner (1980). Reducing the size of the sphere to a very small contact radius of 0.28 nm leads to a prediction of a ΔV‡ of 0.046 nm3 (Hetzel et al. 1976), a value very close to the values found in BPTI and now in HPr.
The positive ΔV‡ value of 0.044 ± 0.008 nm3 of Tyr6 of HPr (Table 2) indicates that the flip requires opening of the space surrounding the Tyr6 ring by approximately 0.044 nm3 at the activation state. Literally, this means that an extra space or a cavity corresponding to the volume of approximately four water molecules is created at a rate 101 to 105 sec−1 around Tyr6. Similar ΔV‡ values for a ring flip within a globular protein were reported previously for two residues of BPTI. It is noteworthy that, despite the variety of micro-environments and types of the individual aromatic rings, the ΔV‡ of 27 ± 3 mL mol−1 for Tyr6 of HPr at 298 K is comparable to that (28 ± 5 mL/mol) of Phe45 and that (36 ± 6 and 51 ± 12 mL mol−1) of Tyr35 of BPTI at 330 K. This observation fits well with the notion that the flip requires a minimum common space or cavity to be created around the ring. This extra space must be created as a result of cooperative thermal fluctuations of atoms surrounding the Tyr6 ring, occurring infrequently (103 to 105 s−1) around Tyr6 at ambient pressure. This means that a cavity is infrequently created in the most buried part of the protein (the core). This model is consistent with the dynamic nature of cavity formation (Kocher et al. 1996) or a mobile defect model such that water occasionally penetrates into the core of the protein (Lumry and Rosenberg 1975; Pain et al. 1987), used to interpret the phenomena of hydrogen exchange of an internal amide group with the bulk water.
We also found that the distribution of ΔH‡ values among the three proteins is rather modest in the range of 71 to 155 kJ mol−1. In a simple picture the ΔH‡ value corresponds to the energy required to create the space or cavity in the core part of a globular protein, the observation may suggest that the infrequent structural fluctuation creating the above-mentioned cavity in the interior of a globular protein is a fairly common process. This view is supported by the observation that these ΔH‡ values roughly coincide with the common activation energies (80–160 kJ mol−1) for hydrogen exchange caused by small amplitude fluctuations of most globular proteins (Woodward et al. 1982). In conclusion, our present result and those previously reported on BPTI and cytochrome c seem to be consistent with the cavity model for the ring flip.
Materials and methods
Sample preparation
HPr from S. carnosus was expressed in E. coli and purified as described previously (Kruse et al. 1993). Freeze-dried HPr was dissolved in 200 μL 99.85% D2O, giving a HPr concentration of 1.5 mM. A small quantity of KO2H was added to the sample to adjust the pH of the solution to pH 7.57 (at 0.1 MPa and 298 K). As internal standard for chemical shift, minimum quantities of 3-(trimethylsilyl)[3,3,2,2–2H] propionate-d4 (TSP) and dioxane were added.
High-pressure NMR measurements
The technical details of the on-line cell variable-pressure NMR method employed here is described elsewhere (Akasaka and Yamada 2001; Yamada et al. 2001). The protein solution is contained in a long quartz capillary, the end of which is a body part (inner diameter of 1 mm, outer diameter of 3 mm) which sits in the coil of the NMR probe for signal detection. The pressure of the sample solution was regulated at a desired value between 3 MPa and 200 MPa with a hand pump remotely located from the 17.6 T magnet (Japan Magnet Technology). 3 MPa was chosen for the lowest pressure of instead of 0.1 MPa, to avoid any effect from air bubbles in the sample solution. A commercial 5-mm 1H-selective NMR probe (Bruker) was used for signal detection without any modification.
NMR spectra at varying pressures were measured on a Bruker DMX-750 spectrometer operating at a 1H frequency of 750.13 MHz. All one-dimensional 1H NMR spectra were recorded with a spectral width of 10 kHz and with 32768 complex data points. Three thousand seventy-two free induction decays were averaged. Water suppression was accomplished by presaturation during the relaxation delay of 1.5 sec. Chemical shifts were measured relatively to the methyl signal of (TSP at 0 ppm or to dioxane at 3.75 ppm). The separation of the two signals was invariant with pressure within experimental error. Data were processed with the XWIN-NMR package (Bruker) running on a Silicon Graphics O2 workstation.
Assignment of tyrosine hydroxyl groups
A sample of 4.3 mM unlabeled HPr in 90% H2O/10% D2O, pH 7.2 was used. NOESY spectra (Jeener et al. 1979) were recorded with a mixing time of 150 msec at a proton resonance frequency of 800.13 MHz with 1024 * 8192 time domain data points in t1 and t2 directions, respectively, and at a temperature of 298 K. Phase-sensitive detection in the t1-direction was obtained using time-proportional phase increments (TPPI) (Marion and Wüthrich 1983). The repetition time was 2.37 sec. Spectra simulations were performed using the program RELAX-JT2 (Görler and Kalbitzer 1997; Görler et al. 1999b; Ried et al. 2004) implemented in the program package AUREMOL (Gronwald and Kalbitzer 2004). As input for the simulations, one of the final three-dimensional solution structures of HPr from S. carnosus together with the corresponding resonance line assignment were used (Görler et al. 1999a). Otherwise, the same parameters, e.g., repetition time, mixing time, digital resolution, etc., that were described for the corresponding experimental spectrum were also used in the simulations. The global correlation time τc = 5.62 nsec of HPr employed in the simulations was calculated from relaxation measurements performed on uniformly 15N enriched HPr at 298 K (Schubel 2000). From the possible motional models as defined in Görler and Kalbitzer (1997) LIPARI_1 was selected for all atom pairs not including a methyl group or an aromatic ring. It represents a simplification of the original motional model defined by Lipari and Szabo (1982a,b), and it is justified in cases where one can assume that the correlation times of the fast internal motions are considerably smaller than the global correlation time. For all atom pairs containing protons from a methyl group a fast-jump model was used where it is assumed that the internal correlation time of the methyl group is much smaller than the global correlation time. For atom pairs containing members from aromatic rings where it can be assumed that the internal correlation time of the jump motion of the ring is much larger than the global correlation time a slow jump approximation was selected. For all atom pairs containing only backbone atoms an average order parameter S2 of 0.95 has been experimentally determined (Schubel 2000). For all atom pairs containing side-chain and main-chain atoms an S2 of 0.80 was used, while for side-chain side-chain interactions an S2 value of 0.65 was assumed. The latter two values were not experimentally determined but taken from the literature (Brünger 1992). Within RELAX-JT2 it is possible to automatically correct for deviations of the molecule from spherical shape. However, in case this option is activated, in the current version of RELAX-JT2 the molecule is treated as a rigid body. Since the three-dimensional structure of HPr can be approximated fairly well with a sphere, this option was not activated.
Calculation of the ring-flip rates and kinetic parameters
The ring-flip rates of Tyr6 were evaluated by simulating experimental line shapes using the density matrix formalism with the program WinDNMR (Reich 1995), taking explicitly the effect of coupling constants into account. The viscosity and temperature dependent change of the T2-relaxation was introduced in the calculation by assuming as a first-order correction for the effects of temperature T and pressure p on the exchange-independent part T2 (T,p) of the transversal relaxation time by
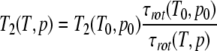 |
(1) |
Here, T0 and p0 are a reference temperature and pressure and τrot the rotational correlation time. When shape changes of the protein are neglected, for the pressure and temperature dependence of τrot holds
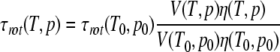 |
(2) |
with V the volume of the protein. T2 can then be approximated as
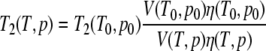 |
(3) |
The viscosity change with temperature or pressure can be obtained from the change of the transversal relaxation times T2i of other aromatic residues, which show no exchange effects. Especially suited are the well-resolved Hδ-resonance of Phe29 and the Hɛ-resonance of Tyr64. The relative viscosity changes can then determined as
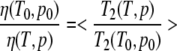 |
(4) |
Before the fit of the data the noise level was decreased by multiplying the FID with an exponential filter leading to an additional line broadening of 1 Hz. The difference in the activation free energy ΔG‡, the activation enthalpy ΔH‡, the activation entropy ΔS‡, and the activation volumes ΔV‡ were obtained by fitting the temperature dependence and pressure dependence of the exchange (or the flip) rate constants k1, −1 to the Eyring equation with
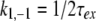 |
(5) |
and
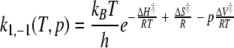 |
(6) |
with kB Boltzmann’s constant, h Planck’s, and R the gas constant. For the fit of the data the coupling constant 3JHδ–Hɛ was taken as 7.4 Hz.
Amide exchange rates
Amide exchange rates were calculated from a series of 1H,15N HSQC spectra of freeze-dried, uniformly 15N enriched HPr protein from S. carnosus dissolved in D2O at pH 7.2. Spectra were recorded at 298 K, the pseudo first-order experimental exchange rate constants kexp,i of the amino acids I were obtained from an exponential fit. Protection factors pi were calculated as
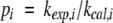 |
(7) |
with kcal,I the exchange rate constants calculated according to Bai et al. (1993).
Acknowledgments
This work was supported by STA (to M.H.), the DAAD (H.R.K), and JSPS (K.A.).
Abbreviations
DSS, 4,4-dimethyl-4-silapentane-sulphonic acid
HPr, histidine-containing phosphocarrier protein
HSQC, heteronuclear single quantum coherence
NOE, nuclear Overhauser effect
NOESY, nuclear Overhauser effect spectroscopy
PEP, phosphoenolpyruvate
PTS, phosphoenolpyruvate-dependent phosphotransferase system
S. carnosus, Staphylococcus carnosus
S. aureus, Staphylococcus aureus
TPPI, time proportional phase incrementation
TSP, 3-(trimethylsilyl)[3,3,2,2-2H] propionate-d4
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.04877104.
References
- Akasaka, K. and Li, H. 2001. Low-lying excited states of proteins from nonlinear pressure shifts in 1H and 15N NMR. Biochemistry 40 8665–8671. [DOI] [PubMed] [Google Scholar]
- Akasaka, K. and Yamada, H. 2001. On-line cell high pressure nuclear magnetic resonance technique: Application to protein studies. In Nuclear magnetic resonance of biological macromolecules, Part A, Methods in enzymology, Vol. 338 (eds. T.L. James et al.), pp. 134–158. Academic Press, New York. [DOI] [PubMed]
- Arnold, M.R., Kremer, W., Luedemann, H.-D., and Kalbitzer, H.R. 2002. 1H NMR parameters of common amino acid residues measured in aqueous solutions of the linear tetrapeptides Gly-Gly-X-Ala at pressures between 0.1 and 200 MPa. Biophys. Chem. 96 129–140. [DOI] [PubMed] [Google Scholar]
- Arnold, M.R., Kalbitzer, H.R., and Kremer, W. 2003. High-sensitivity sapphire cells for high pressure NMR spectroscopy on proteins. J. Magn. Reson. 61 127–131. [DOI] [PubMed] [Google Scholar]
- Bai, Y., Milne, J.S., Mayne, L., and Englander, S.W. 1993. Primary structure effects on peptide group hydrogen exchange. Proteins 17 75–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brünger, A.T. 1992. X-PLOR version 3.1. Yale University Press, New Haven, CT.
- Bundi, A. and Wüthrich, K. 1979. 1H-NMR parameters of the common amino acid residues measured in aqueous solutions of the linear tetrapeptides H-Gly-Gly-X-L-Ala-OH. Biopolymers 18 285–297. [Google Scholar]
- Campbell, I.D., Dobson, C.M., and Williams, R.J. 1975. Proton magnetic resonance studies of the tyrosine residues of hen lysozyme-assignment and conformational mobility. Proc. R. Soc. Lond. Biol. 189 503–509. [DOI] [PubMed] [Google Scholar]
- Campbell, I.D., Dobson, C.M., Moore, G.R., Perkins, S.J., and Williams, R.J.P. 1976. Temperature dependent molecular motion of a tyrosine residue of ferrocytochrome C. FEBS Lett. 70 96–100. [DOI] [PubMed] [Google Scholar]
- Dobson, C.M., Moore, G.R., and Williams, R.J.P. 1975. Assignment of aromatic amino acid PMR resonances of horse ferricytochrome c. FEBS Lett. 51 60–65. [DOI] [PubMed] [Google Scholar]
- Englander, S.W. and Mayne, L. 1992. Protein folding studied using hydrogen-exchange labeling and two-dimensional NMR. Annu. Rev. Biophys. Biomol. Struct. 21 243–265. [DOI] [PubMed] [Google Scholar]
- Frans, A., Mulder, A., Mittermaier, A., Hon, B., Dahlquist, F.W., and Kay, L.E. 2001. Studying excited states of proteins by NMR spectroscopy. Nat. Struct. Biol. 8 932–935. [DOI] [PubMed] [Google Scholar]
- Görler A. and Kalbitzer, H.R. 1997. RELAX: A flexible program for the analysis of NOESY-spectra by back calculation based on the complete relaxation matrix formalism. J. Magn. Reson. 124 177–188. [DOI] [PubMed] [Google Scholar]
- Görler, A., Hengstenberg, W., Kravanja, M., Beneicke, W., Maurer, T., and Kalbitzer H.R. 1999a. Solution structure of the histidine-containing phosphocarrier protein from Staphylococcus carnosus. Appl. Magn. Reson. 17 465–480. [Google Scholar]
- Görler, A., Gronwald, W., Neidig, K.-P., and Kalbitzer, H.R. 1999b. Computer assisted assignment of 13C or 15N edited 3D-NOESY-HSQC spectra using back calculated and experimental spectra. J. Magn. Reson. 137 39–45. [DOI] [PubMed] [Google Scholar]
- Gronwald, W. and Kalbitzer, H.R. 2004. Automated structure determination of proteins by NMR spectroscopy. Progr. NMR Spectros. 44 33–96. [Google Scholar]
- Hetzel, R., Wüthrich, K., Deisenhofer, J., and Huber, H. 1976. Dynamics of the aromatic amino acid residues in globular confirmation of the basic pancreatic trypsin inhibitor (BPTI). II. Semi-empirical energy calculation. Bio-phys. Struct. Mech. 2 159–180. [DOI] [PubMed] [Google Scholar]
- Ishima, R. and Torchia, D.A. 2000. Protein dynamics from NMR. Nat. Struct. Biol. 7 740–743. [DOI] [PubMed] [Google Scholar]
- Jeener, J., Meier, B.H., Bachmann, P., and Ernst, R.R. 1979. Investigation of exchange processes by two-dimensional NMR spectroscopy. J. Chem. Phys. 71 4546–4553. [Google Scholar]
- Kalbitzer, H.R., Görler, A., Li, H., Dubovskii, P., Hengstenberg, W., Kowolik, C., Yamada, H., and Akasaka, K. 2000. 15N and 1H NMR study of histidine containing protein (HPr) from Staphylococcus carnosus at high pressure. Protein Sci. 9 693–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karplus, M. and McCammon, A. 1981. Pressure dependence of aromatic ring rotations in proteins: A collisional interpretation. FEBS Lett. 131 34–36. [Google Scholar]
- Kocher, J.P., Prevost, M., Wodak, S.J., and Lee, B. 1996. Properties of the protein matrix revealed by the free energy of cavity formation. Structure 4 1517–1529. [DOI] [PubMed] [Google Scholar]
- Koradi, R., Billeter, M., and Wüthrich, K. 1996. MOLMOL: A program for display and analysis of macromolecular structures. J. Mol. Graphics 14 51–55. [DOI] [PubMed] [Google Scholar]
- Kramers, H.A. 1940. Brownian motion in a field of force and the diffusion model of chemical reactions. Physica 7 285–304. [Google Scholar]
- Kruse, R., Hengstenberg, W., Beneicke, W., and Kalbitzer, H.R. 1993. Involvement of various amino- and carboxyl-terminal residues in the active site of the histidine-containing protein HPr of the phosphoenolpyruvate-dependent phosphotransferase system of Staphylococcus carnosus: Site-directed mutagenesis with the ptsH gene, biochemical characterization and NMR studies of the mutant proteins. Protein Eng. 6 417–423. [DOI] [PubMed] [Google Scholar]
- Li, H., Yamada, H., and Akasaka, K. 1998. Effect of pressure on individual hydrogen bonds in proteins. Basic pancreatic trypsin inhibitor. Biochemistry 37 1167–1173. [DOI] [PubMed] [Google Scholar]
- ———. 1999. Effect of pressure on the tertiary structure and dynamics of folded basic pancreatic trypsin inhibitor. Biophys. J. 77 2801–2812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipari, G. and Szabo, A. 1982a. Model-free approach to the interpretation of nuclear magnetic resonance relaxation in macromolecules. 1. Theory and range of validity. J. Am. Chem. Soc. 104 4546–4559. [Google Scholar]
- ———. 1982b. Model-free approach to the interpretation of nuclear magnetic resonance relaxation in macromolecules. 2. Analysis of experimental results. J. Am. Chem. Soc. 104 4559–4570. [Google Scholar]
- Lumry, R. and Rosenberg, A. 1975. The mobile defect hypothesis of protein function. Coll. Int. CNRS L’Eau. Syst. Biol. 246 55–63. [Google Scholar]
- Marion, D. and Wüthrich, K. 1983. Application of phase-sensitive two-dimensional correlated spectroscopy (COSY) for measurements of 1H-1H spin-spin coupling constants in proteins. Biochem. Biophys. Res. Commun. 113 967–974. [DOI] [PubMed] [Google Scholar]
- Meiler, J.M., Peti, W., and Griesinger, C. 2003. Dipolar couplings in multiple alignments suggest helical motion in ubiquitin. J. Am. Chem. Soc. 125 8072–8073. [DOI] [PubMed] [Google Scholar]
- Otting, G., Liepinsh, E., and Wüthrich, K. 1993. Disulfide bond isomerization in BPTI and BPT(G36S): An NMR study of correlated mobility in proteins. Biochemistry 13 3571–3582. [DOI] [PubMed] [Google Scholar]
- Pain, R.H. 1987. Protein structure. New light on old defects. Nature 326 247. [DOI] [PubMed] [Google Scholar]
- Postma, P.W., Lengeler, J.W., and Jacobson, G.R. 1993. Phosphoenolpyruvate: carbohydrate phosphotransferase systems of bacteria. Microbiol. Rev. 57 543–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reich, J.P. 1995. WinDNMR: Dynamic spectra for Windows. J. Chem. Educ. 72 1086. [Google Scholar]
- Ried, A., Gronwald, W., Trenner, J., Brunner, K., Neidig, K.-P., and Kalbitzer, H.R. 2004. Improved simulation of NOESY spectra by RELAX-JT2 including effects of J-coupling, tranverse relaxation, and chemical shift anisotropy. J. Biomol. NMR (in press). [DOI] [PubMed]
- Rösch, P., Kalbitzer, H.R., Schmidt-Aderjan, U., and Hengstenberg, W. 1981. 1H nuclear magnetic resonance studies of the structure and mechanism of HPr protein of Staphyloccoccus aureus. Biochemistry 20 1599–1605. [DOI] [PubMed] [Google Scholar]
- Schubel, U. 2000. “Untersuchungen zur Dynamik des HPr-Proteins von Staphylococcus carnosus mit Hilfe von heteronuclearen Kernoverhausereffekt-und Relaxationszeitmessungen.” Diploma thesis, University of Regensburg, Regensburg, Germany.
- Wagner, G. 1980. Activation volumes for the rotational motion of interior aromatic rings in globular proteins determined by high resolution 1H NMR at variable pressure. FEBS Lett. 112 280–284. [DOI] [PubMed] [Google Scholar]
- ———. 1983. Characterization of the distribution of internal motions in the basic pancreatic trypsin inhibitor using a large number of internal NMR probes. Q. Rev. Biophys. 16 1–57. [DOI] [PubMed] [Google Scholar]
- Wagner, G., DeMarco, A., and Wüthrich, K. 1976. Dynamics of the aromatic amino acid residues in the globular conformation of the basic pancreatic trypsin inhibitor (BPTI). I. 1H NMR studies. Biophys. Struct. Mech. 2 139–158. [DOI] [PubMed] [Google Scholar]
- Woodward, C.K., Simon, I., and Tuechsen, E. 1982. Hydrogen exchange and the dynamic structure of proteins. Mol. Cell. Biochem. 48 135–160. [DOI] [PubMed] [Google Scholar]
- Woodward, C.K., Carulla, N., and Barany, G. 2004. Native state hydrogen exchange analysis of protein folding and protein motional dynamics. Methods Enzymol. 380 379–400. [DOI] [PubMed] [Google Scholar]
- Wüthrich, K. and Wagner, G. 1975. NMR investigations of the dynamics of the aromatic residues in the basic pancreatic inhibitor. FEBS Lett. 50 265–268. [DOI] [PubMed] [Google Scholar]
- ———. 1978. Internal motions in globular proteins. Trends Biosci. 3 227–230. [Google Scholar]
- Yamada, H., Nishikawa, K., Honda, M., Shimura, T., Akasaka, K., and Tabayashi, K. 2001. Pressure-resisting cell for high-pressure, high-resolution nuclear magnetic resonance measurements at very high magnetic fields. Rev. Sci. Instrum. 72 1463–1471. [Google Scholar]