

Abstract
The Escherichia coli heat-shock protein ClpB reactivates protein aggregates in cooperation with the DnaK chaperone system. The ClpB N-terminal domain plays an important role in the chaperone activity, but its mechanism remains unknown. In this study, we investigated the effect of the ClpB N-terminal domain on malate dehydrogenase (MDH) refolding. ClpB reduced the yield of MDH refolding by a strong interaction with the intermediate. However, the refolding kinetics was not affected by deletion of the ClpB N-terminal domain (ClpBΔN), indicating that MDH refolding was affected by interaction with the N-terminal domain. In addition, the MDH refolding yield increased 50% in the presence of the ClpB N-terminal fragment (ClpBN). Fluorescence polarization analysis showed that this chaperone-like activity is explained best by a weak interaction between ClpBN and the reversible aggregate of MDH. The dissociation constant of ClpBN and the reversible aggregate was estimated as 45 μM from the calculation of the refolding kinetics. Amino acid substitutions at Leu 97 and Leu 110 on the ClpBN surface reduced the chaperone-like activity and the affinity to the substrate. In addition, these residues are involved in stimulation of ATPase activity in ClpB. Thus, Leu 97 and Leu 110 are responsible for the substrate recognition and the regulation of ATP-induced ClpB conformational change.
Keywords: molecular chaperone, ClpB, chaperone-like activity, protein aggregate, refolding, amino acid substitution, substrate binding site
Heat-shock protein ClpB is a member of the HSP100 family of Escherichia coli heat-shock proteins, and functions in cooperation with the DnaK molecular chaperone system to solubilize protein aggregates and reactivate them (Glover and Lindquist 1998; Goloubinoff et al. 1999; Zolkiewski 1999). ClpB binds denatured protein aggregates, and this binding can stimulate ClpB ATPase activity. ATP-induced ClpB conformational change exposes hydrophobic side chains, thereby allowing DnaK binding and protein refolding (Goloubinoff et al. 1999). The detailed structure of ClpB has been determined and a hexameric (Zolkiewski et al. 1999) or heptameric (Kim et al. 2000) ring-shape structure has been proposed for ClpB assembly. The ClpB sequence contains two ATP-binding domains (NBD1 and NBD2) between N-terminal and C-terminal domains. NBD1 and NBD2 are separated by a linker domain that is critical for chaperone function (Kedzierska et al. 2003; Lee et al. 2003). The N-terminal domain (Barnett et al. 2000; Tek and Zolkiewski 2002), C-terminal domain (Clarke and Eriksson 2000; Cashikar et al. 2002), and linker domain (Kedzierska et al. 2003; Lee et al. 2003) are involved in substrate binding. The crystal structure of Thermus thermophilus ClpB (TClpB) showed that the linker domain is an 85 Å long, leucine-rich coiled-coil peptide resembling a two-bladed propeller (Lee et al. 2003). It has been suggested that large protein aggregates bind between the linker domains of adjacent ClpB molecules and the ATP-induced conformational change of ClpB causes dissociation of the aggregates into smaller size aggregates.
The ClpB N-terminal domain contains about 150 amino acid residues, including two repeated structural domains each of about 70 amino acid residue (Lo et al. 2001). The crystal structure of TClpB showed a large rearrangement of the N-terminal domain, demonstrating that this domain is mobile and does not form a tight interface with the rest of the molecule (Lee et al. 2003). The function of the ClpB N-terminal domain and the nature of the protein aggregates that bind to this domain have not been determined. The N-terminal domain of a Synechococcus ClpB homolog is essential for cell viability (Eriksson et al. 2001), while other studies have indicated that this domain is not essential for resolubilization of protein aggregates and development of thermotolerance (Clarke and Eriksson 2002; Mogk et al. 2003).
The effect of deletion of the N-terminal domain on luciferase refolding in the presence of the DnaK chaperone system was studied (Barnett et al. 2000). When ClpB was present in the refolding buffer, almost 100% of luciferase was refolded, although the refolding yield was only 1%–2% in the presence of ClpB protein with a deleted N-terminal domain (ClpBΔN). Other studies showed that substitution of amino acids in the N-terminal domain shown in Figure 1 ▶ affected chaperone activity of full-length ClpB. Substitution of the conserved residues Thr 7, Asp 103, and Glu 109 with Ala did not affect substrate binding, but the chaperone activity for luciferase refolding was reduced (Liu et al. 2002). Leu 93, Leu 97, and Leu 110 in the hydrophobic groove from helices A5, A6, and A8 were substituted with Gln, and these mutants exhibited severe defects in chaperone activity (Li and Sha 2003). In contrast, TClpB and TClpBΔN showed the same chaperone activity for luciferase refolding from the urea-denatured state (Beinker et al. 2002) suggesting that the TClpB N-terminal domain was not essential for TClpB chaperone activity.
Figure 1.

Three-dimensional structure of ClpBN (PDB code 1KHY; Li and Sha 2003). (A) T7, S84, D103, and E109 substituted to Ala, are shown in the ball-and-stick model in red. (B) L93, L97, and L110 substituted to Gln are shown in the ball-and-stick model in yellow; L14 and L91 substituted to Gln are shown in the ball-and-stick model in green. All figures were drawn with MOLSCRIPT (Kraulis 1991) and Raster3D (Merritt and Bacon 1997).
To investigate the role of the N-terminal domain in ClpB chaperone activity, we have studied the effects of the N-terminal domain on the refolding kinetics of denatured malate dehydrogenase (MDH). We found that ClpB N-terminal domain affected the refolding yield through the interaction to the intermediate of MDH, and discuss the role of this interaction in ClpB activity.
Results and Discussion
Effect of ClpB on refolding of chemically denatured proteins
The effects of ClpB on the refolding of luciferase and MDH from the chemically denatured state were investigated. The effect of HSP104 (the eukaryotic homolog of HSP100) on the refolding of luciferase from the urea-denatured state has been studied previously (Glover and Lindquist 1998). In these studies, denatured luciferase was diluted to 20 nM in refolding buffer, with and without HSP104 at 30°C. In the presence of HSP104, the refolding yield was slightly reduced, but was slightly increased when ATP was also present. GdnHCl was not used as the unfolding agent because residual GdnHCl in the refolding buffer inhibits ClpB ATPase activity (Glover and Lindquist 1998; Jung and Masison 2001). Quantitative analysis was not done in these studies, probably because the yield of spontaneous luciferase refolding from the urea-denatured state was only about 2%. In contrast, we investigated the effect of ClpB on luciferase refolding from the GdnHCl denatured state using optimized refolding conditions, low protein concentration and low temperature (Herbst et al. 1998), in which the maximum yield of luciferase was about 50%. The open circles plotted in Figure 2A ▶ show the refolding kinetics of luciferase dentatured by 5 M GdnHCl. The yield of spontaneous luciferase refolding was 22% after 6 h incubation, which is in the same range as previously reported. We examined the effect of ClpB on the luciferase refolding in the absence of ATP. As shown in the closed circles in Figure 2A ▶, the refolding rate of luciferase was reduced in the presence of ClpB.
Figure 2.
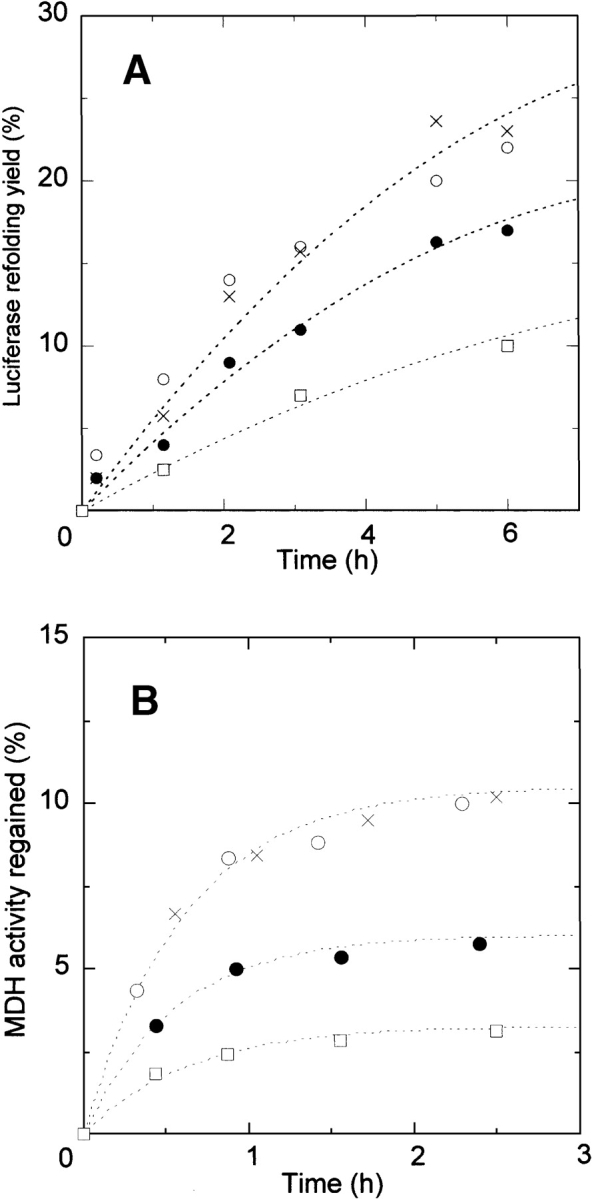
Effect of ClpB and ClpBΔN on protein refolding kinetics from the GuHCl-denatured state. (A) Refolding of luciferase denatured by 5 M GdnHCl. The final luciferase concentration was 61 nM. (B) Refolding of MDH denatured by 3 M GdnHCl. The final MDH monomer concentration was 1.4 μM. Refolding control (○), in the presence of ClpB (•), in the presence of ClpB and 10mM ATP (□), and in the presence of ClpBΔN (×). The concentrations of ClpB and ClpBΔN were 0.3 μM (hexamer).
The same experiments were carried out using the dimeric enzyme MDH, for which the detailed refolding mechanism has been determined (Ranson et al. 1995). The open circles plotted in Figure 2B ▶ show the refolding kinetics of MDH denatured by 3 M GdnHCl. The yield of spontaneous MDH refolding was about 10% after 2.5 h incubation, which is in the same range as previously reported. The lower yield of MDH refolding is due to a branch in the refolding reaction path to a reversible aggregate and then to an irreversible aggregate (Ranson et al. 1995), as shown in equation 1:
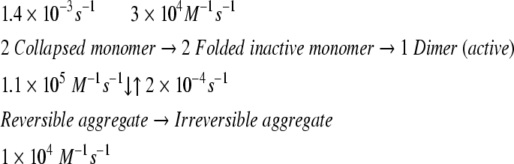 |
(1) |
It has been shown that the GroEL molecular chaperone system (2 μM GroEL, 8 μM GroES, and 10 mM ATP) increases the MDH refolding yield by a factor of 6.5, due to the interaction with the reversible aggregation (Ranson et al. 1995). Substoichiometric amounts of GroEL efficiently increased the MDH refolding yield, indicating that GroEL catalytically reverses the reversible aggregation step, and the rate constant from the intermediate to the collapsed monomer increased to 0.1 sec−1. We examined the effect of ClpB on the MDH refolding in the absence of ATP. As shown in the closed circles plotted in Figure 2B ▶, ClpB interfered with the refolding of MDH as in the case of the luciferase refolding. Previous studies also reported that GroEL alone interfered with the protein refolding in the particular conditions (Buchner et al. 1991; Schmidt and Buchner 1992). This is because the substrate polypeptide, which tightly bound to GroEL in refolding process, was not released from GroEL in the absence of ATP and GroES. Similarly, the effect of ClpB on the protein refolding would be explained by the tight binding to the intermediates of MDH (folded inactive monomer or reversible aggregate) as shown in equation 2:
 |
(2) |
Previous study showed that the self-association of ClpB is weak in the absence of ATP (Zolkiewski et al. 1999), and not only hexamer, but smaller oligomers and monomer (Beinker et al. 2002) are also populated in the ClpB solution in our experimental condition. The higher oligomers would be responsible to the tight binding of the MDH intermediates because the substrate affinity of lower oligomers and monomer should be low due to the smaller number of the binding sites. We have performed the refolding experiments in the presence of ClpB and 10 mM ATP. As shown in the squares plotted in Figure 2 ▶, A and B, we found that the refolding yields of luciferase and MDH were further reduced. This is a contrast to the fact that the protein refolding yield was slightly increased by HSP104 in the presence of ATP (Glover and Lindquist 1998). However, these results cannot be compared because residual GdnHCl in the refolding buffer inhibits ClpB ATPase activity (Glover and Lindquist 1998; Jung and Masison 2001) and the conformational change in ClpB is not induced in our conditions. Therefore, the reduced refolding yield in the presence of ATP would be due to the increased population of higher oligomers in ClpB. This is consistent to our explanation that higher oligomers are involved in the tight binding to the MDH intermediates. The effect of ClpB on the heat-induced MDH aggregate in the presence and absence of ATP has been studied previously (Goloubinoff et al. 1999). Turbidity of an MDH aggregate solution was monitored before and after ClpB addition. In contrast to the studies reported here, those studies found that ClpB alone did not significantly affect MDH aggregates. Instead, turbidity increased in the presence of ClpB and ATP. This turbidity increase may be due to fragmentation of large aggregates by ATP-induced conformational changes.
To examine the contribution of the ClpB N-terminal domain to ClpB binding to the MDH intermediates, we studied the refolding of MDH and luciferase in the presence of ClpBΔN. Neither the refolding kinetics of luciferase nor MDH was affected by ClpBΔN (the crosses plotted in Fig. 2A,B ▶), indicating that deletion of the ClpB N-terminal domain reduces ClpB interaction with the MDH intermediates. When interaction between ClpB and the MDH intermediates is reduced by deletion of the ClpB N-terminal domain, the ClpBΔN oligomer may dissociate thereby decreasing the number of the binding sites. This may explain the reason for the lack of ClpBΔN-intermediates interaction. However, a previous study has shown that self-association of ClpBΔN is stronger than that of wild-type ClpB (Barnett et al. 2000), so reduced ClpBΔN-intermediates interaction cannot be due to ClpBΔN oligomer dissociation. Therefore, we conclude that the ClpB N-terminal domain is responsible for ClpB binding to the MDH intermediates.
Previous studies reported that the chaperone activity of ClpB was significantly reduced by the deletion of N-terminal domain (Barnett et al. 2000), while TClpB and TClpBΔN showed the same chaperone activity (Beinker et al. 2002). However, these results cannot be simply compared because different substrate concentrations were used in each study (25 nM for ClpB study, 180 nM for TClpB study) and luciferase aggregation is a function of protein concentration (Herbst et al. 1997). A larger fraction of the luciferase might be in larger size aggregates and recognized by the ClpB linker domain in the higher luciferase concentration in the TClpB study. This possibility is consistent with the result that the maximum refolding yield in the TClpB study (40%) is much lower than that in the ClpB study (100%). The function of the ClpB N-terminal domain might be more easily observed in conditions of low protein concentration, in which a larger fraction of luciferase is in smaller size aggregates. Another study showed that the ClpB N-terminal domain is not essential for dissociation of aggregates of luciferase and MDH that had been heat treated (Mogk et al. 2003). This effect may be due to a larger fraction of the aggregates produced by heat treatment having a larger size and to the function of the ClpB linker domain.
Effect of ClpBN on MDH refolding
We examined the effect of ClpBN on the refolding of MDH denatured by GdnHCl. In contrast to the effect of ClpB, the MDH refolding yield increased about 50% in the presence of 40 μM ClpBN (Fig. 3A ▶), and increased with increasing concentrations of ClpBN (Fig. 3B ▶). To examine the mechanism of this chaperone-like activity, the MDH refolding experiments were performed in the presence of polyethylene glycol or BSA, which have been reported to prevent the protein aggregation (Jaenicke and Rudolph 1986; Cleland and Randolph 1992) by the nonspecific binding to the folding intermediate. We found that polyethylene glycol did not affect the refolding kinetics of MDH in the experimental condition in Figure 3 ▶. On the other hand, the MDH refolding yield was decreased with the increase of the BSA concentration from 0 to 40 μM in the same condition. The conformation of serum albumin is a sticky partially folded form in reduced condition (Lee and Hirose 1992), and its tight binding to MDH intermediates might be responsible for the reduced refolding yield. These results are distinct from the effect of ClpBN on MDH refolding, and the mechanism of chaperone-like activity of ClpBN cannot be explained by the nonspecific binding. However, unlike the effect of GroEL on MDH refolding, ClpBN needs to be present in greater than equimolar quantities relative to the aggregate substrate for its chaperone-like activity, and the mechanism cannot be explained by ClpBN catalyzing dissociation of reversible MDH aggregates. Previous studies found that molecular chaperones facilitate protein refolding without ATP-induced conformational change by the weak binding to folding intermediates to prevent aggregation (Ayling and Baneyx 1996; Zahn et al. 1996; Ben-Zvi et al. 1998; Chatellier et al. 1998; Tanaka and Fersht 1999; Tanaka et al. 2002). The ClpBN chaperone-like activity may be explained by a similar mechanism. We confirmed that ClpBN binds MDH in the refolding process using fluorescence anisotropy assays (Fig. 4 ▶). An increase in measured anisotropy as a function of macromolecule concentration is a measure of macromolecule binding (Tanaka and Fersht 1999). The ClpBN N-terminal amino acid was labeled with FITC (FITC-ClpBN) and anisotropy measured in the presence of different concentrations of refolded MDH. The anisotropy value of FITC-ClpBN increased immediately after the diluting denatured MDH (within 20 sec), and kept almost the same value for several minutes. The change of the measured anisotropy of FITC-ClpBN increased with increasing MDH concentration (Fig. 4 ▶), indicating binding between MDH and ClpBN.
Figure 3.
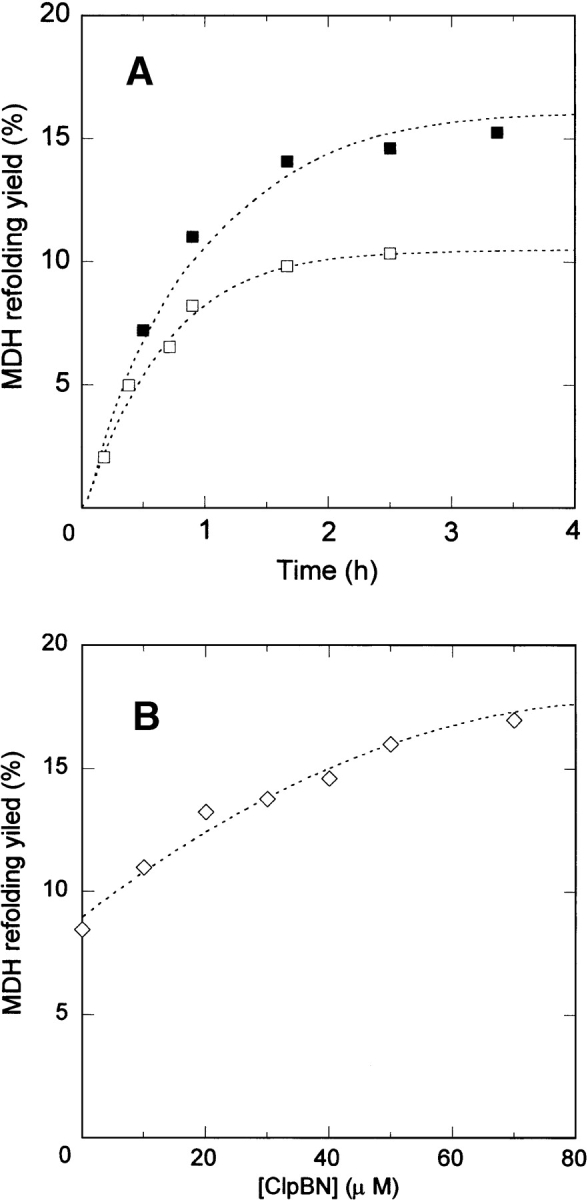
Effect of ClpBN on MDH refolding. (A) Refolding kinetics of MDH in the presence and absence of 40 μM ClpBN. Lines through the data points were calculated from the best fit for the reaction model in equation 3. Refolding control (□) and in the presence of ClpBN (▪). (B) Yield of MDH refolding as a function of ClpBN concentration. The extent of the reactivation was measured 2.5 h after refolding.
Figure 4.

Effect of MDH on fluorescence anisotropy of FITC-ClpBN. Different concentrations of MDH denatured by 3 M GdnHCl were diluted 100-fold in refolding buffer with 0.1 μM FITC-ClpBN. Wild-type ClpBN (○), ClpBNL93Q (×), ClpBNL97Q (□), and ClpBNL110Q (▪). The stable anisotropy value was obtained less than 1 min after refolding. The measured increase from the initial anisotropy value (ΔAnisotoropy) is plotted as a function of the final MDH monomer concentration. Refolding conditions were the same as Figure 2B ▶.
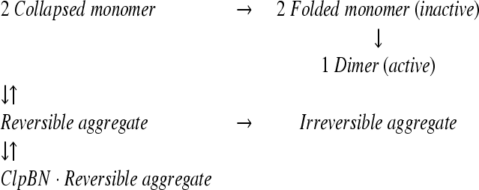
To identify MDH intermediate that binds to ClpBN, the concentration of MDH intermediates in the refolding process was calculated using the kinetic constants determined for MDH refolding kinetics (Ranson et al. 1995). The concentration of the reversible aggregate increased immediately after the initiation of refolding, reached to the maximum (0.5 μM) after 10 sec, and then decreased slowly. This behavior is similar to the change of anisotropy value of FITC-ClpBN in the process of MDH refolding. On the other hand, the concentration of folded monomer slowly increased and reached to its maximum concentration (25 nM) after 4 min. These results indicate that the anisotropy of FITC-ClpBN was increased by the binding with the reversible aggregate of MDH. Therefore, the increased yield of MDH refolding is explained best by the fact that ClpBN interacts with the reversible aggregate MDH, and we propose equation 3 as a mechanism for ClpBN chaperone-like activity on MDH refolding:
 |
(3) |
The kinetics of MDH refolding in the presence of ClpBN shown in equation 3 was calculated, using the kinetic constants shown in equation 1. The value of the dissociation constant (Kd) between the reversible aggregate and ClpBN was determined by numerical analysis of the data in Figure 3A ▶. The theoretical curve showed the best fit to the data for Kd = 45 μM. The affinity of ClpBN to the MDH intermediate is much lower than that of N-terminal domain of ClpB because of the lower number of the binding sites. This would be the reason why the effect of ClpBN on the MDH refolding is opposite to that of ClpB. We have previously investigated the substrate interaction of the molecular chaperone GroEL using its fragment of the substrate binding domain (Tanaka and Fersht 1999). The affinity between the substrate and the fragment of the substrate binding domain was low (Kd = 100–200 μM). Substrate affinity of the GroEL oligomer heptameric ring enables tight substrate binding and recognition of a diverse range of substrates (Ben-Zvi et al. 1998). Similarly, the ClpB N-terminal domain may recognize protein aggregates by the accumulation of weak substrate interactions at multiple sites.
Identification of the ClpB N-terminal domain substrate binding site
To identify the ClpBN residues at the substrate binding site, we examined the effect of ClpBN mutants on MHD refolding. The seven mutated residues in ClpB, performed previously (Liu et al. 2002; Li and Sha 2003), were produced in ClpBN by site-directed mutagenesis. In addition, because Leu 14 and Leu 91 are conserved in the ClpB family (Fig. 1B ▶) and are candidates for the substrate binding site, these two amino acids were changed by mutation to Gln in ClpBN. Therefore, we carried out site-directed mutagenesis on the nine amino acids on the molecular surface shown in Figure 1 ▶. No observable difference was found between the CD spectra of wild-type ClpBN and its mutants (Fig. 5 ▶). Experiments of MDH refolding from the GdnHCl-denatured state were carried out in the presence of 30 μM ClpBN wild-type or mutant protein. The relative values for the MDH refolding yields after 2.5 h incubation are summarized in Figure 6 ▶. This figure shows that the MDH refolding yields in the presence of ClpBNL97Q and ClpBNL110Q were lower than wild-type, suggesting reduced interaction of these mutants with the substrate. The refolding yields in the presence of other mutants were in the same level or slightly higher than the wild type. The increased chaperone-like activity may be because the mutation induced small conformational changes thereby changing the orientation of residues at the substrate binding site. We have performed the fluorescence anisotropy assays to confirm that the mutations affect the interaction between ClpBN and MDH. As shown in Figure 4 ▶, an increase in measured anisotropy was significantly reduced in ClpBNL97Q and ClpBNL110Q, indicating that the substrate binding was reduced in these mutants.
Figure 5.

CD spectra of ClpBN and its mutants (T7A, L97Q, and L110Q). The CD spectra of ClpBN mutants not shown in this figure (L14Q, L91Q, S84A, L93Q, D103A, and E109A) were also identical to that of wild-type ClpBN.
Figure 6.

Effect of ClpBN mutants on MDH refolding from the GuHCl-denatured state. The refolding yield of MDH was measured after 2.5 h incubation in the presence 30 μM ClpBN mutants. Refolding yields are shown relative to the value in the absence of ClpBN. Refolding conditions were the same as Figure 2B ▶.
As shown in Figure 7 ▶, we performed the DSC measurements of these mutants to examine the effect of the mutations on the stability of the N-terminal domain. The thermal unfolding of ClpBN is well approximated by a two-state conformational transition with the mid-point temperature of 73.1°C, as previously reported (Tek and Zolkiewski 2002). The shape of thermal transitions of ClpBL97Q and ClpBL110Q was similar to that of wild type, but their transition temperatures were 66.8°C and 65.8°C, respectively. Therefore, Leu 97 and Leu 110 play a role in stabilizing the ClpBN conformation, and this may explain the reason for the reduced chaperone-like activity. However, the heat absorbance of the unfolding of ClpBL97Q and ClpBL110Q occurred above 55°C, and the destabilization with these mutations does not affect the chaperone-like activity of ClpBN observed in the present experiments. Therefore, Leu 97 and Leu 110 are involved in the substrate binding, and play an important role in the chaperone-like activity in ClpBN. In addition, Figure 4 ▶ shows that L93Q mutation in ClpBN also reduced the substrate affinity in smaller extent. The substrate affinity of ClpBNL14Q and ClpBNL91Q also reduced in the same level as ClpBNL93Q (data not shown). However, the MDH refolding yields was not significantly affected by these mutations, suggesting these residues may play an assistant role in the substrate binding. The substrate affinity of other mutants observed through the fluorescence anisotropy was almost in the same range as that of wild type (data not shown).
Figure 7.

The temperature dependence of the partial heat capacity of the fragment of ClpBN and its mutants (L93Q, L97Q, and L110Q). The buffer scan and a linear baseline in the region of the thermal transition were subtracted from data. For illustrative purposes, the data sets have been offset below the ClpBN data set.
Our result is consistent with the previous result of the effect of single mutations on chaperone activity of full-length ClpB (Li and Sha 2003). The mutant L97Q lost about 75% of its ability as a molecular chaperone, and L110Q lost about 30% of the wild-type molecular chaperone. In addition, the stimulation of ATPase activity of full-length ClpB induced by polypeptide binding was also affected by these mutations. These effects of mutations on the activity of full-length ClpB are explained best by the reduced substrate binding from our results. L93Q has been reported to lose almost 100% of the chaperone activity without change in the stimulation of ATPase activity induced by polypeptide binding. On the other hand, our result showed that the substrate binding in the N-terminal domain was not completely lost by L93Q mutation. Therefore, Leu 93 would be responsible for other function, such as supporting conformational rearrangement in ClpB and/or in a protein substrate (Liu et al. 2002), which is significantly affected by L93Q mutation.
In the studies reported here, we found that the refolding yield of MDH was reduced in the presence of full-length ClpB, whereas ClpBN increased the refolding yield. Fluorescence polarization analysis showed that these results were explained best by the fact that ClpB N-terminal domain interacts with the reversible aggregate of MDH in the refolding process. Interaction between the N-terminal domain and substrate is weak, but is enhanced by multiple binding sites in ClpB oligomers. We found that Leu 97 and Leu 110 in the ClpB N-terminal domain hydrophobic groove are involved in the binding of the protein aggregates.
Materials and methods
Materials
E. coli strain MC1009/pClpB (Squires et al. 1991) was generously provided by Dr. Catherine Squires (Columbia University). Expression vectors for CLpB, ClpBΔN, and ClpBN were generously provided by Prof. Zolkieski (Kansas State University). ClpB and ClpBΔN expression and purification were performed as described previously (Barnett et al. 2000). A QuikChange site-directed mutagenesis kit (Stratagene) was used to generate desired mutations into target genes (using a pair of completely complementary primers) and to amplify the full-length plasmid. ClpBN and its mutants were expressed in E. coli BL21(DE3)plys, purified by affinity chromatography on a Ni-NTA-agarose column and cleaved from the histidine tail by thrombin digestion, essentially as described previously (Tek and Zolkiewski 2002). The cleaved protein was further purified by size-exclusion HPLC (Superdex 200, Pharmacia) in 50 mM Tris (pH 7.5), 200 mM KCl.
Refolding experiments
Refolding experiments for firefly (Photinus pyralis) luciferase (Promega) and mitochondrial MDH (Sigma) were performed as described previously. Briefly, 6.1 μM denatured luciferase in 5 M GdnHCl was diluted 100-fold in refolding buffer (100 mM potassium phosphate [pH 7.8], 1 mM EDTA, 1 mM DTE) at 5°C (Herbst et al. 1998), and 140 μM (monomer concentration) denatured MDH in 3 M GdnHCl was diluted 100-fold in refolding buffer (50 mM TEA [pH 7.5], 50 mM KCl, 20 mM MgCl2, 2 Mm DTT) at 30°C (Ranson et al. 1995). The refolding yield was estimated from the enzymatic activity. Numerical analysis of kinetic data was performed with the Gepasi software program (Mendes 1997).
Spectroscopic measurements
Fluorescence anisotropy measurements were made with a Shimadzu RF2000 spectrofluorimeter (Kyoto, Japan) with an excitation wavelength of 490 nm and an emission wavelength of 520 nm. The ClpB N-terminal amino group was labeled with fluorescein isothiocyanate (FITC) for 4 h at pH 8.0, and excess FITC was removed using a PD10 column (Amersham Pharmacia). The final protein concentration was determined with the BCA protein assay and the concentration of FITC was determined by absorbance at 490 nm (68,000 M−1 cm−1). The labeling ratio for fluorescent derivatives of RCMLA was confirmed to be 1.0. Anisotropy values of 0.1 μM FITC-ClpBN were measured at 30-sec intervals during MDH refolding. CD measurements were made with a Jasco-720 spectropolarimeter using a 1-mm path length cell.
DSC
Calorimetric measurements were performed by Nano-DSC II Model 6100 (Calorimetry Science Co.). Most experiments were done at a scan rate of 2.0°C/min and protein concentrations of 0.7–1.5 mg/mL. All data analysis of baseline subtraction, concentration normalization, and deconvolution was performed with software from Calorimetry Science.
Acknowledgments
We thank Dr. C. Squires, Prof. M. Zolkiewski for providing the expression vectors for ClpB and its mutants, and Mr. T. Kuroita (Toyobo Co., Ltd.) for technical assistance. Financial support for this study was provided by the Asahi Glass Foundation.
Abbreviations
ClpBN, N-terminal domain of ClpB
ClpBΔN, ClpB protein with a deleted N-terminal domain
MDH, malate dehydrogenase
GdnHCl, guanidine hydrochloride
Kd, dissociation constant
Article published online ahead of print. Article and publication date are at http://www.proteinscience.org/cgi/doi/10.1110/ps.04780704.
References
- Ayling, A. and Baneyx, F. 1996. Influence of the GroE molecular chaperone machine on the in vitro refolding of Escherichia coli β-galactosidase. Protein Sci. 5 478–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnett, M.E., Zolkiewska, A., and Zolkiewski, M. 2000. Structure and activity of ClpB from Escherichia coli. Role of the amino- and carboxyl-terminal domains. J. Biol. Chem. 275 37565–37571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beinker, P., Schlee, S., Groemping, Y., Seidel, R., and Reinstein, J. 2002. The N terminus of ClpB from Thermus thermophilus is not essential for the chaperone activity. J. Biol. Chem. 277 47160–47166. [DOI] [PubMed] [Google Scholar]
- Ben-Zvi, A.P., Chatellier, J., Fersht, A.R., and Goloubinoff, P. 1998. Minimal and optimal mechanisms for GroE-mediated protein folding. Proc. Natl. Acad. Sci. 95 15275–15280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchner, J., Schmidt, M., Fuchs, M., Jaenick, R., Rudolph, R., Schmid, F.X., and Kiefhaber, T. 1991. GroE facilitates refolding of citrate synthase by suppressing aggregation. Biochemistry 30 1586–1591. [DOI] [PubMed] [Google Scholar]
- Cashikar, A.G., Schirmer, E.C., Hattendorf, D.A., Glover, J.R., Ramakrishnan, M.S., Ware, D.M., and Lindquist, S.L. 2002. Defining a pathway of communication from the C-terminal peptide binding domain to the N-terminal ATPase domain in a AAA protein. Mol. Cell 9 751–760. [DOI] [PubMed] [Google Scholar]
- Chatellier, J., Hill, F., Lund, P.A., and Fersht, A.R. 1998. In vivo activities of GroEL minichaperones. Proc. Natl. Acad. Sci. 94 3576–3578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke, A.K. and Eriksson, M.-J. 2000. The truncated form of the bacterial heat shock protein ClpB/HSP100 contributes to development of thermotolerance in the cyanobacterium Synechococcus sp. strain PCC 7942. J. Bacteriol. 182 7092–7096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleland, J.L. and Randolph, T.W. 1992. Mechanism of polyethylene golycol interaction with the molten globule folding intermediate of bovine carbonic anhydrase B. J. Biol. Chem. 267 3147–3153. [PubMed] [Google Scholar]
- Eriksson, M.-J., Schelin, J., Miskiewicz, E., and Clarke, A.K. 2001. Novel form of ClpB/HSP100 protein in the cyanobacterium Synechococcus. J. Bacteriol. 183 7392–7396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glover, J.R. and Lindquist, S. 1998. Hsp104, Hsp70, and Hsp40: A novel chaperone system that rescues previously aggregated proteins. Cell 94 73–82. [DOI] [PubMed] [Google Scholar]
- Goloubinoff, P., Mogk, A., Ben-Zvi, A.P., Tomoyasu, T., and Bukau, B. 1999. Sequential mechanism of solubilization and refolding of stable protein aggregates by a bichaperone network. Proc. Natl. Acad. Sci. 96 13732–13737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbst, R., Schafer, U., and Seckler, R. 1997. Equilibrium intermediates in the reversible unfolding of firefly (Photinus pyralis) luciferase. J. Biol. Chem. 272 7099–7105. [DOI] [PubMed] [Google Scholar]
- Herbst, R., Gast, K., and Seckler, R. 1998. Folding of firefly (Photinus pyralis) luciferase: Aggregation and reactivation of unfolding intermediates. Biochemistry 37 6586–6597. [DOI] [PubMed] [Google Scholar]
- Janicke, R. and Rudolph, R. 1986. Refolding and association of lolgomeric proteins. Methods Enzymol. 131 218–250. [DOI] [PubMed] [Google Scholar]
- Jung, G. and Masison, D.C. 2001. Guanidine hydrochloride inhibits Hsp104 activity in vivo: A possible explanation for its effect in curing yeast prions. Curr. Microbiol. 43 7–10. [DOI] [PubMed] [Google Scholar]
- Kedzierska, S., Akoev, V., Barnett, M.E., and Zolkiewski, M. 2003. Structure and function of the middle domain of ClpB from Escherichia coli. Biochemistry 42 14242–14248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, K.I., Cheong, G.-W., Park, S.-C., Ha, J.-S., Woo, K.M., Choi, S.J., and Chung, C.H. 2000. Heptameric ring structure of the heat-shock protein ClpB, a protein-activated ATPase in Escherichia coli. J. Mol. Biol. 303 655–666. [DOI] [PubMed] [Google Scholar]
- Kraulis, J. 1991. MOLSCRIPT: A program to produce both detailed and schematic plots of protein structures. J. Appl. Crystallogr. 24 946–950. [Google Scholar]
- Lee, J.Y. and Hirose, M. 1992. Partially folded state of the disulfide-reduced form of human serum albumin as an intermediate for reversible denaturation. J. Biol. Chem. 267 14753–14758. [PubMed] [Google Scholar]
- Lee, S., Sowa, M.E., Watanabe, Y., Sigler, P.B., Chiu, W., Yoshida, M., and Tsai, F.T.F. 2003. The structure of ClpB: A molecular chaperone that rescues proteins from an aggregated state. Cell 115 229–240. [DOI] [PubMed] [Google Scholar]
- Li, J. and Sha, B. 2003. Crystal structure of the E. coli Hsp100 ClpB N-terminal domain. Structure 11 323–328. [DOI] [PubMed] [Google Scholar]
- Liu, Z., Tek, V., Akoev, V., and Zolkiewski, M. 2002. Conserved amino acid residues within the amino-terminal domain of ClpB are essential for the chaperone activity. J. Mol. Biol. 321 111–120. [DOI] [PubMed] [Google Scholar]
- Lo, H., Baker, T.A., and Sauer, R.T. 2001. Characterization of the N-terminal repeat domain of Escherichia coli ClpA—A class I Clp/HSP100 ATPase. Protein Sci. 10 551–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendes, P. 1997. Biochemistry by numbers: Simulation of biochemical pathways with Gepasi 3. Trends Biochem. Sci. 22 361–363. [DOI] [PubMed] [Google Scholar]
- Merritt, E.A. and Bacon, D.J. 1997. Raster3D: Photorealistic molecular graphics. Method Enzymol. 277 505–524. [DOI] [PubMed] [Google Scholar]
- Mogk, A., Schlieker, C., Strub, C., Rist, W., Weibezahn, J., and Bukau, B. 2003. Roles of individual domains and conserved motifs of the AAA+ chaperone ClpB in oligomerization, ATP hydrolysis, and chaperone activity. J. Biol. Chem. 278 17615–17624. [DOI] [PubMed] [Google Scholar]
- Ranson, N.A., Dunster, N.J., Burston, S.G., and Clarke, A.R. 1995. Chaperonins can catalyse the reversal of early aggregation steps when a protein misfolds. J. Mol. Biol. 250 581–586. [DOI] [PubMed] [Google Scholar]
- Schmidt, M. and Buchner, J. 1992. Interaction of GroE with an all-β-protein. J. Biol. Chem. 267 16829–16833. [PubMed] [Google Scholar]
- Squires, C.L., Pedersen, S., Ross, B.M., and Squires, C. 1991. ClpB is the Escherichia coli heat shock protein F84.1. J. Bacteriol. 173 4257–4262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka, N. and Fersht, A.R. 1999. Identification of substrate binding site of GroEL minichaperone in solution. J. Mol. Biol. 292 173–180. [DOI] [PubMed] [Google Scholar]
- Tanaka, N., Nakao, S., Wadai, D. Ikeda, S., Chatellier, J., and Kunugi, S. 2002. The substrate binding domain of DnaK facilitates slow protein refolding. Proc. Natl. Acad. Sci. 99 15398–15403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tek, V. and Zolkiewski, M. 2002. Stability and interactions of the amino-terminal domain of ClpB from Escherichia coli. Protein Sci. 11 1192–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zahn, R., Buckle, A.M., Perrett, S., Johnson, C.M., Corrales, F.J., Golbik, R., and Fersht, A.R. 1996. Chaperone activity and structure of monomeric polypeptide binding domains of GroEL. Proc. Natl. Acad. Sci. 93 15024–15029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zolkiewski, M. 1999. ClpB cooperates with DnaK, DnaJ, and GrpE in suppressing protein aggregation. A novel multi-chaperone system from Escherichia coli. J. Biol. Chem. 274 28083–28086. [DOI] [PubMed] [Google Scholar]
- Zolkiewski, M., Kessel, M., Ginsburg, S., and Maurizi, M.R. 1999. Nucleotide-dependent oligomerization of ClpB from Escherichia coli. Protein Sci. 8 1899–1903. [DOI] [PMC free article] [PubMed] [Google Scholar]